

Abstract
The frequently reported amino acid covariation of the highly polymorphic human immunodeficiency virus type 1 (HIV-1) exterior envelope glycoprotein V3 region has been assumed to reflect fitness epistasis between residues. However, nonrandom association of amino acids, or linkage disequilibrium, has many possible causes, including population subdivision. If the amino acids at a set of sequence sites differ in frequencies between subpopulations, then analysis of the whole population may reveal linkage disequilibrium even if it does not exist in any subpopulation. HIV-1 has a complex population structure, and the effects of this structure on linkage disequilibrium were investigated by estimating within- and among-subpopulation components of variance in linkage disequilibrium. The amino acid covariation previously reported is explained by differences in amino acid frequencies among virus subpopulations in different patients and by nonsystematic disequilibrium among patients. Disequilibrium within patients appears to be entirely due to differences in amino acid frequencies among sampling time points and among chemokine coreceptor usage phenotypes of virus particles, but not source tissues. Positive selection explains differences in allele frequencies among time points and phenotypes, indicating that these differences are adaptive rather than due to genetic drift. However, the absence of a correlation between linkage disequilibrium and phenotype suggests that fitness epistasis is an unlikely cause of disequilibrium. Indeed, when population structure is removed by analyzing sequences from a single time point and phenotype, no disequilibrium is detectable within patients. These results caution against interpreting amino acid covariation and coevolution as evidence for fitness epistasis.
LINKAGE disequilibrium refers to the nonrandom association of alleles among loci or the nonrandom association of residues among molecular sequence sites. The departure of alleles from random association is of considerable interest because it reflects important population genetic processes (reviewed by Slatkin 2008) and may have important consequences for the efficiency of natural selection and the evolution of recombination (Felsenstein 1988; Kondrashov 1993). But, although linkage disequilibrium is easy to measure, ascertaining its causes is not. Disequilibrium may be generated by interactions among alleles at different loci in their effects on fitness, known as fitness epistasis (e.g., Kimura 1956; Lewontin and Kojima 1960; Felsenstein 1965; Karlin and Feldman 1970). Genetic drift may also cause disequilibrium simply because sampling a finite number of haplotypes will generate nonrandom associations (Hill and Robertson 1968; Ohta and Kimura 1969; Hudson 1985; Slatkin 1994). Similarly, population bottlenecks may create disequilibrium because of the chance loss of some haplotypes. Other forces, such as inbreeding, genomic inversions, and gene conversion, may also generate disequilibrium (see Slatkin 2008). Finally, population subdivision may produce linkage disequilibrium if subpopulations differ in allele frequencies. In this situation, even if subpopulations exhibit linkage equilibrium, disequilibrium may be evident at the whole population level (Mitton and Koehn 1973; Nei and Li 1973). In the extreme case, if the alleles fixed at a set of loci differ between two subpopulations, neither subpopulation will exhibit disequilibrium, but the alleles will be seen to be in disequilibrium at the whole population level. Additionally, if there is gene flow between such subpopulations, then disequilibrium will also be evident within subpopulations (Li and Nei 1974; Slatkin 1975).
The first step in determining the causes of linkage disequilibrium is to test for the effects of population subdivision (Slatkin 2008). If population subdivision can be ruled out, or is a minor contributor, then other forces such as epistasis or genetic drift may be considered. Ohta (1982) describes a method of partitioning the total variance in linkage disequilibrium into within- and among-subpopulation components that is analogous to Wright's (1940) measures of population subdivision for single loci, FIS and FST. This method is commonly used to determine how much of disequilibrium is attributable to population structure (Slatkin 2008).
Considerable linkage disequilibrium, or covariation, among amino acids has been reported for a number of human immunodeficiency virus type 1 (HIV-1) proteins encoded by the gag, nef, tat, and pol genes (Hoffman et al. 2003; Rhee et al. 2007; Wang and Lee 2007; Liu et al. 2008; Myers and Pillay 2008). However, disproportionate attention has been focused on the third variable region (V3) of the exterior envelope glycoprotein, gp120, encoded by the env gene (Korber et al. 1993; Bickel et al. 1996; Gilbert et al. 2005; Poon et al. 2007; Travers et al. 2007). V3 has been a focus of attention because it is the main determinant of which cell types are infected by HIV-1 (Hwang et al. 1991) and because it is the primary target for neutralizing antibodies (Zolla-Pazner 2004). The motivation for these studies has been the discovery of functional interactions among residues that may aid in vaccine development, thereby explicitly or implicitly assuming that the observed covariation is due to fitness epistasis. However, HIV-1 has a complex population structure, which may contribute to the observed linkage disequilibrium.
The basic population unit of HIV-1 is the virus population within a patient. These populations are themselves structured geographically into major clades, called “subtypes,” which are nested within “groups” (Gao et al. 1999). Within patients, the virus population may be subdivided among host tissues and among foci of infection within host organs (e.g., Wong et al. 1997; Frost et al. 2001). In addition, because of the rapid evolution of HIV-1, the viral population within a patient may also be structured temporally, with DNA sequences sampled at intervals of months or years often exhibiting significantly different site-specific frequencies (e.g., Bonhoeffer et al. 1995; Wolinsky et al. 1996; Shankarappa et al. 1999).
The virus population within a patient may also be subdivided among host cell types. An HIV-1 particle (virion) enters a cell through interactions between gp120 on the virion surface and two cell-surface receptors: the CD4 receptor and one of two chemokine coreceptors, either CCR5 or CXCR4 (reviewed by Wyatt and Sodroski 1998). Binding to CD4 causes conformational changes to gp120 that expose V3 for coreceptor binding (Huang et al. 2005, 2007). And since target cell types vary in their expression of chemokine coreceptors, macrophages expressing predominantly CCR5 and T cells expressing predominantly CXCR4, the coreceptor bound by V3 determines the type of cell infected. V3 determines which coreceptor is bound (Dittmar et al. 1997; Speck et al. 1997) through the amino acid composition of the crown, or tip, of the V3 structure (Cormier and Dragic 2002). Therefore, the virus population infecting a patient may be subdivided among host cell types on the basis of the coreceptor usage phenotype imparted by V3.
Studies of V3 amino acid covariation have invariably used only one or a few sequences from each of many patients to deal with the statistical nonindependence of multiple sequences from the same patient. Therefore, these studies have not been designed to rule out the possibility that the linkage disequilibrium observed is caused by population subdivision among and within patients. Some of these studies have also attempted to control for the lack of independence among the viral sequences from different patients due to phylogenetic relationships caused by transmission histories (Poon et al. 2007; Travers et al. 2007). However, because the phylogenetic methods employed assume the independent evolution of sequence sites and do not take into account the substantial recombination in HIV-1 (Levy et al. 2004), these approaches are unlikely to be valid. Here, I have investigated the effects of population subdivision on V3 amino acid covariation by estimating components of variance in linkage disequilibrium. I show that the majority of the disequilibrium observed at the global population level is due to differences in amino acid frequencies among patients. These differences among patients are, in turn, due mainly to differences in amino acid frequencies among time points and coreceptor usage phenotypes within patients. In addition, none of the disequilibrium appears to be associated with coreceptor usage phenotype, suggesting that fitness epistasis is not a cause of disequilibrium. These results caution against interpreting residue covariation or coevolution as evidence for fitness epistasis.
METHODS
Sequence data set:
Analyses were restricted to HIV-1 subtype B, the most commonly sequenced subtype and the main subject of previous studies of V3 amino acid covariation. Sequences were downloaded from the HIV Sequence Database (www.hiv.lanl.gov). The criteria for inclusion were that the sequences (1) were from an identified patient in the database (with a “patient ID”), (2) had the typical V3 length of 35 amino acids, (3) had cysteines at both termini (these are absolutely conserved in functional V3), and (4) did not contain undetermined residues. A small minority of the resulting sequences (0.5%) could not be aligned with the remaining sequences without the addition of alignment gaps; these sequences were removed to avoid ambiguous alignments. On October 30, 2008, these criteria resulted in 35,883 sequences from 3297 patients. For the purpose of comparison with previous studies, only one sequence per patient was used to identify linkage disequilibrium in the global population. Analyses of the effects of population subdivision among and within patients, which required at least 20 sequences per patient, involved 63 different patients. Sequences aligned unambiguously and did not require alignment gaps; this was confirmed by eye and by the automatic sequence alignment program MUSCLE (Edgar 2004).
Measuring and testing linkage disequilibrium:
Linkage disequilibrium was measured in the usual manner, with the coefficient of linkage disequilibrium
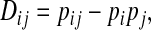 |
(1) |
where pij is the observed frequency of sequences containing the amino acids Ai and Bj at sites A and B (the haplotype or gametic frequency), and pi and pj are the observed frequencies of these amino acids at the individual sites (Weir 1996). Dij may be interpreted as the deviation of the haplotype frequency from its expected frequency under linkage equilibrium. The statistical significance of linkage disequilibrium at a pair of sequence sites was determined with a chi-square test for multiple alleles at each site,
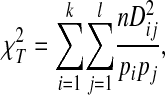 |
(2) |
where k and l are the numbers of amino acids at each site and n is the number of sequences (sample size) (Weir 1996). The degrees of freedom for this test are (k − 1)(l − 1). To control for inflation of the type 1 error rate, α, due to testing multiple pairs of polymorphic sites, the familywise error rate, α/c, where c is the number of tests, was used as the level of significance (Weir 1996). Comparisons were made between all possible pairs of polymorphic V3 amino acid sites. Since the V3 sequences analyzed are 35 amino acids long and the two terminal amino acid sites are absolutely conserved, there were a maximum of 33(32)/2 = 528 possible pairs of polymorphic sites. Tests were made even more conservative by using α = 0.001. These tests are sensitive to alleles with low frequencies, producing spurious significant results (Weir and Hill 1986; Awadalla et al. 1999). Therefore, tests were restricted to amino acids with a minimum site-specific frequency of 10%, as in Awadalla et al. (1999). Previous studies of V3 amino acid covariation have reported detecting unrealistically high numbers of significant covariations (Korber et al. 1993; Bickel et al. 1996), possibly because of this effect.
Variance components of linkage disequilibrium:
Ohta (1982) describes a commonly used method to partition the total variance in linkage disequilibrium into within- and among-subpopulation components. These variance components are analogous to Wright's (1940) measures of population subdivision for single loci, FIS and FST:
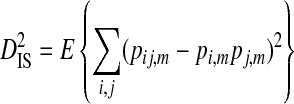 |
(3) |
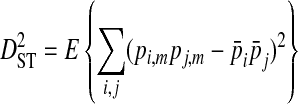 |
(4) |
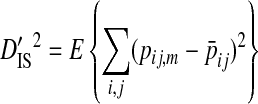 |
(5) |
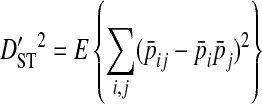 |
(6) |
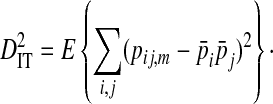 |
(7) |
In these equations, pij,m, pi,m, and pj,m are the haplotype and site-specific frequencies of amino acids Ai and Bj at sites A and B in subpopulation m, 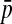 is the mean across subpopulations weighted by sample size, summation is taken over all i and j, and the expectation, E, is the weighted average of the sum of squared deviations across subpopulations (Whittam et al. 1983).
is the mean across subpopulations weighted by sample size, summation is taken over all i and j, and the expectation, E, is the weighted average of the sum of squared deviations across subpopulations (Whittam et al. 1983).
The deviation term in 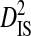 , (pij,m − pi,mpj,m), is the coefficient of linkage disequilibrium for a pair of amino acids within a subpopulation, and therefore
, (pij,m − pi,mpj,m), is the coefficient of linkage disequilibrium for a pair of amino acids within a subpopulation, and therefore 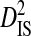 is the within-subpopulation component of variance in linkage disequilibrium. The deviation term in
is the within-subpopulation component of variance in linkage disequilibrium. The deviation term in 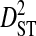 is the deviation of the product of amino acid frequencies within a subpopulation relative to the product of frequencies for the whole population.
is the deviation of the product of amino acid frequencies within a subpopulation relative to the product of frequencies for the whole population. 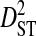 is therefore the among-subpopulation component of variance in linkage disequilibrium and represents the variance due to differences in amino acid frequencies among subpopulations.
is therefore the among-subpopulation component of variance in linkage disequilibrium and represents the variance due to differences in amino acid frequencies among subpopulations. 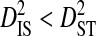 indicates that some of the linkage disequilibrium observed in the whole population is due to differences in amino acid frequencies among subpopulations, as opposed to being due simply to disequilibrium within subpopulations, in which case
indicates that some of the linkage disequilibrium observed in the whole population is due to differences in amino acid frequencies among subpopulations, as opposed to being due simply to disequilibrium within subpopulations, in which case 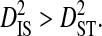
The deviation term in 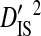 is the deviation of the haplotype frequency within a subpopulation relative to that of the whole population, and as such
is the deviation of the haplotype frequency within a subpopulation relative to that of the whole population, and as such 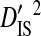 represents the variance due to differences in haplotype frequencies among subpopulations. The deviation term in
represents the variance due to differences in haplotype frequencies among subpopulations. The deviation term in 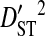 is the coefficient of linkage disequilibrium for a pair of amino acids for the whole population, and
is the coefficient of linkage disequilibrium for a pair of amino acids for the whole population, and 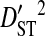 is therefore the variance in linkage disequilibrium for the whole population. And the deviation term in
is therefore the variance in linkage disequilibrium for the whole population. And the deviation term in 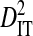 is the deviation of the haplotype frequency in a subpopulation from its expected frequency based on the amino acids frequencies in the whole population, and, as such,
is the deviation of the haplotype frequency in a subpopulation from its expected frequency based on the amino acids frequencies in the whole population, and, as such, 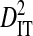 represents the total variance in linkage disequilibrium. Note that
represents the total variance in linkage disequilibrium. Note that 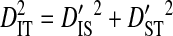 , but that
, but that 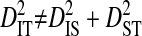 (Ohta 1982).
(Ohta 1982). 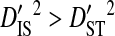 indicates that the disequilibrium within subpopulations is nonsystematic among subpopulations, whereas
indicates that the disequilibrium within subpopulations is nonsystematic among subpopulations, whereas 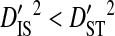 indicates that the disequilibrium is systematic. Nonsystematic disequilibrium means that the disequilibrium within subpopulations differs among subpopulations. Note, however, that that there need not be disequilibrium within subpopulations to generate
indicates that the disequilibrium is systematic. Nonsystematic disequilibrium means that the disequilibrium within subpopulations differs among subpopulations. Note, however, that that there need not be disequilibrium within subpopulations to generate 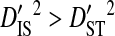 , since differences in amino acid frequencies among subpopulations may also cause differences in haplotype frequencies that produce this inequality. If subpopulations are identical, in the sense that they occupy identical environments, nonsystematic disequilibrium indicates that genetic drift within and among subpopulations is the cause of the disequilibrium, whereas systematic disequilibrium indicates adaptation involving epistasis as the cause (Ohta 1982). However, if subpopulations are not identical, because they occupy different environments, then nonsystematic disequilibrium may indicate either genetic drift or epistatic adaptation to local environments as the cause of the disequilibrium. Systematic disequilibrium, in this case, would indicate epistatic adaptation to the global environment, but not to local environments, as the cause of the disequilibrium (Table 1).
, since differences in amino acid frequencies among subpopulations may also cause differences in haplotype frequencies that produce this inequality. If subpopulations are identical, in the sense that they occupy identical environments, nonsystematic disequilibrium indicates that genetic drift within and among subpopulations is the cause of the disequilibrium, whereas systematic disequilibrium indicates adaptation involving epistasis as the cause (Ohta 1982). However, if subpopulations are not identical, because they occupy different environments, then nonsystematic disequilibrium may indicate either genetic drift or epistatic adaptation to local environments as the cause of the disequilibrium. Systematic disequilibrium, in this case, would indicate epistatic adaptation to the global environment, but not to local environments, as the cause of the disequilibrium (Table 1).
TABLE 1.
Causes of linkage disequilibrium based on whether disequilibrium within subpopulations is systematic or nonsystematic among identical or different subpopulations, as determined by the linkage disequilibrium variance components D′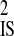 and D′
and D′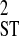
| Linkage disequilibrium
|
||
|---|---|---|
| Subpopulations | Systematic 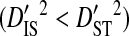
|
Nonsystematic 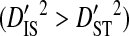
|
| Identical | Local epistasis | Genetic drift |
| Different | Global epistasis | Genetic drift or local epistasis |
Testing for positive selection:
Positive selection was detected by testing whether the mean nonsynonymous nucleotide distance (dN) exceeds the mean synonymous distance (dS) in pairwise sequence comparisons (Nei and Kumar 2000). Distances were estimated using the modified Nei–Gojobori method with the Jukes–Cantor model of nucleotide evolution and a nucleotide transition-to-transversion ratio of 2 (estimated using the Kimura two-parameter model of nucleotide evolution). Standard errors (SE) of distances were estimated using 500 bootstrap samples of the data. Distances were calculated between groups of sequences, such as between the sequences belonging to different chemokine coreceptor usage phenotypes. Statistical significance was determined using the Z-test. Analyses were carried out using the computer application MEGA 4.0 (Tamura et al. 2007). Phylogeny-based methods of testing for positive selection were not used because they are not appropriate for these data; the high rate of recombination in HIV-1 cannot be accommodated by phylogeny reconstruction methods and results in a high rate of false positives (Lemey et al. 2006).
RESULTS
Population subdivision among patients:
V3 is highly polymorphic (Figure 1). Using one sequence from each of the 3297 patients in the data set, statistically significant linkage disequilibrium was detected for 10 pairs of sites that were also identified when analyzing all sequences from 51 patients, each with a minimum of 100 sequences sampled (8600 sequences in total) (Table 2). Covariation between amino acids at these sites has been commonly reported (Korber et al. 1993; Bickel et al. 1996; Gilbert et al. 2005; Poon et al. 2007; Travers et al. 2007). These sites include three sites (11, 13, and 25) that are among the most polymorphic and that have been implicated in determining chemokine coreceptor usage (de Jong et al. 1992; Fouchier et al. 1992; Hung et al. 1999; Pastore et al. 2006).
Figure 1.—

V3 polymorphism. Amino acids with a minimum site-specific frequency of 1% are shown in order of decreasing frequency (top to bottom). Frequencies were calculated from a data set containing one sequence from each of 3297 patients.
TABLE 2.
Linkage disequilibrium variance components within and among patients
| Sites | 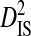 |
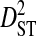 |
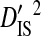 |
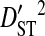 |
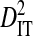 |
|---|---|---|---|---|---|
| 10, 32 | 0.00704 | 0.32857 | 0.33299 | 0.00144 | 0.33443 |
| 11, 13 | 0.00664 | 0.35998 | 0.36786 | 0.00506 | 0.37292 |
| 11, 25 | 0.00571 | 0.36850 | 0.37848 | 0.00109 | 0.37957 |
| 13, 14 | 0.00492 | 0.33667 | 0.32644 | 0.01206 | 0.33850 |
| 13, 25 | 0.00925 | 0.33989 | 0.35016 | 0.00342 | 0.35358 |
| 14, 22 | 0.00291 | 0.43311 | 0.42846 | 0.00548 | 0.43394 |
| 14, 25 | 0.00427 | 0.37203 | 0.38222 | 0.00221 | 0.38443 |
| 22, 25 | 0.00819 | 0.45563 | 0.46276 | 0.00458 | 0.46735 |
| 29, 32 | 0.00349 | 0.27981 | 0.28444 | 0.00264 | 0.28708 |
| 32, 34 | 0.00858 | 0.32199 | 0.32142 | 0.00075 | 0.32217 |
Sites shown are those with statistically significant linkage disequilibrium in both a data set containing one sequence from each of 3297 patients and a data set containing at least 100 sequences from each of 51 patients (8600 sequences in total). Variance components were estimated from the 51-patients data set.
Using the 51-patients data set, statistically significant linkage disequilibrium was detected for 48 pairs of sites. Linkage disequilibrium variance components were estimated for these data with the sequences from each patient identified as a separate subpopulation. Variance components for these site pairs show consistently 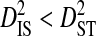 , with mean
, with mean 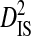 (0.00438) nearly two orders of magnitude lower than mean
(0.00438) nearly two orders of magnitude lower than mean 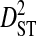 (0.34336). This indicates that, for every pair of sites, the linkage disequilibrium detected for the whole population is due overwhelmingly to differences in site-specific amino acid frequencies among the virus subpopulations infecting patients.
(0.34336). This indicates that, for every pair of sites, the linkage disequilibrium detected for the whole population is due overwhelmingly to differences in site-specific amino acid frequencies among the virus subpopulations infecting patients. 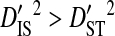 consistently among pairs of sites as well, with mean
consistently among pairs of sites as well, with mean 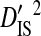 (0.34654) more than two orders of magnitude higher than mean
(0.34654) more than two orders of magnitude higher than mean 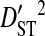 (0.00274) and equal to 99% of the mean total variance in linkage disequilibrium,
(0.00274) and equal to 99% of the mean total variance in linkage disequilibrium, 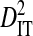 (0.34928). This shows that the disequilibrium within patients is mainly nonsystematic among patients. Nonsystematic disequilibrium is also evident from the lack of overlap among patients in site pairs with significant disequilibrium (data not shown). Table 2 shows variance components for the 10 site pairs with significant disequilibrium also detected when analyzing the data set consisting of one sequence from each of 3297 patients.
(0.34928). This shows that the disequilibrium within patients is mainly nonsystematic among patients. Nonsystematic disequilibrium is also evident from the lack of overlap among patients in site pairs with significant disequilibrium (data not shown). Table 2 shows variance components for the 10 site pairs with significant disequilibrium also detected when analyzing the data set consisting of one sequence from each of 3297 patients.
Therefore, the linkage disequilibrium observed for the entire subtype B population is explained by differences in amino acid frequencies among patients and nonsystematic disequilibrium among patients. However, nonsystematic disequilibrium among patients cannot automatically be attributed to genetic drift because patients are not identical environments (Table 1). Patients differ in various aspects of their immune systems and in the tissue sources, sampling times (relative to initial infection), and chemokine coreceptor usage phenotypes of their sampled V3 sequences. Nonsystematic linkage disequilibrium among patients could arise from further population subdivision within patients among tissues, sampling times, and coreceptor usage phenotypes.
Population subdivision among source tissues within patients:
To test the effect of population subdivision among source tissues within patients, sequences from 7 patients were analyzed. Each of these patients had at least 30 sequences sampled from each of two distinct tissues (no patient had 30 sequences sampled from each of more than two distinct tissues). Three of these patients are from the 51-patient data set used to test the effect of population subdivision among patients. Tissue sources labeled “blood,” “plasma,” peripheral blood mononuclear cells (“PBMC”), and “serum” in the HIV-1 sequence database were grouped into the single tissue category, blood. And tissue sources labeled “semen,” “seminal cells,” and “seminal plasma” were grouped into the single category, semen. There is low total variance in linkage disequilibrium (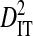 ) in 5 of the 7 patients (Table 3). Each of these 5 patients had sequences sampled from blood and either semen or lymph node. For 1 of these patients, no site pairs exhibited statistically significant disequilibrium. This is consistent with the low variance within patients when patients were analyzed as subpopulations in the 51-patients data set (
) in 5 of the 7 patients (Table 3). Each of these 5 patients had sequences sampled from blood and either semen or lymph node. For 1 of these patients, no site pairs exhibited statistically significant disequilibrium. This is consistent with the low variance within patients when patients were analyzed as subpopulations in the 51-patients data set (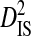 ; Table 2). The remaining 2 patients, which had samples taken from blood and cerebral spinal fluid, exhibited considerable total variance in disequilibrium, at levels similar to the total variance in the 51-patients data set (compare
; Table 2). The remaining 2 patients, which had samples taken from blood and cerebral spinal fluid, exhibited considerable total variance in disequilibrium, at levels similar to the total variance in the 51-patients data set (compare 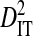 between Tables 2 and 3). Nevertheless, for all patients
between Tables 2 and 3). Nevertheless, for all patients 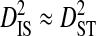 and
and 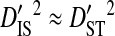 , indicating that population subdivision among tissues contributes little to the linkage disequilibrium of the total population infecting a patient.
, indicating that population subdivision among tissues contributes little to the linkage disequilibrium of the total population infecting a patient.
TABLE 3.
Linkage disequilibrium variance components within and among source tissues within patients
|
N
|
Mean
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Patient ID | Blood | Semen | Lymph node | CSFa | Site pairs | 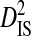 |
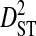 |
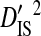 |
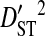 |
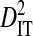 |
| 10139351 | 268 | 92 | 0 | 0 | 2 | 0.02405 | 0.00840 | 0.00827 | 0.01870 | 0.02698 |
| 10144196 | 64 | 34 | 0 | 0 | 23 | 0.07584 | 0.01198 | 0.01593 | 0.07119 | 0.08712 |
| 10150807 | 40 | 42 | 0 | 0 | 0 | — | — | — | — | — |
| 10149482 | 292 | 0 | 30 | 0 | 2 | 0.06463 | 0.05190 | 0.05449 | 0.07280 | 0.12729 |
| 10149484 | 219 | 0 | 32 | 0 | 4 | 0.02059 | 0.06076 | 0.06256 | 0.02304 | 0.08561 |
| 10149719 | 34 | 0 | 0 | 34 | 15 | 0.11742 | 0.05943 | 0.05494 | 0.15412 | 0.20906 |
| 10149720 | 33 | 0 | 0 | 32 | 15 | 0.08597 | 0.15845 | 0.13178 | 0.17020 | 0.30198 |
The numbers of sequences from each tissue and of site pairs with statistically significant linkage disequilibrium are shown (N). Variance components are means across site pairs.
Cerebral spinal fluid.
Population subdivision among sampling times within patients:
To test the effect of population subdivision due to sampling times within patients, data from 5 patients were analyzed. Each of these patients had ≥50 sequences sampled in each of ≥2 years. Three of these patients are from the 51-patient data set, and 2 are from the data set used to test for an effect of tissue source. The total variance in linkage disequilibrium (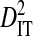 ) was moderate to high for 3 patients and undefined for the 2 patients with no statistically significant disequilibrium (Table 4). The total variance does not appear to be related to the total number of years between samples. However,
) was moderate to high for 3 patients and undefined for the 2 patients with no statistically significant disequilibrium (Table 4). The total variance does not appear to be related to the total number of years between samples. However, 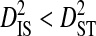 and
and 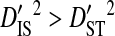 consistently for all significant site pairs. These inequalities are modest in 2 of the patients. For the patient with the highest total variance, these inequalities are larger, indicating that a substantial amount of the variance in disequilibrium within patients may be due to changes in allele frequencies over time and that the disequilibrium within time points is mostly nonsystematic among time points. Tests for positive selection between the first and last time point sample for each patient show that the mean nonsynonymous nucleotide distance (dN) is significantly greater than the synonymous distance (dS) for the patient with the highest total variance only (Table 4). This indicates that differences in allele frequencies between time points are likely caused by positive selection.
consistently for all significant site pairs. These inequalities are modest in 2 of the patients. For the patient with the highest total variance, these inequalities are larger, indicating that a substantial amount of the variance in disequilibrium within patients may be due to changes in allele frequencies over time and that the disequilibrium within time points is mostly nonsystematic among time points. Tests for positive selection between the first and last time point sample for each patient show that the mean nonsynonymous nucleotide distance (dN) is significantly greater than the synonymous distance (dS) for the patient with the highest total variance only (Table 4). This indicates that differences in allele frequencies between time points are likely caused by positive selection.
TABLE 4.
Linkage disequilibrium variance components within and among time point samples within patients
|
N
|
Mean
|
||||||||
|---|---|---|---|---|---|---|---|---|---|
| Patient ID | Samples | Yr | Sequences | Site pairs | 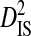 |
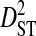 |
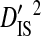 |
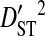 |
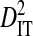 |
| 10149483 | 2 | 1 | 154 | 0 | — | — | — | — | — |
| 10149484 | 2 | 1 | 158 | 4 | 0.02712 | 0.13702 | 0.15073 | 0.02765 | 0.17838 |
| 10149482 | 3 | 4 | 306 | 3 | 0.04680 | 0.09851 | 0.11768 | 0.04139 | 0.15907 |
| 10160923** | 2 | 4 | 105 | 29 | 0.00841 | 0.55047 | 0.39583 | 0.17540 | 0.57123 |
| 10160924 | 2 | 4 | 113 | 0 | — | — | — | — | — |
The numbers of samples, of years between first and last samples, of total sequences sampled, and of site pairs with statistically significant linkage disequilibrium are shown (N). Variance components are means across site pairs. **dN (SE) = 0.1648 (0.0349); dS (SE) = 0.0055 (0.0021); H0, dN = dS; Z = 3.21; P < 0.01.
Population subdivision among phenotypes within patients:
Linkage disequilibrium within patients may also be caused by population subdivision among chemokine coreceptor usage phenotypes. Virions may use CCR5 exclusively (R5 phenotype), CXCR4 exclusively (X4 phenotype), or use both coreceptors (R5X4 phenotype). Because coreceptor use determines the target cells that may be infected, these phenotypes may represent partially isolated viral subpopulations. Only three patients had a minimum of 20 sequences from each of at least two of the three phenotypes. Two patients contained R5 and R5X4 sequences and the third contained R5 and X4 sequences. None of these patients was used in previous analyses. In all three patients it was generally the case that 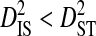 and
and 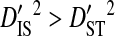 for each site pair with statistically significant disequilibrium (Table 5), although the inequalities are not nearly as large as in the analysis of variance components among patients (Table 2). The inequalities were much larger for the patient with R5 and X4 sequences than for the other two patients, possibly because of the greater isolation between R5 and X4 phenotype subpopulations (R5 and R5X4 both use CCR5). Indeed, for the patient with R5 and X4 sequences, the within-phenotype variance component,
for each site pair with statistically significant disequilibrium (Table 5), although the inequalities are not nearly as large as in the analysis of variance components among patients (Table 2). The inequalities were much larger for the patient with R5 and X4 sequences than for the other two patients, possibly because of the greater isolation between R5 and X4 phenotype subpopulations (R5 and R5X4 both use CCR5). Indeed, for the patient with R5 and X4 sequences, the within-phenotype variance component, 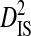 , is 0 for the majority of 38 significant site pairs because one or both sites of a pair are fixed for a different amino acid in each phenotype subpopulation (data not shown). This result suggests that the linkage disequilibrium observed within patients harboring more than one coreceptor usage phenotype is to some extent due to differences in amino acid frequencies among phenotypes, especially between R5 and X4. Tests for positive selection between phenotypes within patients show that dN is significantly greater than dS for the patient harboring R5 and X4 phenotypes only (Table 5), indicating that positive selection explains the differences in allele frequencies between these phenotypes.
, is 0 for the majority of 38 significant site pairs because one or both sites of a pair are fixed for a different amino acid in each phenotype subpopulation (data not shown). This result suggests that the linkage disequilibrium observed within patients harboring more than one coreceptor usage phenotype is to some extent due to differences in amino acid frequencies among phenotypes, especially between R5 and X4. Tests for positive selection between phenotypes within patients show that dN is significantly greater than dS for the patient harboring R5 and X4 phenotypes only (Table 5), indicating that positive selection explains the differences in allele frequencies between these phenotypes.
TABLE 5.
Linkage disequilibrium variance components within and among coreceptor usage phenotypes within patients
|
N
|
Mean
|
||||||||
|---|---|---|---|---|---|---|---|---|---|
| Patient ID | R5 | X4 | R5X4 | Site pairs | 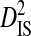 |
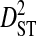 |
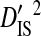 |
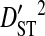 |
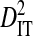 |
| 10156657 | 54 | 0 | 39 | 7 | 0.05468 | 0.14497 | 0.10273 | 0.06356 | 0.16629 |
| 10156658 | 35 | 0 | 37 | 23 | 0.07850 | 0.19826 | 0.15365 | 0.10270 | 0.25635 |
| 7129** | 32 | 20 | 0 | 38 | 0.01047 | 0.42015 | 0.31326 | 0.11340 | 0.42666 |
The numbers of sequences of each phenotype and of site pairs with statistically significant linkage disequilibrium are shown (N). Variance components are means across site pairs. **dN (SE) = 0.1644 (0.0408); dS (SE) = 0.0357 (0.0221); H0, dN = dS; Z = 2.74; P < 0.01.
The nonsystematic linkage disequilibrium observed among patients (Table 2) could arise if patients differ in the predominant coreceptor usage phenotype of their virus populations and if the disequilibrium within phenotypes is nonsystematic among phenotypes. However, there is only weak evidence for nonsystematic disequilibrium among phenotypes (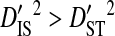 ; Table 5). The lack of strong evidence for nonsystematic disequilibrium among phenotypes suggests that disequilibrium is not correlated with V3 function and therefore that fitness epistasis is an unlikely cause of linkage disequilibrium.
; Table 5). The lack of strong evidence for nonsystematic disequilibrium among phenotypes suggests that disequilibrium is not correlated with V3 function and therefore that fitness epistasis is an unlikely cause of linkage disequilibrium.
Population subdivision among patients within phenotypes:
If fitness epistasis were a major cause of linkage disequilibrium in V3, then most of the variance in disequilibrium for a coreceptor usage phenotype would be within, rather than among, patients harboring that phenotype (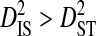 ). This would indicate that the disequilibrium is associated with the phenotype rather than with differences in allele frequencies among patients. It would also be expected that the disequilibrium within patients would be systematic among patients for a given phenotype (
). This would indicate that the disequilibrium is associated with the phenotype rather than with differences in allele frequencies among patients. It would also be expected that the disequilibrium within patients would be systematic among patients for a given phenotype (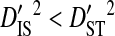 ). To test these predictions, the total variance in disequilibrium was estimated for individual phenotypes and partitioned among patients. Data sets were constructed for each phenotype for which at least 2 patients each had ≥30 sequences sampled. These data sets could be constructed for the R5 and R5X4 phenotypes, but not for the X4 phenotype. Thirteen patients were used in these analyses, all of which contained R5 sequences, and 2 of which also contained R5X4 sequences. Three of the patients are from the data set used to test for an effect of phenotype within patients (Table 5), and 1 is from the 51-patients data set. These analyses show that for site pairs with statistically significant disequilibrium,
). To test these predictions, the total variance in disequilibrium was estimated for individual phenotypes and partitioned among patients. Data sets were constructed for each phenotype for which at least 2 patients each had ≥30 sequences sampled. These data sets could be constructed for the R5 and R5X4 phenotypes, but not for the X4 phenotype. Thirteen patients were used in these analyses, all of which contained R5 sequences, and 2 of which also contained R5X4 sequences. Three of the patients are from the data set used to test for an effect of phenotype within patients (Table 5), and 1 is from the 51-patients data set. These analyses show that for site pairs with statistically significant disequilibrium, 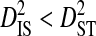 and
and 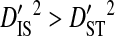 consistently for the R5 phenotype and nearly always for the R5X4 phenotype (Table 6). This is opposite to what would be expected if epistasis were causing most of the disequilibrium. Values for the variance components are similar to those observed when partitioning the variance in the whole population among patients (Table 2). Therefore, the disequilibrium observed within these phenotypes from data pooled across patients is mainly due to differences in amino acid frequencies among patients and nonsystematic disequilibrium among patients. In accordance with this result, comparisons among patients within each phenotype show virtually no overlap in the identities of site pairs with significant disequilibrium (data not shown). This result suggests that the linkage disequilibrium observed between V3 amino acid sites does not reflect functional interactions related to coreceptor usage and is therefore unlikely to be caused by fitness epistasis.
consistently for the R5 phenotype and nearly always for the R5X4 phenotype (Table 6). This is opposite to what would be expected if epistasis were causing most of the disequilibrium. Values for the variance components are similar to those observed when partitioning the variance in the whole population among patients (Table 2). Therefore, the disequilibrium observed within these phenotypes from data pooled across patients is mainly due to differences in amino acid frequencies among patients and nonsystematic disequilibrium among patients. In accordance with this result, comparisons among patients within each phenotype show virtually no overlap in the identities of site pairs with significant disequilibrium (data not shown). This result suggests that the linkage disequilibrium observed between V3 amino acid sites does not reflect functional interactions related to coreceptor usage and is therefore unlikely to be caused by fitness epistasis.
TABLE 6.
Linkage disequilibrium variance components within and among patients within coreceptor usage phenotypes
|
N
|
Mean
|
|||||||
|---|---|---|---|---|---|---|---|---|
| Phenotype | Patients | Sequences | Site pairs | 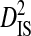 |
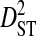 |
 |
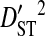 |
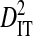 |
| R5 | 13 | 513 | 24 | 0.00756 | 0.34647 | 0.34176 | 0.01316 | 0.35493 |
| R5X4 | 2 | 76 | 35 | 0.05923 | 0.23340 | 0.20491 | 0.07959 | 0.28450 |
The numbers of patients, of sequences for each phenotype, and of site pairs with statistically significant linkage disequilibrium are shown (N). Variance components are means across site pairs.
Population subdivision among patients independent of within-patient subdivision:
The above analyses show that linkage disequilibrium within patients is at least partly attributable to population subdivision among sequences sampled in different years and among coreceptor usage phenotypes. To analyze the residual disequilibrium among and within patients after controlling for time and phenotype, variance components were estimated for sequences sampled in a single year from a single phenotype within individual patients. Only 3 patients had samples of at least 20 sequences from a single year and phenotype, and for all 3 patients the phenotype was R5. These 3 patients were also used in the previous analysis of population subdivision within and among patients within phenotypes (Table 6). For this data set, 14 site pairs exhibit statistically significant disequilibrium (Table 7). Total variances in disequilibrium, 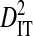 , are of similar magnitude or higher than those observed for the 51 patients, and, as in the analysis of the 51 patients,
, are of similar magnitude or higher than those observed for the 51 patients, and, as in the analysis of the 51 patients, 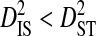 and
and 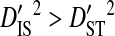 consistently across site pairs. This indicates that the disequilibrium at the whole population level is largely due to differences in amino acid frequencies among patients and possibly to nonsystematic disequilibrium among patients. Indeed, the within-patient variance component,
consistently across site pairs. This indicates that the disequilibrium at the whole population level is largely due to differences in amino acid frequencies among patients and possibly to nonsystematic disequilibrium among patients. Indeed, the within-patient variance component, 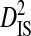 , is 0 for all but one site pair because, in each of these, one or both sites of a pair are fixed for a different amino acid in different patients. This confirms that the disequilibrium for the whole population (the 3 patients) is largely due to differences in amino acid frequencies among the patients.
, is 0 for all but one site pair because, in each of these, one or both sites of a pair are fixed for a different amino acid in different patients. This confirms that the disequilibrium for the whole population (the 3 patients) is largely due to differences in amino acid frequencies among the patients.
TABLE 7.
Linkage disequilibrium variance components within and among patients for R5 sequences sampled in a single year
| Sites | 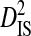 |
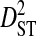 |
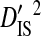 |
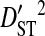 |
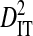 |
|---|---|---|---|---|---|
| 2, 11 | 0.00000 | 0.38123 | 0.31723 | 0.06400 | 0.38123 |
| 2, 29 | 0.00113 | 0.34031 | 0.27653 | 0.06400 | 0.34053 |
| 10, 13 | 0.00000 | 0.52894 | 0.37342 | 0.15553 | 0.52894 |
| 10, 14 | 0.00000 | 0.63533 | 0.44092 | 0.19441 | 0.63533 |
| 10, 20 | 0.00000 | 0.63533 | 0.44092 | 0.19441 | 0.63533 |
| 10, 25 | 0.00000 | 0.56790 | 0.39873 | 0.16917 | 0.56790 |
| 11, 29 | 0.00000 | 0.55926 | 0.40051 | 0.15875 | 0.55926 |
| 13, 14 | 0.00000 | 0.52894 | 0.37342 | 0.15553 | 0.52894 |
| 13, 20 | 0.00000 | 0.52894 | 0.37342 | 0.15553 | 0.52894 |
| 13, 25 | 0.00000 | 0.44859 | 0.31658 | 0.13201 | 0.44859 |
| 14, 20 | 0.00000 | 0.63533 | 0.44092 | 0.19441 | 0.63533 |
| 14, 25 | 0.00000 | 0.56790 | 0.39873 | 0.16917 | 0.56790 |
| 20, 25 | 0.00000 | 0.56790 | 0.39873 | 0.16917 | 0.56790 |
| 22, 25 | 0.00000 | 0.62974 | 0.55596 | 0.07378 | 0.62974 |
Data are from three patients, each with at least 20 sequences sampled (64 sequences in total). Sites shown exhibit statistically significant linkage disequilibrium.
Note that the sequences from all 3 patients were from the same phenotype, and therefore the differences among patients cannot be attributed to differences in phenotype. However, the differences among these patients may be attributed to differences in time of sampling since initial infection and to differences in immune selection on V3. Although 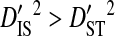 for all site pairs, the inequalities are smaller than those observed for the 51-patients data set, and no disequilibrium could be detected within individual patients, suggesting that this inequality is due to differences in amino acid frequencies among patients rather than to nonsystematic disequilibrium among patients. This result shows that, in the absence of population subdivision within patients, the linkage disequilibrium observed for V3 sequences pooled from different patients is caused by differences in amino acid frequencies among patients and not by disequilibrium within patients. Therefore, this result confirms that the disequilibrium observed within patients in the earlier analyses of this study is the result of population subdivision within patients.
for all site pairs, the inequalities are smaller than those observed for the 51-patients data set, and no disequilibrium could be detected within individual patients, suggesting that this inequality is due to differences in amino acid frequencies among patients rather than to nonsystematic disequilibrium among patients. This result shows that, in the absence of population subdivision within patients, the linkage disequilibrium observed for V3 sequences pooled from different patients is caused by differences in amino acid frequencies among patients and not by disequilibrium within patients. Therefore, this result confirms that the disequilibrium observed within patients in the earlier analyses of this study is the result of population subdivision within patients.
DISCUSSION
The substantial linkage disequilibrium, or amino acid covariation, reported from analyses of one or a few V3 sequences from each of many patients (Korber et al. 1993; Bickel et al. 1996; Gilbert et al. 2005; Poon et al. 2007; Travers et al. 2007) can be explained by population subdivision among and within patients. Most of this disequilibrium is attributable to differences in amino acid frequencies among patients and among time points and coreceptor usage phenotypes within patients. Within phenotypes, most of the variance in disequilibrium is explained by differences in amino acid frequencies among patients and nonsystematic disequilibrium among patients. This suggests that the disequilibrium is not associated with V3 function and therefore is unlikely to be caused by fitness epistasis. The analysis of sequences from a single year and the same phenotype within each of several patients showed that the total variance in linkage disequilibrium is explained by differences in amino acid frequencies among patients, with no significant disequilibrium detected within these patients. This confirms the role of differences in amino acid frequencies among virus subpopulations infecting different patients in generating disequilibrium at the whole population level and the role of within-patient population subdivision in generating disequilibrium within patients.
Frost et al. (2001) report evidence of population subdivision among foci of infection within the spleen affecting the nucleotide diversity of the V1/V2 region of the HIV-1 env gene. Population subdivision at this small scale, within a tissue type, is in contrast to the finding in the present study that subdivision among source tissues does not contribute to the total variance in linkage disequilibrium. A possible explanation for this difference is that Frost et al. may have detected stochastic effects of subdivision (e.g., founder effects and genetic drift) on synonymous nucleotide differences among subpopulations, whereas, in the case of V3 amino acid disequilibrium, similar selection across tissues may overwhelm the stochastic effects of subdivision among tissues.
Genetic drift and other stochastic forces alone are unlikely explanations for the effects of population subdivision on linkage disequilibrium in V3 for several reasons. First, genetic drift is not observed for V3 under severe serial population bottlenecks in culture, in contrast to other similar-sized HIV-1 protein regions (Yuste et al. 2000). This is an important observation because HIV-1 appears to undergo a severe population bottleneck during interpatient transmission (Derdeyn et al. 2004). Second, shortly after initial infection, V3 quickly evolves toward the sequence with the most common amino acid at each site for the R5 phenotype (Zhang et al. 1993; da Silva 2006), indicating strong selection by CCR5. This is not surprising considering that V3 is the main determinant of which chemokine coreceptor is used by a virion (Dittmar et al. 1997; Speck et al. 1997) through amino acid variation in its crown (Cormier and Dragic 2002) and considering that V3 modulates the use of the coreceptor (de Jong et al. 1992; Hung et al. 1999) and thereby affects the rate-limiting step in cellular infection (Platt et al. 2005). Third, a wide variety of comparative sequence analysis methods have been used to show that the V3 region is under strong positive selection (e.g., Bonhoeffer et al. 1995; Yamaguchi and Gojobori 1997; Nielsen and Yang 1998; Gerrish 2001; Williamson 2003; Templeton et al. 2004; da Silva 2006). Evidence of strong selection on V3 is consistent with the observation in the present study of positive selection between time points and between coreceptor usage phenotypes within patients.
Fitness interactions, or fitness epistasis, among V3 amino acids could be reasonably hypothesized given that amino acids at several sites appear to be involved in determining coreceptor usage (de Jong et al. 1992; Fouchier et al. 1992; Hung et al. 1999; Pastore et al. 2006). Furthermore, structural analyses have suggested interactions between some V3 sites that may affect V3 structural conformation and thereby coreceptor usage (Rosen et al. 2006; Cardozo et al. 2007; Gorry et al. 2007), although none of these interactions has been demonstrated through functional analyses or fitness assays. If fitness epistasis related to coreceptor tropism causes linkage disequilibrium in V3, the disequilibrium would be predicted to correlate with coreceptor usage phenotype. In other words, there should be significant disequilibrium within phenotypes and this disequilibrium should be nonsystematic among phenotypes. However, there is no disequilibrium within phenotypes, apart from that caused by differences in amino acid frequencies and nonsystematic disequilibrium among patients. Therefore, there is no evidence for fitness epistasis related to coreceptor usage causing linkage disequilibrium in V3.
However, there are two factors that may obscure an association between linkage disequilibrium and coreceptor usage phenotype. First, other gp120 regions, such as V1/V2 (e.g., Pastore et al. 2006), also affect coreceptor tropism. This may weaken any existing association between fitness epistasis among V3 residues and phenotype. Second, positive epistasis between beneficial mutations may cause the interacting residues to quickly spread to fixation within a phenotype subpopulation, thus eliminating polymorphism from the interacting sites. Such epistasis does not generate lasting linkage disequilibrium within a phenotype subpopulation and therefore may not result in an association between disequilibrium and phenotype. Instead, such a scenario may produce variance in disequilibrium and nonsystematic disequilibrium among phenotypes due to differences in amino acid frequencies among phenotypes. However, the weak evidence for nonsystematic disequilibrium among phenotypes (Table 5) argues against this possibility.
The conclusions of this study caution against interpreting correlations of residues among sequence sites as evidence for functional interactions and fitness epistasis. Such correlations may simply reflect differences in amino acid frequencies among subpopulations, although fitness epistasis that leads to the fixation of different residues in different subpopulations cannot be ruled out by the method employed here. Linkage disequilibrium has many possible causes, and the first step in ascertaining a cause is to examine the effect of population structure.
Acknowledgments
I acknowledge the Discipline of Genetics and the School of Molecular and Biomedical Science at the University of Adelaide for their support.
References
- Awadalla, P., A. Eyre-Walker and J. M. Smith, 1999. Linkage disequilibrium and recombination in hominid mitochondrial DNA. Science 286 2524–2525. [DOI] [PubMed] [Google Scholar]
- Bickel, P. J., P. C. Cosman, R. A. Olshen, P. C. Spector, A. G. Rodrigo et al., 1996. Covariability of V3 loop amino acids. AIDS Res. Hum. Retroviruses 12 1401–1411. [DOI] [PubMed] [Google Scholar]
- Bonhoeffer, S., E. C. Holmes and M. A. Nowak, 1995. Causes of HIV diversity. Nature 376 125. [DOI] [PubMed] [Google Scholar]
- Cardozo, T., T. Kimura, S. Philpott, B. Weiser, H. Burger et al., 2007. Structural basis for coreceptor selectivity by the HIV type 1 V3 loop. AIDS Res. Hum. Retroviruses 23 415–426. [DOI] [PubMed] [Google Scholar]
- Cormier, E. G., and T. Dragic, 2002. The crown and stem of the V3 loop play distinct roles in human immunodeficiency virus type 1 envelope glycoprotein interactions with the CCR5 coreceptor. J. Virol. 76 8953–8957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Silva, J., 2006. Site-specific amino acid frequency, fitness and the mutational landscape model of adaptation in human immunodeficiency virus type 1. Genetics 174 1689–1694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Jong, J. J., A. de Ronde, W. Keulen, M. Tersmette and J. Goudsmit, 1992. Minimal requirements for the human immunodeficiency virus type 1 V3 domain to support the syncytium-inducing phenotype: analysis by single amino acid substitution. J. Virol. 66 6777–6780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derdeyn, C. A., J. M. Decker, F. Bibollet-Ruche, J. L. Mokili, M. Muldoon et al., 2004. Envelope-constrained neutralization-sensitive HIV-1 after heterosexual transmission. Science 303 2019–2022. [DOI] [PubMed] [Google Scholar]
- Dittmar, M. T., A. McKnight, G. Simmons, P. R. Clapham, R. A. Weiss et al., 1997. HIV-1 tropism and co-receptor use. Nature 385 495–496. [DOI] [PubMed] [Google Scholar]
- Edgar, R. C., 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32 1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenstein, J., 1965. The effect of linkage on directional selection. Genetics 52 349–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenstein, J., 1988. Sex and the evolution of recombination, pp. 74–86 in The Evolution of Sex, edited by R. E. Michod and B. R. Levin. Sinauer Associates, Sunderland, MA.
- Fouchier, R. A., M. Groenink, N. A. Kootstra, M. Tersmette, H. G. Huisman et al., 1992. Phenotype-associated sequence variation in the third variable domain of the human immunodeficiency virus type 1 gp120 molecule. J. Virol. 66 3183–3187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frost, S. D. W., M.-J. Dumaurier, S. Wain-Hobson and A. J. L. Brown, 2001. Genetic drift and within-host metapopulation dynamics of HIV-1 infection. Proc. Natl. Acad. Sci. USA 98 6975–6980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao, F., E. Bailes, D. L. Robertson, Y. Chen, C. M. Rodenburg et al., 1999. Origin of HIV-1 in the chimpanzee Pan troglodytes troglodytes. Nature 397 436–441. [DOI] [PubMed] [Google Scholar]
- Gerrish, P., 2001. The rhythm of microbial adaptation. Nature 413 299–302. [DOI] [PubMed] [Google Scholar]
- Gilbert, P. B., V. Novitsky and M. Essex, 2005. Covariability of selected amino acid positions for HIV type 1 subtypes C and B. AIDS Res. Hum. Retroviruses 21 1016–1030. [DOI] [PubMed] [Google Scholar]
- Gorry, P. R., R. L. Dunfee, M. E. Mefford, K. Kunstman, T. Morgan et al., 2007. Changes in the V3 region of gp120 contribute to unusually broad coreceptor usage of an HIV-1 isolate from a CCR5 Δ32 heterozygote. Virology 362 163–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill, W. G., and A. Robertson, 1968. Linkage disequilibrium in finite populations. Theor. Appl. Genet. 38 226–231. [DOI] [PubMed] [Google Scholar]
- Hoffman, N. G., C. A. Schiffer and R. Swanstrom, 2003. Covariation of amino acid positions in HIV-1 protease. Virology 314 536. [DOI] [PubMed] [Google Scholar]
- Huang, C.-c., M. Tang, M.-Y. Zhang, S. Majeed, E. Montabana et al., 2005. Structure of a V3-containing HIV-1 gp120 core. Science 310 1025–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, C.-c., S. N. Lam, P. Acharya, M. Tang, S.-H. Xiang et al., 2007. Structures of the CCR5 N terminus and of a tyrosine-sulfated antibody with HIV-1 gp120 and CD4. Science 317 1930–1934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson, R. R., 1985. The sampling distribution of linkage disequilibrium under an infinite allele model without selection. Genetics 109 611–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung, C. S., N. Vander Heyden and L. Ratner, 1999. Analysis of the critical domain in the V3 loop of human immunodeficiency virus type 1 gp120 involved in CCR5 utilization. J. Virol. 73 8216–8226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang, S. S., T. J. Boyle, H. K. Lyerly and B. R. Cullen, 1991. Identification of the envelope V3 loop as the primary determinant of cell tropism in HIV-1. Science 253 71–74. [DOI] [PubMed] [Google Scholar]
- Karlin, S., and M. W. Feldman, 1970. Linkage and selection: two locus symmetric viability model. Theor. Popul. Biol. 1 39–71. [DOI] [PubMed] [Google Scholar]
- Kimura, M., 1956. A model of a genetic system which leads to closer linkage by natural selection. Evolution 10 278–287. [Google Scholar]
- Kondrashov, A. S., 1993. Classification of hypotheses on the advantage of amphimixis. J. Hered. 84 372–387. [DOI] [PubMed] [Google Scholar]
- Korber, B. T., R. M. Farber, D. H. Wolpert and A. S. Lapedes, 1993. Covariation of mutations in the V3 loop of human immunodeficiency virus type 1 envelope protein: an information theoretic analysis. Proc. Natl. Acad. Sci. USA 90 7176–7180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemey, P., A. Rambaut and O. G. Pybus, 2006. HIV evolutionary dynamics within and among hosts. AIDS Rev. 8 125–140. [PubMed] [Google Scholar]
- Levy, D. N., G. M. Aldrovandi, O. Kutsch and G. M. Shaw, 2004. Dynamics of HIV-1 recombination in its natural target cells. Proc. Natl. Acad. Sci. USA 101 4204–4209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewontin, R. C., and K.-i. Kojima, 1960. The evolutionary dynamics of complex polymorphisms. Evolution 14 458–472. [Google Scholar]
- Li, W.-H., and M. Nei, 1974. Stable linkage disequilibrium without epistasis in subdivided populations. Theor. Popul. Biol. 6 173–183. [DOI] [PubMed] [Google Scholar]
- Liu, Y., E. Eyal and I. Bahar, 2008. Analysis of correlated mutations in HIV-1 protease using spectral clustering. Bioinformatics 24 1243–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitton, J. B., and R. K. Koehn, 1973. Population genetics of marine pelecypods. III. Epistasis between functionally related isoenzymes of Mytilus edulis. Genetics 73 487–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers, R. E., and D. Pillay, 2008. Analysis of natural sequence variation and covariation in human immunodeficiency virus type 1 integrase. J. Virol. 82 9228–9235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nei, M., and S. Kumar, 2000. Molecular Evolution and Phylogenetics. Oxford University Press, London/New York/Oxford.
- Nei, M., and W.-H. Li, 1973. Linkage disequilibrium in subdivided populations. Genetics 75 213–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen, R., and Z. Yang, 1998. Likelihood models for detecting positively selected amino acid sites and applications to the HIV-1 envelope gene. Genetics 148 929–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta, T., 1982. Linkage disequilibrium due to random genetic drift in finite subdivided populations. Proc. Natl. Acad. Sci. USA 79 1940–1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta, T., and M. Kimura, 1969. Linkage disequilibrium at steady state determined by random genetic drift and recurrent mutation. Genetics 63 229–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastore, C., R. Nedellec, A. Ramos, S. Pontow, L. Ratner et al., 2006. Human immunodeficiency virus type 1 coreceptor switching: V1/V2 gain-of-fitness mutations compensate for V3 loss-of-fitness mutations. J. Virol. 80 750–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platt, E. J., J. P. Durnin and D. Kabat, 2005. Kinetic factors control efficiencies of cell entry, efficacies of entry inhibitors, and mechanisms of adaptation of human immunodeficiency virus. J. Virol. 79 4347–4356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poon, A. F., F. I. Lewis, S. L. Pond and S. D. Frost, 2007. An evolutionary-network model reveals stratified interactions in the V3 loop of the HIV-1 envelope. PLoS Comput. Biol. 3 e231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee, S.-Y., T. F. Liu, S. P. Holmes and R. W. Shafer, 2007. HIV-1 subtype B protease and reverse transcriptase amino acid covariation. PLoS Comput. Biol. 3 e87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen, O., M. Sharon, S. R. Quadt-Akabayov and J. Anglister, 2006. Molecular switch for alternative conformations of the HIV-1 V3 region: implications for phenotype conversion. Proc. Natl. Acad. Sci. USA 103 13950–13955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankarappa, R., J. B. Margolick, S. J. Gange, A. G. Rodrigo, D. Upchurch et al., 1999. Consistent viral evolutionary changes associated with the progression of human immunodeficiency virus type 1 infection. J. Virol. 73 10489–10502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slatkin, M., 1975. Gene flow and selection in a two-locus system. Genetics 81 787–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slatkin, M., 1994. Linkage disequilibrium in growing and stable populations. Genetics 137 331–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slatkin, M., 2008. Linkage disequilibrium – understanding the evolutionary past and mapping the medical future. Nat. Rev. Genet. 9 477–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speck, R. F., K. Wehrly, E. J. Platt, R. E. Atchison, I. F. Charo et al., 1997. Selective employment of chemokine receptors as human immunodeficiency virus type 1 coreceptors determined by individual amino acids within the envelope V3 loop. J. Virol. 71 7136–7139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura, K., J. Dudley, M. Nei and S. Kumar, 2007. MEGA4: molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol. Biol. Evol. 24 1596–1599. [DOI] [PubMed] [Google Scholar]
- Templeton, A. R., R. A. Reichert, A. E. Weisstein, X.-F. Yu and R. B. Markham, 2004. Selection in context: patterns of natural selection in the glycoprotein 120 region of human immunodeficiency virus 1 within infected individuals. Genetics 167 1547–1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travers, S. A. A., D. C. Tully, G. P. McCormack and M. A. Fares, 2007. A study of the coevolutionary patterns operating within the env gene of the HIV-1 group M subtypes. Mol. Biol. Evol. 24 2787–2801. [DOI] [PubMed] [Google Scholar]
- Wang, Q., and C. Lee, 2007. Distinguishing functional amino acid covariation from background linkage disequilibrium in HIV protease and reverse transcriptase. PLoS ONE 2 e814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weir, B. S., 1996. Genetic Data Analysis II: Methods for Discrete Population Genetic Data. Sinauer Associates, Sunderland, MA.
- Weir, B. S., and W. G. Hill, 1986. Nonuniform recombination within the human beta-globin gene cluster. Am. J. Hum. Genet. 38 776–781. [PMC free article] [PubMed] [Google Scholar]
- Whittam, T. S., H. Ochman and R. K. Selander, 1983. Geographic components of linkage disequilibrium in natural populations of Escherichia coli. Mol. Biol. Evol. 1 67–83. [DOI] [PubMed] [Google Scholar]
- Williamson, S., 2003. Adaptation in the env gene of HIV-1 and evolutionary theories of disease progression. Mol. Biol. Evol. 20 1318–1325. [DOI] [PubMed] [Google Scholar]
- Wolinsky, S. M., B. T. Korber, A. U. Neumann, M. Daniels, K. J. Kunstman et al., 1996. Adaptive evolution of human immunodeficiency virus-type 1 during the natural course of infection. Science 272 537–542. [DOI] [PubMed] [Google Scholar]
- Wong, J. K., C. C. Ignacio, F. Torriani, D. Havlir, N. J. Fitch et al., 1997. In vivo compartmentalization of human immunodeficiency virus: evidence from the examination of pol sequences from autopsy tissues. J. Virol. 71 2059–2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright, S., 1940. Breeding structure of populations in relation to speciation. Am. Nat. 74 232–248. [Google Scholar]
- Wyatt, R., and J. Sodroski, 1998. The HIV-1 envelope glycoproteins: fusogens, antigens, and immunogens. Science 280 1884–1888. [DOI] [PubMed] [Google Scholar]
- Yamaguchi, Y., and T. Gojobori, 1997. Evolutionary mechanisms and population dynamics of the third variable envelope region of HIV within single hosts. Proc. Natl. Acad. Sci. USA 94 1264–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuste, E., C. Lopez-Galindez and E. Domingo, 2000. Unusual distribution of mutations associated with serial bottleneck passages of human immunodeficiency virus type 1. J. Virol. 74 9546–9552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, L. Q., P. MacKenzie, A. Cleland, E. C. Holmes, A. J. Brown et al., 1993. Selection for specific sequences in the external envelope protein of human immunodeficiency virus type 1 upon primary infection. J. Virol. 67 3345–3356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zolla-Pazner, S., 2004. Identifying epitopes of HIV-1 that induce protective antibodies. Nat. Rev. Immunol. 4 199–210. [DOI] [PMC free article] [PubMed] [Google Scholar]