

Abstract
Genomic selection (GS) using high-density single-nucleotide polymorphisms (SNPs) is promising to improve response to selection in populations that are under artificial selection. High-density SNP genotyping of all selection candidates each generation, however, may not be cost effective. Smaller panels with SNPs that show strong associations with phenotype can be used, but this may require separate SNPs for each trait and each population. As an alternative, we propose to use a panel of evenly spaced low-density SNPs across the genome to estimate genome-assisted breeding values of selection candidates in pedigreed populations. The principle of this approach is to utilize cosegregation information from low-density SNPs to track effects of high-density SNP alleles within families. Simulations were used to analyze the loss of accuracy of estimated breeding values from using evenly spaced and selected SNP panels compared to using all high-density SNPs in a Bayesian analysis. Forward stepwise selection and a Bayesian approach were used to select SNPs. Loss of accuracy was nearly independent of the number of simulated quantitative trait loci (QTL) with evenly spaced SNPs, but increased with number of QTL for the selected SNP panels. Loss of accuracy with evenly spaced SNPs increased steadily over generations but was constant when the smaller number individuals that are selected for breeding each generation were also genotyped using the high-density SNP panel. With equal numbers of low-density SNPs, panels with SNPs selected on the basis of the Bayesian approach had the smallest loss in accuracy for a single trait, but a panel with evenly spaced SNPs at 10 cM was only slightly worse, whereas a panel with SNPs selected by forward stepwise selection was inferior. Panels with evenly spaced SNPs can, however, be used across traits and populations and their performance is independent of the number of QTL affecting the trait and of the methods used to estimate effects in the training data and are, therefore, preferred for broad applications in pedigreed populations under artificial selection.
THE goal of genomic selection (GS), as described by Meuwissen et al. (2001), is to exploit linkage disequilibrium between quantitative trait loci (QTL) and high-density markers across the genome for breeding value estimation in genetic improvement programs for livestock. To implement GS, first, effects of high-density single-nucleotide polymorphisms (HD-SNPs) are estimated on the basis of individuals that are genotyped and phenotyped for a quantitative trait (training). Then, genome-assisted breeding values (GEBVs) of selection candidates are predicted by applying the estimated marker effects to their marker genotypes. Several simulation studies (Meuwissen et al. 2001; Solberg et al. 2006; Habier et al. 2007) have revealed the potential of GS to improve response to selection by allowing estimation of breeding values on selection candidates without requiring phenotypic data on the individuals themselves. One of the main challenges of GS is that the number of markers (50,000 is common) is often much greater than the number of phenotypes available to estimate their effects. Methods to deal with this can be classified (Xu 2007) into variable selection methods, such as stepwise regression (Habier et al. 2007; Piyasatian et al. 2007), partial least squares (Moser et al. 2007; Tier et al. 2007), principle component regression (Benjamin and Nicola 2004; Woolaston et al. 2007), and machine learning methods (Long et al. 2007), and into shrinkage methods, such as the Bayesian methods of Meuwissen et al. (2001) and Xu (2003), LASSO (Xu 2007), and Bayesian variable selection (George and McCulloch 1993), which utilize prior information. The simulation studies of Meuwissen et al. (2001) and Habier et al. (2007), which compared least-squares, stepwise, and ridge regression, along with Bayesian methods, have found that the BayesB method of Meuwissen et al. (2001) gives the highest accuracy of GEBVs. When implementing GS with shrinkage methods such as BayesB, in principle all markers used for training must also be used for prediction, and thus selection candidates must be genotyped for all HD-SNPs. Genotyping selection candidates for HD-SNP panels may, however, not be cost effective when the number of selection candidates is high or the economic benefit per selection candidate is low compared to the cost of genotyping, as is the case when selection candidates have low reproduction rates, such as cows in cattle breeding programs, or in general for livestock species such as pigs, poultry, fish, or sheep. Current genotyping costs per individual are considerably lower for low-density SNP (LD-SNP) panels than for HD-SNP panels. Thus, there is much interest in developing methods to implement GS using LD-SNP panels. The most common strategy that has been proposed to develop LD-SNP panels is to employ variable selection methods to identify a small set of markers that are predictive of trait phenotype or breeding value. A potential problem with variable selection for development of an LD-SNP panel, however, is that selected HD-SNPs might be different for each quantitative trait and population, thereby increasing the number of SNPs that must be genotyped when GS is implemented for the multiple-trait breeding programs in livestock. In addition, the effectiveness of this approach may depend on the number of QTL that affect the trait; larger numbers of SNPs will be needed for traits with larger numbers of QTL. To overcome these limitations, we propose to use evenly spaced LD-SNPs on the entire genome to obtain GEBVs of selection candidates. In this approach, training individuals are genotyped for HD-SNPs, whereas their descendants, including selection candidates, are genotyped for evenly spaced LD-SNPs. By utilizing cosegregation of HD-SNPs with LD-SNPs within a family, HD-SNP alleles are tracked from training individuals to selection candidates by estimating probabilities of descent of HD-SNP alleles from the training individuals to their descendants on the basis of LD-SNP genotypes of the descendants and HD-SNP haplotypes of their ancestors in the training data. These probabilities are then used to predict GEBVs on the selection candidates without having to genotype them for HD-SNPs. A similar approach was proposed for genomic selection by Hayes and Goddard (2008) and Goddard (2008), for association mapping in plants by Yu et al. (2008), and for inferring HD genotypes in a human pedigree by Burdick et al. (2006). Note that the use of cosegregation within families to impute missing SNP genotypes, as proposed here, is different from the strategy that is employed in human genetics, in which TAG SNPs are used to identify haplotype blocks that segregate across the population (Johnson et al. 2001; Patil et al. 2001; Marchini et al. 2007; Servin and Stephens 2007). In contrast to these studies, the proposed strategy identifies haplotype blocks on a within-family basis. Although this does require availability of pedigree information, an advantage is that haplotype blocks are much greater within families than across the population and, therefore, require much lower marker densities to trace alleles at markers that are not genotyped.
The objective of this article was to evaluate the loss of accuracy for GEBVs that are predicted by using LD-SNPs based on the sparse marker approach and to evaluate the impact of the number of QTL that affect the trait. Accuracy of an LD-SNP panel based on selected HD-SNPs was included for comparison. Simulated data were used to estimate accuracies of GEBVs for the various GS methods.
THEORY
The evenly spaced LD-SNP approach (ELD-GS) proposed here can be outlined as follows: (1) Estimate the effects of HD-SNP alleles in an ancestral training population using a method such as BayesB, (2) estimate HD-SNP haplotypes of training individuals, (3) estimate probabilities of descent of marker alleles (PDMs) to trace HD-SNP alleles from ancestors to selection candidates by using LD-SNP genotypes and pedigree information, and (4) predict GEBVs on selection candidates. Methods for each of these steps are described in the following.
Estimation of the effects of HD-SNP alleles:
The statistical model to estimate effects of HD-SNP alleles using the training population can be written as
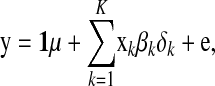 |
(1) |
where 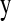 is the vector of trait phenotypes, μ is the overall mean,
is the vector of trait phenotypes, μ is the overall mean, 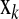 is the column vector of HD-SNP genotypes, βk is the effect and δk a 0/1-indicator variable, all for HD-SNP k, and
is the column vector of HD-SNP genotypes, βk is the effect and δk a 0/1-indicator variable, all for HD-SNP k, and 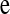 is the vector of random residual effects with mean zero and variance
is the vector of random residual effects with mean zero and variance 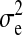 . The HD-SNP genotype of an individual in
. The HD-SNP genotype of an individual in 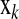 is coded as the number of copies of one HD-SNP allele it carries, i.e., 0, 1, or 2. In BayesB (Meuwissen et al. 2001), βk is treated as random with prior
is coded as the number of copies of one HD-SNP allele it carries, i.e., 0, 1, or 2. In BayesB (Meuwissen et al. 2001), βk is treated as random with prior 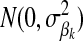 . The prior distribution for δk, indicating whether HD-SNP k is included in the model (i.e., δk = 1) or not, is the probability that HD-SNP k has a nonzero effect, which is predefined here to be 0.05. If δk = 1, then the prior distribution for
. The prior distribution for δk, indicating whether HD-SNP k is included in the model (i.e., δk = 1) or not, is the probability that HD-SNP k has a nonzero effect, which is predefined here to be 0.05. If δk = 1, then the prior distribution for 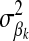 was a scaled inverse chi square with ν = 4.2 d.f. and scale S = 0.0429, as used by Meuwissen et al. (2001). For the error variance,
was a scaled inverse chi square with ν = 4.2 d.f. and scale S = 0.0429, as used by Meuwissen et al. (2001). For the error variance, 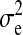 , the prior distribution was also a scaled inverse chi square with ν = 4.2 and S = 0.52 having expected value of 1 and finite variance. Markov chain Monte Carlo (MCMC) sampling was used to infer model parameters, where μ, βk, and
, the prior distribution was also a scaled inverse chi square with ν = 4.2 and S = 0.52 having expected value of 1 and finite variance. Markov chain Monte Carlo (MCMC) sampling was used to infer model parameters, where μ, βk, and  were sampled with a Gibbs step and δk and
were sampled with a Gibbs step and δk and 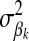 with a Metropolis–Hastings step. The MCMC sampler was run for 10,000 iterations with a burn-in of 1000 iterations.
with a Metropolis–Hastings step. The MCMC sampler was run for 10,000 iterations with a burn-in of 1000 iterations.
Estimation of HD-SNP haplotypes:
Alleles at the HD-SNPs are traced from training individuals to later generations using LD-SNPs, as is described in the next section. To do this, HD-SNP haplotypes of training individuals have to be derived first. In this study, it was assumed that HD-SNP haplotypes of training individuals are known. In practice, HD-SNP haplotypes of training individuals can be estimated if parents of the training individuals are also genotyped for HD-SNPs, noting that the offspring of training individuals will be genotyped only for LD-SNPs.
Estimation of PDMs to trace HD-SNP alleles:
Genotypes at LD-SNPs of training individuals and their descendants, including selection candidates, are used to estimate the probability of descent for each HD-SNP allele of nonfounders (PDM). Two PDMs are estimated for each allele at each HD-SNP of a LD-genotyped descendant, which indicate the probabilities that the HD-SNP allele originates from the parent's maternal (grandmaternal origin) or paternal (grandpaternal origin) allele. The PDMs of descendant i at HD-SNP k are denoted by 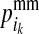 and
and 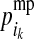 for the maternal allele and by
for the maternal allele and by  and
and 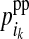 for the paternal allele. To estimate these PDMs, first, ordered genotypes and segregation indicators of alleles at the LD-SNPs of the LD-genotyped descendants are sampled with an overlapping blocking Gibbs algorithm. The order of an LD-SNP genotype specifies which allele was transmitted from the mother and which one from the father. The segregation indicator of a given allele is 0 if it is of grandmaternal origin and 1 if it is of grandpaternal origin. In this approach, training individuals are treated as founders and their descendants as nonfounders. The LD-SNP haplotypes of training individuals and thus the order of their LD-SNP genotypes are assumed known (see previous section). The blocking in this Gibbs algorithm is by individuals to reduce cutset sizes during peeling (Elston and Stewart 1971) and reverse peeling (Heath 1997), which both are performed within blocks. Each block contains a sire, its mates, their offspring, and the parents of the sire and the mates. The blocks are overlapping, because sires and dams also occur as offspring in other blocks, a strategy also described by Thomas et al. (2000) and Abraham et al. (2007). The Gibbs sampler was run for 1000 iterations with a burn-in of 100 iterations. Realizations of the Gibbs sampler are used to estimate the joint probability distribution of segregation indicators on the maternal (m) and paternal (p) haplotypes of a nonfounder for every adjacent LD-SNP pair. These probabilities are denoted by Pr
for the paternal allele. To estimate these PDMs, first, ordered genotypes and segregation indicators of alleles at the LD-SNPs of the LD-genotyped descendants are sampled with an overlapping blocking Gibbs algorithm. The order of an LD-SNP genotype specifies which allele was transmitted from the mother and which one from the father. The segregation indicator of a given allele is 0 if it is of grandmaternal origin and 1 if it is of grandpaternal origin. In this approach, training individuals are treated as founders and their descendants as nonfounders. The LD-SNP haplotypes of training individuals and thus the order of their LD-SNP genotypes are assumed known (see previous section). The blocking in this Gibbs algorithm is by individuals to reduce cutset sizes during peeling (Elston and Stewart 1971) and reverse peeling (Heath 1997), which both are performed within blocks. Each block contains a sire, its mates, their offspring, and the parents of the sire and the mates. The blocks are overlapping, because sires and dams also occur as offspring in other blocks, a strategy also described by Thomas et al. (2000) and Abraham et al. (2007). The Gibbs sampler was run for 1000 iterations with a burn-in of 100 iterations. Realizations of the Gibbs sampler are used to estimate the joint probability distribution of segregation indicators on the maternal (m) and paternal (p) haplotypes of a nonfounder for every adjacent LD-SNP pair. These probabilities are denoted by Pr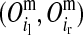 and Pr
and Pr , where
, where 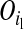 and
and 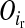 are segregation indicators, respectively, for the left and the right LD-SNP of an adjacent pair for individual i. For example, Pr
are segregation indicators, respectively, for the left and the right LD-SNP of an adjacent pair for individual i. For example, Pr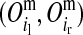 indicates the probability of the grandparental origin for the 2-SNP haplotype received from the mother at an adjacent LD-SNP pair, and in particular whether the alleles on those 2-SNP haplotypes were transmitted without recombination between the two adjacent LD-SNPs. Note that through peeling, information from all LD-SNPs and from the whole pedigree is used to sample the segregation indicators of an adjacent LD-SNP pair. With the segregation indicator for a given LD-SNP allele being either 0 or 1, there are four possible combinations of segregation indicators for a pair of LD-SNPs on a haplotype. Given one of these combinations, the conditional PDM can be calculated for the maternal and the paternal allele of a descendant at every HD-SNP in the interval of the flanking LD-SNPs using formulas shown in Table 1, where recombination frequencies are obtained by Haldane's map function. The PDMs used to trace HD-SNP alleles are finally derived by weighting the conditional PDM for each combination of segregation indicators for the flanking LD-SNPs with their corresponding probabilities, i.e., Pr
indicates the probability of the grandparental origin for the 2-SNP haplotype received from the mother at an adjacent LD-SNP pair, and in particular whether the alleles on those 2-SNP haplotypes were transmitted without recombination between the two adjacent LD-SNPs. Note that through peeling, information from all LD-SNPs and from the whole pedigree is used to sample the segregation indicators of an adjacent LD-SNP pair. With the segregation indicator for a given LD-SNP allele being either 0 or 1, there are four possible combinations of segregation indicators for a pair of LD-SNPs on a haplotype. Given one of these combinations, the conditional PDM can be calculated for the maternal and the paternal allele of a descendant at every HD-SNP in the interval of the flanking LD-SNPs using formulas shown in Table 1, where recombination frequencies are obtained by Haldane's map function. The PDMs used to trace HD-SNP alleles are finally derived by weighting the conditional PDM for each combination of segregation indicators for the flanking LD-SNPs with their corresponding probabilities, i.e., Pr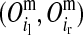 and Pr
and Pr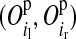 .
.
TABLE 1.
Conditional probabilities of descent, Pr(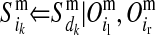 ) and Pr(
) and Pr(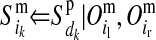 ), of the maternal allele (
), of the maternal allele ( ) of individual i from the maternal (
) of individual i from the maternal (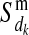 ) and paternal (
) and paternal (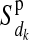 ) allele of its mother d at HD-SNP k, depending on segregation indicators at the left (
) allele of its mother d at HD-SNP k, depending on segregation indicators at the left (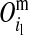 ) and the right (
) and the right (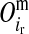 ) LD-SNP of an adjacent pair on the maternal haplotype of individual i
) LD-SNP of an adjacent pair on the maternal haplotype of individual i
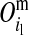 |
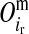 |
Pr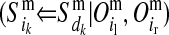
|
Pr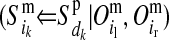
|
|---|---|---|---|
| 0 | 0 | (1 – θlk)(1 – θkr)/(1 – θlr) | θlkθkr/(1 – θlr) |
| 0 | 1 | (1 – θlk)θkr/θlr | θlk(1 – θkr)/θlr |
| 1 | 0 | θlk(1 – θkr)/θlr | (1 – θlk)θkr/θlr |
| 1 | 1 | θlkθkr/(1 – θlr) | (1 – θlk)(1 – θkr)/(1 – θlr) |
θlk (θkr), recombination frequency between HD-SNP k and the left (right) LD-SNP; θlr, recombination frequency between left and right LD-SNPs; 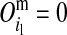 (1) means grandmaternal (grandpaternal) origin.
(1) means grandmaternal (grandpaternal) origin.
Prediction of GEBVs:
The PDMs are used to estimate effects of the maternal and paternal HD-SNP alleles of LD-genotyped selection candidates by using them to weight estimates of effects of alleles at HD-SNPs of the individual's mother and father. For example, the effect of the maternal allele of individual i at HD-SNP k, 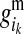 , is estimated by
, is estimated by
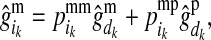 |
(2) |
where 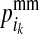 (
( ) is the PDM for the maternal allele of i, i.e., the probability that the allele, which i received from its mother d, is the mother's maternal (paternal) allele; and
) is the PDM for the maternal allele of i, i.e., the probability that the allele, which i received from its mother d, is the mother's maternal (paternal) allele; and 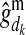 and
and 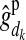 are respectively the estimated effects of the maternal and the paternal allele of d at HD-SNP k. The effect of the paternal allele of i is estimated correspondingly. With multiple generations of LD genotyping, this equation is applied recursively, starting with individuals with known HD-SNP haplotypes such as the training individuals, for which allele effect estimates can be obtained as
are respectively the estimated effects of the maternal and the paternal allele of d at HD-SNP k. The effect of the paternal allele of i is estimated correspondingly. With multiple generations of LD genotyping, this equation is applied recursively, starting with individuals with known HD-SNP haplotypes such as the training individuals, for which allele effect estimates can be obtained as
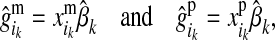 |
(3) |
where 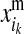 and
and 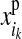 denote the maternal and the paternal allele, respectively, and
denote the maternal and the paternal allele, respectively, and 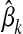 is the effect at HD-SNP k estimated by BayesB using the training data. Finally, the GEBV of i is predicted with
is the effect at HD-SNP k estimated by BayesB using the training data. Finally, the GEBV of i is predicted with
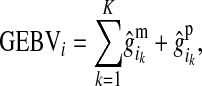 |
(4) |
where the summation is over all K HD-SNPs.
SIMULATION
To evaluate the effectiveness of the proposed ELD-GS approach, simulation was used to evaluate the loss in accuracy compared to HD-GS and also to compare it to use of LD-SNP panels derived by marker selection approaches. Simulations started with a base population of 500 individuals that were randomly mated, including selfing (thus effective population size was 500), for 1000 discrete generations and then reduced to a size of 100 individuals (Figure 1, generation −1062). Thereafter, random mating was used for another 50 generations. The larger population size for the first 1000 generations takes into account that historical effective population sizes for livestock populations are assumed to be larger than they are today (Hayes et al. 2003). The population was increased over the next 10 generations to obtain a population of 500 males and 500 females in generation −2. The following 3 generations (−1, 0, and 1) were obtained by randomly mating 50 sires to 500 dams in each discrete generation. Each female had 1 male and 1 female offspring and thus each sire had 10 sons and 10 daughters. The 1000 individuals in generation 1 were phenotyped for the quantitative trait and genotyped at 1000 HD-SNPs to use them for training. The last 3 generations (2, 3, and 4) were obtained by mating 10 males with 100 females to produce 200 offspring per generation. This reduction in population size was done to reduce computing time for sampling LD-SNP genotypes and segregation indicators with the Gibbs sampler. For the comparison of the alternative GS methods, individuals in generations 2, 3, and 4 were assumed to be genotyped (i) for the HD-SNP panel consisting of the 1000 HD-SNPs used for training, (ii) for evenly spaced LD-SNP panels, and (iii) for LD-SNP panels, based on subsets of selected HD-SNPs, as is explained in more detail below.
Figure 1.—

Simulated population.
The genome was simulated with 10 chromosomes of 1 M, each having 2000 evenly spaced SNPs in generation −1062. Thus, the initial SNP spacing was 0.05 cM. A total of 100, 500, or 1000 QTL were randomly distributed among SNPs. In generation −1062, all loci were simulated to be biallelic with allele frequencies 0.5 and in Hardy–Weinberg and linkage equilibrium. Effects at the QTL were sampled from a gamma distribution with shape 0.4 and scale 1.66 as used by Meuwissen et al. (2001). A mutation rate of 2.5 × 10−5 was used for both SNPs and QTL and recombinations were modeled according to a binomial map function, where the maximum number of uniformly and independently distributed crossovers on a chromosome of 1 M was four (Karlin 1984), i.e., assuming interference. Initial allele frequencies differ from similar simulations conducted by, e.g., Meuwissen et al. (2001), who started with a population that was fixed for all loci, and mutation rate was larger than in practice. Starting with a segregating population and using this high mutation rate, however, allowed us to approach, but not reach, mutation–drift equilibrium with a stationary U-shaped distribution of allele frequencies after 1000 compared to >100,000 generations of random mating. Moreover, this high mutation rate ensured that enough loci are segregating for statistical analyses after 1000 generations. It can be shown by simulation that the simulated disequilibrium for an effective population size of 500 individuals after 1000 generations of random mating corresponds well to the expected disequilibrium in mutation–drift equilibrium calculated with the formula given by Ohta and Kimura (1969). With this formula it can also be shown that, even at small distances between loci, mutation rate has only a small effect on disequilibrium in mutation–drift equilibrium for an effective population size of 500. To develop the HD-SNP panel for genotyping, using the SNPs still segregating in generation 0, each chromosome was first divided into 100 bins with an equal number of SNPs in each bin. Then, within each bin the SNP with frequency closest to 0.5 was selected, giving a total of 1000 segregating HD-SNPs for analyses. The evenly spaced LD-SNPs were chosen from HD-SNPs by taking the first HD-SNP on each chromosome and then SNPs that were at least 10 cM apart and had a minor allele frequency >0.2 in generation 0. In another scenario, LD-SNPs were chosen to be 20 cM apart.
Heritability of the quantitative trait was set to 0.5 by rescaling effects of QTL that were still segregating (minor allele frequency, MAF >0) in generation 0. Phenotypes were calculated as the sum of genotypic effects of an individual plus a residual effect sampled from a standard normal distribution.
Genomic selection aims to exploit linkage disequilibrium between QTL and HD-SNPs and thus the accuracy of GEBVs that was used to compare alternative GS methods depends on the amount of linkage disequilibrium, which was measured here by r2 for each pair of QTL and HD-SNP. Because MAF can skew r2, only QTL and HD-SNPs with minor allele frequencies >0.05 were used to analyze linkage disequilibrium. Average r2 was estimated for bins of length 0.5 cM and compared with expected r2 calculated with E(r2) = 1/(1 + 4 × Ne × θ) (Sved 1969), where Ne is the effective population size and θ is the recombination frequency.
The loss in accuracy of GEBVs for ELD-GS compared to the accuracy obtained by HD-GS depends on the precision of estimated PDMs, which was analyzed by estimating the mean absolute difference between true segregation indicators of HD-SNPs alleles and PDMs. The more this measure is <0.5, the more information LD-SNPs provided to estimate PDMs.
In practice, offspring of selection candidates are not available when the first selection decisions based on GEBVs are made and thus they cannot provide information to estimate PDMs of their parents. Therefore, PDMs of individuals in generations 2 and 3 were estimated without genotype information of their simulated descendants.
In the following, the proposed ELD-GS approach is referred to as ELD-10 and ELD-20 for LD-SNP spacings of 10 and 20 cM. In addition, ELD-10+ and ELD-20+ were analyzed in which parents of selection candidates were assumed to be also HD genotyped, after they were selected using LD-GS. Having HD genotypes on parents of selection candidates can improve the accuracy of GEBVs for two reasons. First, HD-SNP haplotypes of the parents can be derived so that genetic effects of HD-SNP alleles can be estimated with Equation 3, thereby removing the uncertainty about the state of the HD-SNP allele of parents resulting from previous generations. In this study, HD-SNP haplotypes of the parents were not estimated, but assumed known. Second, the order of LD-SNP genotypes of the parents can be derived, which aids estimation of PDMs.
The ELD-GS approaches were also compared with results from two methods to select a subset of LD-SNPs from HD-SNPs based on the training data: BayesB (BB) and forward stepwise least-squares regression (FSS). In the BB approach, the 110 or 40 HD-SNPs that were fitted most frequently in the MCMC algorithm of BayesB in the training data were used for LD-SNP genotyping in what is denoted by SLD-BB-110 or SLD-BB-40. The number 110 corresponds to the number of markers used in the ELD-GS approach with 10-cM marker distance. Selection of 40 SNPs reflects that most selection programs consider multiple traits, requiring a smaller number of selected markers per trait to end up with a final LD panel comparable to what we propose for the ELD-GS approach. In LD-GS approaches that are referred to as SLD-FSS, FSS was implemented as described by Habier et al. (2007), in which two-sided t-statistics are calculated to decide whether to include an HD-SNP in the LD-SNP panel or to remove it. The algorithm starts without HD-SNP in the LD-SNP panel and includes one if the P-value is lower than the significance level and removes one if the P-value is greater. In the method referred to as SLD-FSS-0.01, the significance level was set to 0.01. In strategy SLD-FSS-110, exactly 110 SNPs were selected by FSS, comparable to the number of SNPs used for ELD-GS and SLD-BB-110. For the SLD-GS approaches, GEBVs were predicted by
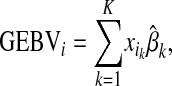 |
(5) |
where 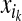 is the genotype of individual i and
is the genotype of individual i and 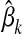 is the effect at HD-SNP k estimated either by BayesB or by FSS. If a marker is not selected for the LD-SNP panel, then
is the effect at HD-SNP k estimated either by BayesB or by FSS. If a marker is not selected for the LD-SNP panel, then 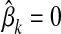 .
.
RESULTS
The numbers of segregating (MAF > 0.01) QTL after 1060 generations for 100, 500, and 1000 QTL initially simulated were 49 (±2), 220 (±4), and 501 (±5). The MAF of segregating QTL tended to be uniformly distributed with mean 0.24 (±0.01). Average map distance between adjacent HD-SNPs was 1 cM and average MAF of HD-SNPs was 0.41 (±0.01). In scenarios with an LD-SNP spacing of 10 cM, 110 SNPs with an average MAF of 0.42 (±0.03) were selected, with 11 SNPs per 1-M chromosome. In the scenario with a LD-SNP spacing of 20 cM, 60 SNPs were selected, 6 SNPs per chromosome. The extent of linkage disequilibrium between pairs of QTL and HD-SNPs generated after 1063 generations is depicted in Figure 2. Average r2 was substantial at short distances but decreased exponentially with increasing distance between loci. Average r2's were 0.38, 0.20, 0.14, and 0.04 at distances of 0.25, 0.75, 1.25 and 5 cM. Average LD at distances <2 cM was lower than expected on the basis of Ne = 100, because historical Ne was >100. Appreciable disequilibrium did not exist between loci separated by >10 cM or between nonsyntenic loci.
Figure 2.—

Linkage disequilibrium between QTL and HD-SNPs measured by r2 against distance in centimorgans (cM) obtained from one replicate in which 500 QTL were simulated. The solid line is the average r2 estimated for bins of length 0.5 cM. The dashed line is the predicted r2 based on the equation E(r2) = 1/(1 + 4 × 100 × θ), where 100 is the effective population size and θ is the recombination frequency.
Accuracies of GEBVs in generations 1–4 obtained by the high-density BayesB (HD-BB) and ELD strategies are shown in Figure 3 for the scenario with 500 simulated QTL. The accuracy in generation 1 was identical for all five methods, because generation 1 was the training generation and GEBVs of training individuals were predicted with Equation 5, using HD-SNPs. In generations after training, the accuracy of HD-BB decreased at a decreasing rate from 0.64 (±0.010) to 0.55 (±0.014) but was always greater than that of any ELD strategy, as expected. The absolute loss of accuracy for the ELD strategies compared to the accuracy of HD-BB was considerably higher for an LD-SNP spacing of 20 cM than for 10 cM, which goes along with a much higher mean absolute difference (MAD) of PDMs for 20 cM than for 10 cM, as shown in Table 2. The absolute loss in accuracy was 0.03 for ELD-10 and 0.08 for ELD-20 in generation 2, and then increased steadily to 0.07 and 0.15 for ELD-10 and ELD-20 in generation 4. Because HD-SNP haplotypes were assumed known for training individuals, the accuracy of ELD-10 was identical to that of ELD-10+ in generation 2, as it was for ELD-20 and ELD-20+. In the following generations, the absolute loss in accuracy remained relatively constant for ELD-10+ and ELD-20+, in contrast to the ELD-10 and -20 strategies. The MAD of PDMs for ELD-10+ and ELD-20+ (not shown here) remained constant after generation 2.
Figure 3.—

Accuracies of GEBVs for 500 simulated QTL obtained by genomic selection using high-density genotyping (HD-BB) and low-density genotypic strategies using evenly spaced SNPs at 10- (ELD-10, ELD-10+) or 20-cM intervals (ELD-20 and ELD-20+). Strategies denoted by + indicate that parents were also high-density genotyped. Genomic selection training was based on 1000 individuals each having 1000 HD-SNP genotypes. Results are based on 25 replicates.
TABLE 2.
Mean absolute difference between true segregation indicators and probabilities of descent of high-density SNP alleles for equally spaced low-density (LD) SNPs, depending on the spacing of LD-SNPs, the generation after training, and the number of simulated QTL (25 replicates)
| No. of simulated QTL | LD-SNP spacing (cM) | Generation
|
||
|---|---|---|---|---|
| 2 | 3 | 4 | ||
| 100 | 10 | 0.146 | 0.151 | 0.153 |
| 500 | 10 | 0.146 | 0.152 | 0.157 |
| 1000 | 10 | 0.145 | 0.152 | 0.156 |
| 500 | 20 | 0.249 | 0.265 | 0.272 |
SE < 0.002.
Table 3 shows the loss of accuracy for ELD-10 and ELD-10+ as a percentage of the accuracy of HD-BB for different numbers of simulated QTL. For ELD-10 for 500 simulated QTL, the proportional loss of accuracy increased from 4.4% in generation 2 to 13.9% in generation 4, whereas the loss for ELD-10+ did not increase significantly over generations. Doubling the number of simulated QTL to 1000 had limited impact on the accuracy of HD-GS or on the proportional losses for ELD-10 and ELD-10+. Reducing the number of simulated QTL to 100 increased accuracies for all methods and reduced the decline in accuracies over generations (Table 3). But the proportional losses in accuracy for ELD-10 and ELD-10+ were similar to those observed for 500 QTL. The observed robustness of proportional losses in accuracy for the ELD approaches is consistent with the consistency of the MAD of PDMs across numbers of QTL simulated that is shown in Table 2.
TABLE 3.
Accuracy of GEBVs with high-density (HD) genotyping and loss of accuracy using approaches based on genotyping evenly spaced low-density (ELD) SNPs, as a percentage of the accuracy of HD-BayesB (HD-BB) in generations after training and for a trait with 100, 500, and 1000 simulated QTL
| No. of simulated QTL | Generation
|
|||
|---|---|---|---|---|
| Method | 2 | 3 | 4 | |
| HD-BB | 70.9 (±1.64) | 69.0 (±1.55) | 65.1 (±1.96) | |
| 100 | ELD-10 | 4.4 (±3.16) | 9.5 (±3.50) | 15.8 (±4.60) |
| ELD-10+ | 4.4 (±3.16) | 5.0 (±3.08) | 4.5 (±3.63) | |
| HD-BB | 64.4 (±1.03) | 58.2 (±1.41) | 55.3 (±1.37) | |
| 500 | ELD-10 | 4.4 (±3.29) | 8.6 (±4.82) | 13.9 (±5.43) |
| ELD-10+ | 4.4 (±3.29) | 5.7 (±3.89) | 5.2 (±3.71) | |
| HD-BB | 63.2 (±1.20) | 57.1 (±1.69) | 56.1 (±1.79) | |
| 1000 | ELD-10 | 4.6 (±2.98) | 8.6 (±3.81) | 14.2 (±4.90) |
| ELD-10+ | 4.6 (±2.98) | 4.5 (±3.12) | 6.0 (±3.86) | |
One thousand individuals each having 1000 HD-SNPs were used to estimate HD-SNP effects with BayesB and LD-SNPs at intervals of 10 cM were used to trace HD-SNP alleles (±SE, based on 25 replicates). ELD-10+ indicates that individuals used for breeding in each generation were also genotyped for HD-SNPs.
Table 4 shows the proportional loss in accuracy for the LD-GS strategies with selected HD-SNPs on the LD panel, compared to HD-GS. Selecting 110 SNPs using BayesB (strategy SLD-BB-110) had the lowest loss, in all cases, and was also slightly better than the ELD strategies. Proportional losses for SLD-BB-110 ranged from 0.7 to 3.0% and decreased over generations but increased with the number of QTL. The increase in the loss of accuracy with number of QTL occurred for all SLD strategies, except for SLD-FSS-110, for which proportional losses were greatest for 100 QTL and smallest for 500 QTL. Strategy SLD-BB-40 was superior to ELD-10, ELD-10+, and the two SLD-FSS methods with 100 QTL, but inferior to ELD-10+ with 1000 QTL. With 500 QTL, SLD-BB-40 had greater proportional losses than ELD-10 and ELD-10+ in generation 2, but similar losses to ELD-10+ afterwards. The loss of SLD-FSS-0.01 also increased with number of QTL, especially from 500 to 1000 QTL, but trends over generations differed by number of QTL: With 100 simulated QTL, the loss was constant over generations, but the loss clearly increased with 1000 simulated QTL and also from generation 3 to 4 with 500 QTL. Strategy SLD-FSS-110 was inferior to all other methods and its loss always increased over generations.
TABLE 4.
Loss of accuracy of GEBVs with genotyping of selected low-density (SLD) SNPs as a percentage of the accuracy obtained by high-density BayesB in generations after training and with 100, 500, and 1000 simulated QTL
| No. of simulated QTL | Generation
|
|||
|---|---|---|---|---|
| Method | 2 | 3 | 4 | |
| SLD-BB-110 | 1.1 (±3.2) | 0.8 (±3.2) | 0.7 (±4.2) | |
| 100 | SLD-BB-40 | 3.1 (±3.1) | 2.0 (±3.2) | 1.7 (±4.2) |
| SLD-FSS-0.01 | 9.0 (±2.4) | 8.1 (±3.3) | 8.7 (±4.0) | |
| SLD-FSS-110 | 18.2 (±3.2) | 23.1 (±3.2) | 24.9 (±4.2) | |
| SLD-BB-110 | 2.5 (±3.2) | 1.9 (±4.8) | 1.5 (±5.4) | |
| 500 | SLD-BB-40 | 7.8 (±3.2) | 6.5 (±4.8) | 4.02 (±5.0) |
| SLD-FSS-0.01 | 10.7 (±2.7) | 10.6 (±4.0) | 12.1 (±4.1) | |
| SLD-FSS-110 | 12.5 (±2.8) | 14.2 (±2.8) | 18.8 (±4.3) | |
| SLD-BB-110 | 3.0 (±3.3) | 2.6 (±3.8) | 2.3 (±4.9) | |
| 1000 | SLD-BB-40 | 9.2 (±3.3) | 9.7 (±3.7) | 8.4 (±4.9) |
| SLD-FSS-0.01 | 12.8 (±3.2) | 17.7 (±4.1) | 21.0 (±4.6) | |
| SLD-FSS-110 | 13.7 (±3.5) | 18.4 (±3.8) | 22.0 (±4.2) | |
One thousand individuals were used to estimate HD-SNP effects (±SE, based on 25 replicates). SLD-BB-110 and SLD-BB-40 refer to selection of 110 and 40 SNPs using BayesB; SLD-FSS-0.01 and SLD-FSS-110 refer to selection of SNPs by forward stepwise selection with a significance level of 0.01 or with a total of 110 SNPs selected.
DISCUSSION
The first objective of this study was to evaluate the loss of accuracy of GEBVs obtained by the ELD-GS approach. Losses were small in the first generation of LD-SNP genotyping (<5% at an LD-SNP density of 10 cM) but increased over generations. Genotyping individuals that were used for breeding in each generation also for the HD-SNP panel removed the increase in the loss of accuracy over generations. When the LD-SNP spacing was increased from 10 to 20 cM, the loss in accuracy nearly doubled regardless of whether parents were genotyped at HD-SNPs or not. The loss of accuracy with the ELD strategies was nearly independent of the number of QTL simulated. In the following, the main factors affecting accuracy and the loss of accuracy with ELD-GS are discussed.
Estimation of the effects of HD-SNP alleles:
The magnitude of accuracies of GEBVs with any GS strategy, including ELD-GS, depends to a large extent on the method used to estimate HD-SNP effects. In this study BayesB was used, because it gave the highest accuracy of GEBVs in comparison to alternative methods (Meuwissen et al. 2001; Habier et al. 2007), and Calus et al. (2008) showed that when SNP density is high, a Bayesian approach similar to BayesB was as good as the HAP-IBD method of Meuwissen and Goddard (2001) that utilizes information from both linkage disequilibrium and cosegregation. The accuracy of GEBVs obtained by BayesB in this study for 100 simulated QTL can be compared with accuracies found in Meuwissen et al. (2001), Solberg et al. (2006), and Habier et al. (2007), because the number of 50 segregating QTL was similar in all these studies. The decline in accuracies in generations following training was, however, lower in this study, because QTL positions were simulated differently. Here, QTL were randomly distributed across the genome, whereas they were located at the center of adjacent HD-SNP intervals in the other studies. Thus, on average higher disequilibrium is expected here between markers and QTL, because they can be located closer together.
Accuracies of all GS methods depend on linkage disequilibrium and the SNP density used for training. The average amount of disequilibrium at a given distance was higher in this study than in recent reports that measured disequilibrium with r2 in cattle (McKay et al. 2007; de Roos et al. 2008; Sargolzaei et al. 2008) and chicken (Andreescu et al. 2007). For example, de Roos et al. (2008) found average r2 values of 0.35, 0.22, 0.14, and 0.06 at distances 0.01, 0.04, 0.1, and 1 cM, whereas similar r2 values were obtained here at 0.25, 0.75, 1.25, and 3.25 cM. Although the simulated disequilibrium was higher than in recent reports, results and conclusions are transferable into practice, because the average distance between HD-SNPs was only 1 cM in this study, whereas panels with average distances of <0.05 cM are available in practice. Thus, the amount of usable disequilibrium in our simulated data should be comparable to that available in practice. The number of HD-SNP effects estimated in this study was smaller than that in practice (1000 vs. 50,000 HD-SNPs). This, however, should not affect conclusions about the HD-GS and ELD-GS approaches, because Fernando et al. (2008) showed that BayesB results in high accuracies of GEBVs with 60,000 HD-SNPs.
Accuracies of BayesB decreased rapidly in the first two generations after training (Figure 3), which is mainly attributed to the decay of additive-genetic relationships captured by HD-SNPs (Habier et al. 2007). The accuracy was greatest and the decay over generations was lowest for 100 simulated QTL, indicating that more genetic variation is captured by disequilibrium than for a greater number of QTL. Another reason for the decline of accuracy in early generations after training is the decay of disequilibrium between QTL and HD-SNPs due to recombinations. Information from both disequilibrium and cosegregation can be used for training to avoid the rapid decline of accuracy after training.
Estimation of HD-SNP haplotypes:
HD-SNP haplotypes of training individuals, which were assumed known here, must be inferred in practice from the HD-SNP genotypes of their parents. With HD-SNPs, this should be possible with high accuracy even if a training individual has no other close relatives in the training data. In addition, paternal half-sib families may be used for training, which not only allows HD-SNP haplotypes to be estimated more accurately, but also reduces the number of sires that must be genotyped.
Estimation of PDMs to trace HD-SNP alleles:
The loss of accuracy of ELD-GS is due to the incomplete prediction of grandparental origins of HD-SNP alleles using PDMs. The precision of PDMs depends on how accurate haplotypes of adjacent LD-SNP pairs can be traced using joint probabilities of segregation indicators and on how accurate the grandparental origin of a HD-SNP allele can be predicted conditional on the segregation indicators of flanking LD-SNPs. The accuracy to trace LD-SNP haplotypes is determined by the allele frequencies of LD-SNPs, the spacing between them, and the family structure. Intermediate allele frequencies are most informative for the prediction of segregation indicators at a single locus. This could be improved by using more informative microsatellites instead of SNPs, but high genotyping costs would not justify that. Furthermore, with a smaller LD-SNP spacing, flanking LD-SNPs provide more information to infer segregation indicators at a certain locus, which is important if the genotypes of parents and offspring are not informative at that locus. In addition, LD-SNP haplotypes can be inferred and traced better if family sizes are larger. The accuracy to predict the grandparental origin of a HD-SNP allele conditional on the segregation indicators is determined by the spacing of LD-SNPs and the occurrence of recombinations in the interval of flanking LD-SNPs. The larger the spacing of LD-SNPs is, the lower the precision of PDMs, because recombinations are more likely to occur between adjacent LD-SNPs. Precision of PDMs decreases even if no recombination is observed between adjacent LD-SNPs, because the possibility of a recombination is taken into account. If recombination did occur between adjacent LD-SNPs, the precision of PDMs is reduced more. With an odd number of crossovers, uncertainty about the grandparental origin of an allele increases more for HD-SNPs that are located near the center of the flanking LD-SNPs. With an even number of crossovers in the LD-SNP interval, PDMs can predict the wrong grandparental origin for some HD-SNPs in the interval. This worst-case scenario occurs if the alleles at adjacent LD-SNPs have identical grandparental origin, but the flanked HD-SNP allele has opposite origin. Recombination in the LD-SNP interval is expected to have a greater impact on accuracy of the GEBV when the flanked HD-SNP allele has large effect. This happens if the linked QTL has a large effect and its disequilibrium with the HD-SNP is high.
As expected, precision of PDMs, measured as the mean absolute difference of true segregation indicators and PDMs (Table 2), was independent of the number of simulated QTL and higher with a smaller LD-SNP spacing. The latter can be observed by comparing ELD-10+ and ELD-20+ in Table 2. Without HD genotyping of the parents of selection candidates, precision of PDMs declined over generations. The reason is that estimation of segregation indicators at SNPs in selection candidates depends on information about the order of LD-SNP genotypes of the parents of selection candidates. In generation 2, the order of LD-SNP genotypes of parents (training individuals in generation 1) was assumed known, but in later generations this order had to be inferred from observed, unordered LD-SNP genotypes. Information about the order of LD-SNPs genotypes of training individuals reduces to an asymptote over generations such that precision of segregation indicators will decrease less in later generations and finally the precision is expected to remain constant. For strategies ELD-10+ and ELD-20+ the order of LD-SNPs of parents is known, because they are HD genotyped and the precision of PDMs remains constant over generations (results not shown). The effect of a decreasing precision of PDMs over generations on loss of accuracy cannot be determined here, because the increasing loss for ELD-10 and ELD-20 is also due to the uncertainty of PDMs in previous generations.
Precision of PDMs does not depend on the number of HD-SNPs if LD-SNPs are not a subset of HD-SNPs. As is realistic, LD-SNPs were part of the HD-SNPs in this study, and thus when HD-SNP density is higher than simulated here, the mean absolute difference of PDMs is expected to be slightly lower than found here. The reason is that LD-SNPs have PDMs with higher precision than HD-SNPs that are not LD-SNPs. Therefore, as the density of the HD-SNPs increases, the fraction of LD-SNPs decreases and, thus, precision will be somewhat lower than shown in Table 2. For example, when the number of HD-SNPs was increased from 1000 to 4000, the range of the mean absolute difference increased from 0.146–0.159 to 0.158–0.169. The loss of accuracy with ELD also increased by 1–2%. By increasing the number of HD-SNPs from 1000 to 4000, the fraction of LD-SNPs among HD-SNPs decreased from 11 to 2.75%. Thus with a higher density of HD-SNPs, precision of PDMs is not expected to decrease such that loss of accuracy would increase significantly. This increased loss with 4000 HD-SNPs, however, might not be caused by lower precision of PDMs; recombination between adjacent LD-SNPs might also have a greater impact on loss of accuracy than with a lower density of HD-SNPs. The explanation is that with higher marker density, disequilibrium between QTL and HD-SNPs is expected to be greater, such that there are fewer HD-SNPs explaining more genetic variation than with lower density of HD-SNPs. In practice, this may become more important in the future when SNP density further increases above 50,000.
The optimal spacing of LD-SNPs cannot be determined without further comprehensive studies, taking into account genotyping costs, accuracy of GEBVs, and population structures. However, it is unlikely that genotyping costs continue to decrease below a certain number of LD-SNPs. For example, genotyping cost for a LD-SNP spacing of 10 and 20 cM might not differ much, if at all. The loss of accuracy, in contrast, was twice as high for 20 cM than for 10 cM (Figure 3). Consequently, a spacing of 10 cM may be more cost effective than a 20-cM spacing, so that 330 LD-SNPs would be used for livestock species. Panels of 384 SNPs are currently available on a cost-effective basis. To trace alleles with similar precision for all regions of the genome, LD-SNPs should be evenly spaced. Alternatively, some LD-SNPs could be selected that are located closer to potential QTL positions, which could increase the accuracy of GEBV based on LD-SNP panels. The disadvantage is that a suboptimal spacing may reduce the accuracy of GEBV for other traits.
Prediction of GEBVs:
The loss of accuracy (absolute and proportional) with ELD-10 and ELD-20 clearly increased from one generation to the next, because uncertainty about the grandparental origin of HD-SNP alleles accumulates over generations (Figure 3, Table 3). It will continue to increase in subsequent generations, unless HD-SNP haplotypes of parents are obtained by genotyping the parents for HD-SNPs. In that case, the loss of accuracy will be much lower, as illustrated by results for strategies ELD-10+ and ELD-20+ in Figure 3 and Table 3, noting that HD-SNP haplotypes of the parents were assumed known here. Moreover, because the precision of PDMs is constant over generations for these two strategies and the accuracy of GEBVs obtained by BayesB will not decrease notably after information from genetic relationships captured by markers has vanished (Habier et al. 2007), their proportional loss of accuracy will remain constant.
The proposed ELD-GS approach uses three types of information to predict GEBVs of selection candidates. First, both linkage disequilibrium between QTL and HD-SNPs and additive-genetic relationships captured by HD-SNPs are used to estimate effects of HD-SNP alleles. Then, cosegregation of LD-SNPs is used to trace the effects of HD-SNP alleles across generations. The accuracy of the ELD-GS approach could be improved by using the information from cosegregation not only for tracing, but also to estimate HD-SNP effects if individuals in later generations also have phenotypes.
LD-SNP panels based on selected HD-SNPs:
The second objective of this study was to evaluate the loss of accuracy for LD-SNP panels on the basis of selected HD-SNPs. Strategy SLD-BB-110, which used the top 110 SNPs on the basis of BayesB analysis of the HD-SNP training data, was superior to all other LD-GS methods, and the SLD-FSS-110 strategy, which picked 110 SNPs on the basis of forward least-squares regression, was always worst. Strategy SLD-FSS-0.01, which picked between 30 and 35 SNPs that met the 0.01 significance criterion, had lower accuracy than both SLD-BB-40 and ELD-10+, but was better than ELD-10 if the number of QTL was low. For all SLD strategies, except SLD-FSS-110, the loss of accuracy increased with the number of QTL. The reason for this is that the selected SNPs explain less genetic variation if genetic effects are spread over more QTL, such that the accuracy that is attained is lower. Accuracy is also reduced if the SNPs that are selected are false positives. This was the case for strategy SLD-FSS-110, especially when the number of QTL was low, which explains why SLD-FSS-110 had the highest loss at 100 QTL (Table 3). Furthermore, the increasing loss over generations for SLD-FSS-0.01 at a higher number of QTL shows that this approach is less able to capture information from disequilibrium than LD-GS methods based on effects estimated by BayesB, for which the loss decreased over generations (Table 3). This can be concluded because the impact of disequilibrium on accuracy of GEBVs increases over generations, whereas that of genetic relationships declines (Habier et al. 2007).
Application of LD-GS in breeding:
The full potential of GS can be exploited only if it is cost effective in all four selection paths that contribute to typical genetic improvement programs in livestock. When based on loss of accuracy compared to HD-GS, inclusion of SNPs that were fitted most frequently in BayesB on an LD-SNP panel seems to be the method of choice for a single trait. The number of selected SNPs should not be too small; otherwise the loss in accuracy can be substantial, especially in the first generation after training and if the number of QTL is large. This can be seen by comparing results for SLD-BB-110 with those for SLD-BB-40 in Table 3. An important disadvantage of marker-selection approaches, however, is that different SNPs are likely to be selected for different traits such that an LD-SNP panel developed for one trait would probably work less well for another trait. If each economic trait requires its own LD-SNP panel, costs for development and genotyping may not be lower than for the HD-SNP panel. The ELD-GS strategies, in contrast, are trait independent, because the information from cosegregation of LD-SNP alleles can be utilized with one LD-SNP panel for all traits. It is even likely that LD-SNP panels can be developed that work well across populations, breeds, and generations, whereas marker-selection approaches may require different panels in different breeds and across generations. Moreover, the panel used for ELD-GS is independent from the statistical model used to estimate genetic effects. In addition, it can trace not only effects of QTL or markers, but also dominance and epistatic effects. Another advantage of ELD-GS is that it selects on the whole genome. Marker-selection approaches, in contrast, may select on certain portions of the genome, which could have consequences for fixation of undesirable alleles or loss of favorable alleles in portions of the genome that are not covered by the selected SNPs. Finally, the ELD-SNP panels should also be suitable for parentage verification and traceability, allowing a single panel to meet multiple needs.
To limit the loss of accuracy using ELD-GS, parents that are used for breeding could be HD genotyped each generation. The reduced loss of accuracy of ELD-10+ over ELD-10 demonstrates the benefit of this. In this case, parents are genotyped twice, first using the LD-SNP panel as selection candidates and then using the HD-SNP panel to infer HD-SNP haplotypes. This has to be taken into account when calculating genotyping costs. The HD genotyping may not be needed for all parents, in particular those that have low economic benefit in a breeding program, for example, production cows in a dairy cattle improvement program. In addition, depending on the family structure, HD genotypes may be derived with sufficient accuracy on some parents on the basis of LD genotypes. Further optimization is warranted to determine which individuals should be HD genotyped.
Because the actual number of QTL is unknown in reality, an LD-GS approach should perform well regardless of the number of QTL. The ELD strategies appear to have this advantage. Strategy SLD-BB-110 always had the lowest loss of accuracy but loss increased with the number of QTL. Strategies SLD-BB-40 and SLD-FSS-0.01 lost substantial accuracy at a high number of QTL. Economic analyses are required to determine which LD-GS approach is optimal for each of the four selection paths on a case-by-case basis. Genotyping technology may be improved in the future such that HD-SNP panels will become comparable in cost to LD-SNP panels, but until then ELD-GS will contribute to utilizing the potential of GS in livestock.
Conclusions:
The number of markers that selection candidates are genotyped for to predict GEBVs using HD-SNP panels can be reduced substantially with limited loss of accuracy in comparison to HD genotyping of selection candidates. For a specific trait, the loss in accuracy is smallest if the markers that are fitted most frequently in the MCMC algorithm of BayesB are included on a LD-SNP panel. The loss in accuracy is low for this approach across generations following training and the number of QTL affecting the quantitative trait. Strategies that select markers using FSS, in contrast, are inferior to marker selection using BayesB and their loss of accuracy depends greatly on the number of QTL, which is unknown in reality. Strategies that select markers on the basis of the data, however, result in SNP panels that are trait dependent. In contrast, the ELD strategies that place LD-SNPs evenly spaced across the genome and use these to trace HD-SNPs from HD-genotyped ancestors to selection candidates are trait independent. The loss of accuracy for ELD-GS is also independent of the number of QTL and smallest if HD-SNP haplotypes of the parents of selection candidates are derived each generation. In this case, the loss of accuracy in later generations is constant. Further economic analyses are needed to determine which LD-GS approach is cost effective for specific breeding applications.
Acknowledgments
D. Habier acknowledges financial support from the Deutsche Forschungsgemeinschaft. This research was further supported by the U.S. Department of Agriculture, National Research Initiative grant USDA-NRI-2007-35205-17862 and by State of lowa Hatch and Multi-State Research Funds.
References
- Abraham, K. J., L. R. Totir and R. L. Fernando, 2007. Improved techniques for sampling complex pedigrees with the Gibbs sampler. Genet. Sel. Evol. 39 27–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreescu, C., S. Avendano, S. R. Brown, A. Hassen, S. J. Lamont et al., 2007. Linkage disequilibrium in related breeding lines of chickens. Genetics 177 2161–2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamin, H. D., and C. J. Nicola, 2004. Principal component analysis for selection of optimal SNP-sets that capture intragenic genetic variation. Genet. Epidemiol. 26 11–21. [DOI] [PubMed] [Google Scholar]
- Burdick, J. T., W.-M. Chen, G. R. Abecasis and V. G. Cheung, 2006. In silico method for inferring genotypes in pedigrees. Nat. Genet. 38 1002–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calus, M. P. L., T. H. E. Meuwissen, A. P. W. de Roos and R. F. Veerkamp, 2008. Accuracy of genomic selection using different methods to define haplotypes. Genetics 178 553–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Roos, A. P. W., B. J. Hayes, R. J. Spelman and M. E. Goddard, 2008. Linkage disequilibrium and persistence of phase in Holstein-Friesian, Jersey and Angus cattle. Genetics 179 1503–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elston, R. C., and J. Stewart, 1971. A general model for the genetic analysis of pedigree data. Hum. Hered. 21 523–542. [DOI] [PubMed] [Google Scholar]
- Fernando, R. L., D. Habier, C. Stricker, J. C. M. Dekkers and L. R. Totir, 2008. Genomic selection. Acta Agric. Scand. Sect. A Anim. Sci. 57 192–195. [Google Scholar]
- George, E. J., and R. E. McCulloch, 1993. Variable selection via Gibbs sampling. J. Am. Stat. Assoc. 91 883–904. [Google Scholar]
- Goddard, M. E., 2008. The use of high density genotyping in animal health. Animal genomics for animal health. Dev. Biol. 132 383–390. [DOI] [PubMed] [Google Scholar]
- Habier, D., R. L. Fernando and J. C. M. Dekkers, 2007. The impact of genetic relationship information on genome-assisted breeding values. Genetics 177 2389–2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes, B., and M. E. Goddard, 2008. Artificial selection method and reagents. Patent Application No. WO/2008/074101.
- Hayes, B. J., P. M. Visscher, H. C. McPartlan and M. E. Goddard, 2003. Novel multilocus measure of linkage disequilibrium to estimate past effective population size. Genome Res. 13 635–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heath, S. C., 1997. Markov chain Monte Carlo segregation and linkage analysis for oligogenic models. Am. J. Hum. Genet. 61 748–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson, G. C. L., L. Esposito, B. J. Barratt, A. N. Smith, J. Heward et al., 2001. Haplotype tagging for the identification of common disease genes. Nat. Genet. 29 233–237. [DOI] [PubMed] [Google Scholar]
- Karlin, S., 1984. Theoretical aspects of genetic map functions in recombination processes, pp. 209–228 in Human Population Genetics: The Pittsburgh Symposium, edited by A. Chakravarti. Van Nostrand Reinhold, New York.
- Long, N., D. Gianola, G. J. M. Rosa, S. Weigel and S. Avendano, 2007. Machine learning classification procedure for selecting SNPs in genomic selection: application to early mortality in broilers. J. Anim. Breed. Genet. 124 377–389. [DOI] [PubMed] [Google Scholar]
- Marchini, J., B. Howie, S. Myers, G. McVean and P. Donnelly, 2007. A new multipoint method for genome-wide association studies by imputation of genotypes. Nat. Genet. 39 906–913. [DOI] [PubMed] [Google Scholar]
- McKay, S. D., R. D. Schnabel, B. M. Murdoch, L. K. Matukumalli, J. Aerts et al., 2007. Whole genome linkage disequilibrium maps in cattle. BMC Genet. 8 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meuwissen, T. H. E., and M. E. Goddard, 2001. Prediction of identity by descent probabilities from marker-haplotypes. Genet. Sel. Evol. 33 605–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meuwissen, T. H. E., B. J. Hayes and M. E. Goddard, 2001. Prediction of total genetic value using genome-wide dense marker maps. Genetics 157 1819–1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moser, G., B. Tier, R. E. Crump, J. Soelkner, K. R. Zenger et al., 2007. Estimation of molecular breeding values in genome wide selection using supervised dimension reduction based on partial least squares. Papers and Abstracts From the Workshop on QTL and Marker-Assisted Selection, March 22–23, 2007, Toulouse, France, edited by A. Legarra.
- Ohta, T., and M. Kimura, 1969. Linkage disequilibrium at steady state determined by random genetic drift and recurrent mutation. Genetics 63 229–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil, N., A. J. Berno, D. A. Hinds, W. A. Barrett, J. M. Doshi et al., 2001. Blocks of limited haplotype diversity revealed by high-resolution scanning of human chromosome 21. Science 294 1719–1723. [DOI] [PubMed] [Google Scholar]
- Piyasatian, N., R. L. Fernando and J. C. M. Dekkers, 2007. Genomic selection for marker-assisted improvement in line crosses. Theor. Appl. Genet. 115 665–674. [DOI] [PubMed] [Google Scholar]
- Sargolzaei, M., F. S. Schenkel, G. B. Jansen and L. R. Schaeffer, 2008. Extent of linkage disequilibrium in Holstein cattle in North America. J. Dairy Sci. 91 2106–2117. [DOI] [PubMed] [Google Scholar]
- Servin, B., and M. Stephens, 2007. Imputation-based analysis of association studies: candidate regions and quantitative traits. PLoS Genet. 3 e114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solberg, T. R., A. Sonesson, J. Wooliams and T. H. E. Meuwissen, 2006. Genomic selection using different marker types and density. 8th World Congress on Genetics Applied to Livestock Production, August 13–18, 2006, Belo Horizonte, Minas Gerais, Brazil.
- Sved, J. A., 1969. Linkage disequilibrium and homozygosity of chromosome segments in finite populations. Theor. Popul. Biol. 2 125–141. [DOI] [PubMed] [Google Scholar]
- Thomas, A., A. Gutin, V. Abkevich and A. Bansal, 2000. Multilocus linkage analysis by blocked Gibbs sampling. Stat. Comput. 10 259–269. [Google Scholar]
- Tier, B., J. Cavanagh, R. Crump, M. Khatkar, G. Moser et al., 2007. Genome wide selection: experiences from the Australian Dairy Industry. The 3rd International Conference on Quantitative Genetics, August 2007, Hangzhou, China.
- Woolaston, A. F., B. Tier and R. D. Murison, 2007. Principal components regression of SNP data to predict genetic merit. Papers and Abstracts From the Workshop on QTL and Marker-Assisted Selection, March 22–23, 2007, Toulouse, France, edited by A. Legarra.
- Xu, S., 2003. Estimating polygenic effects using markers of the entire genome. Genetics 163 789–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, S., 2007. An empirical Bayes method for estimating epistatic effects of quantitative trait loci. Biometrics 63 513–521. [DOI] [PubMed] [Google Scholar]
- Yu, J., J. B. Holland, M. D. McMullen and E. S. Buckler, 2008. Genetic design and statistical power of nested association mapping in maize. Genetics 178 539–551. [DOI] [PMC free article] [PubMed] [Google Scholar]