

Abstract
The availability of genomewide dense markers brings opportunities and challenges to breeding programs. An important question concerns the ways in which dense markers and pedigrees, together with phenotypic records, should be used to arrive at predictions of genetic values for complex traits. If a large number of markers are included in a regression model, marker-specific shrinkage of regression coefficients may be needed. For this reason, the Bayesian least absolute shrinkage and selection operator (LASSO) (BL) appears to be an interesting approach for fitting marker effects in a regression model. This article adapts the BL to arrive at a regression model where markers, pedigrees, and covariates other than markers are considered jointly. Connections between BL and other marker-based regression models are discussed, and the sensitivity of BL with respect to the choice of prior distributions assigned to key parameters is evaluated using simulation. The proposed model was fitted to two data sets from wheat and mouse populations, and evaluated using cross-validation methods. Results indicate that inclusion of markers in the regression further improved the predictive ability of models. An R program that implements the proposed model is freely available.
GENOMEWIDE dense marker maps are now available for many species in plants and animals (e.g., WANG et al. 2005). An important challenge is how this information should be incorporated into statistical models for prediction of genetic values in animal and plant breeding programs or prediction of diseases.
A standard quantitative genetic model assumes that genetic 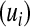 and environmental
and environmental 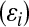 effects act additively, to produce phenotypic outcomes
effects act additively, to produce phenotypic outcomes 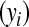 according to the rule
according to the rule 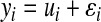 . The information set now available for predicting genetic values may include, in addition to phenotypic records, a pedigree, molecular markers, or both.
. The information set now available for predicting genetic values may include, in addition to phenotypic records, a pedigree, molecular markers, or both.
Several methodologies have been proposed for incorporating dense marker data into regression models. A distinction can be made between methods that explicitly regress phenotypic records on markers via the regression function 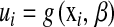 , where
, where 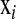 is a vector of marker covariates and
is a vector of marker covariates and 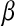 is a vector of regression coefficients, e.g.,
is a vector of regression coefficients, e.g., 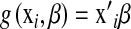 , and those that view genetic values as a function of the subject and use marker information to build a (co)variance structure between subjects. The first group of methods includes standard Bayesian regression (BR) with random coefficients, i.e., a Bayesian model where regression coefficients are assigned the same Gaussian prior, and other shrinkage methods such as Bayes A or Bayes B of Meuwissen et al. (2001), and specifications described in Gianola et al. (2003). The second type of approach was suggested by Gianola et al. (2006) and Gianola and van Kaam (2008), who proposed using reproducing kernel Hilbert spaces regression (RKHS), with the information set consisting of SNP (single-nucleotide polymorphism) genotypes, possibly supplemented by genealogies. As discussed in De los Campos et al. (2009), in this approach, marker information is used to create a prior (co)variance structure between genomic values,
, and those that view genetic values as a function of the subject and use marker information to build a (co)variance structure between subjects. The first group of methods includes standard Bayesian regression (BR) with random coefficients, i.e., a Bayesian model where regression coefficients are assigned the same Gaussian prior, and other shrinkage methods such as Bayes A or Bayes B of Meuwissen et al. (2001), and specifications described in Gianola et al. (2003). The second type of approach was suggested by Gianola et al. (2006) and Gianola and van Kaam (2008), who proposed using reproducing kernel Hilbert spaces regression (RKHS), with the information set consisting of SNP (single-nucleotide polymorphism) genotypes, possibly supplemented by genealogies. As discussed in De los Campos et al. (2009), in this approach, marker information is used to create a prior (co)variance structure between genomic values, 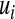 ,
, 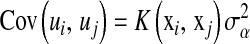 , where
, where 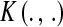 is some positive-definite function and
is some positive-definite function and 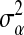 is a parameter to be estimated from the data.
is a parameter to be estimated from the data.
The two types of approaches lead to predictions of genomic values for quantitative traits. An advantage of explicitly regressing phenotypes on marker covariates is that the model can produce information about genomic regions that may affect the trait of interest. However, a main difficulty is that the number of regression coefficients (p) is typically large, even larger than the number of records (n), with p ≫ n. Therefore, a crucial aspect is how this methodology can cope with the curse of dimensionality and with colinearity.
With whole-genome scans, many markers are likely to be located in regions that are not involved in the determination of traits of interest. On the other hand, some markers may be in linkage disequilibrium with some QTL or in regions harboring genes involved in the infinitesimal component of the trait. This suggests that differential shrinkage of marker effects should be a feature of the model, as noted by Meuwissen et al. (2001). Tibshirani (1996) proposed a regression method (least absolute shrinkage and selection operator, LASSO) that combines the good features of subset selection (i.e., variable selection) with the shrinkage produced by BR. Recently, Park and Casella (2008) presented a Bayesian version of the LASSO method (Bayesian LASSO, BL) and suggested a Gibbs sampler for its implementation. Alternatives to the Gibbs sampler of Park and Casella are discussed in Hans (2008). While the BL described by Park and Casella is appealing for the reasons mentioned above, it does not accommodate pedigree information or regression on (co)variates other than the markers for which a different shrinkage approach may be desired.
Several authors have considered combining pedigree and marker data into a single model in the context of QTL analysis (e.g., Fernando and Grossman 1989; Bink et al. 2002, 2008). Here, in this spirit, the BL is modified and extended to accommodate pedigree information as well as covariates other than markers.
The main objectives of this article are to (1) discuss the use of BL and related methods in the context of linear regression of quantitative traits on molecular markers, (2) evaluate the sensitivity of BL with respect to the choice of the prior for the regularization parameter, (3) extend the BL so that pedigrees or regressions on covariates other than markers can also be included in the model, and (4) evaluate the methodology using data from a self-pollinated wheat population and an outcross mouse population. The article is organized as follows: the first section, bayesian lasso, introduces the BL as presented in Park and Casella (2008) and discusses connections between BL and closely related methods, such as those proposed by Meuwissen et al. (2001) or variants proposed by other authors. monte carlo study evaluates the sensitivity of BL with respect to the choice of prior for the regularization parameter. bayesian regression coupled with lasso presents an extension of BL, treating effects of different types of regressors with different priors. In data analysis, the proposed methodology is applied to two data sets representing a collection of wheat lines and a population of mice. concluding remarks are provided in the final section of the article. An R function (R Development Core Team 2008) that fits the model and data sets used in this article are made available (see supporting information, File S1 and File S2).
THE BAYESIAN LASSO
Tibshirani (1996) proposed using the sum of the absolute values of the regression coefficients (or L1 norm) as a penalty in regression models, to simultaneously produce variable selection and shrinkage of coefficients; the proposed methodology was termed LASSO. In LASSO, estimates are obtained by solving the constrained optimization problem
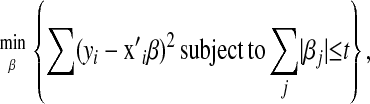 |
(1) |
where 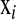 is a vector of covariates,
is a vector of covariates, 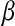 is the corresponding vector of regression coefficients, and t is an arbitrary positive constant. Above, it is assumed that data are centered, i.e.,
is the corresponding vector of regression coefficients, and t is an arbitrary positive constant. Above, it is assumed that data are centered, i.e., 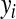 has zero mean. Optimization problem (1) is equivalent to
has zero mean. Optimization problem (1) is equivalent to
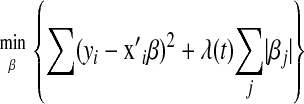 |
(2) |
(e.g., Tibshirani 1996), for some value of the smoothing parameter 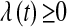 . It is known that the solution to (2) may involve zeroing out some elements of
. It is known that the solution to (2) may involve zeroing out some elements of 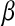 , and there are many ways of illustrating why this may be so. One manner is to examine the shape of the feasible set in (1) (e.g., Tibshirani 1996); another way is to consider the Bayesian interpretation of the LASSO. From (2), it follows that the solution can be viewed as the posterior mode in a Bayesian model with Gaussian likelihood,
, and there are many ways of illustrating why this may be so. One manner is to examine the shape of the feasible set in (1) (e.g., Tibshirani 1996); another way is to consider the Bayesian interpretation of the LASSO. From (2), it follows that the solution can be viewed as the posterior mode in a Bayesian model with Gaussian likelihood, 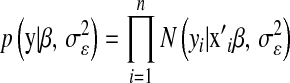 , and a prior on
, and a prior on 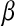 that is the product of p independent, zero-mean, double-exponential (DE) densities; that is,
that is the product of p independent, zero-mean, double-exponential (DE) densities; that is, 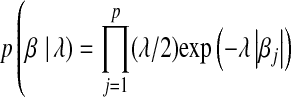 . In contrast, BR is obtained by assuming the same likelihood and a prior on
. In contrast, BR is obtained by assuming the same likelihood and a prior on 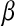 that is the product of p independent normal densities; that is,
that is the product of p independent normal densities; that is, 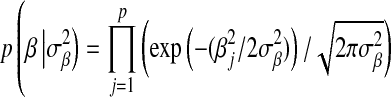 , where
, where 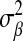 is a variance parameter common to all regression coefficients. The difference between these two priors is illustrated in Figure 1: the DE density places more mass at zero and has thicker tails than the Gaussian distribution. From this perspective, relative to BR, LASSO produces stronger shrinkage of regression coefficients that are close to zero and less shrinkage of those with large absolute values.
is a variance parameter common to all regression coefficients. The difference between these two priors is illustrated in Figure 1: the DE density places more mass at zero and has thicker tails than the Gaussian distribution. From this perspective, relative to BR, LASSO produces stronger shrinkage of regression coefficients that are close to zero and less shrinkage of those with large absolute values.
Figure 1.—

Densities of a normal and of a double-exponential distribution (both with null mean and with unit variance).
Parameter 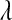 , sometimes referred to as a regularization parameter, plays a central role: as it approaches zero, the solution to (2) tends to ordinary least squares, while large values of
, sometimes referred to as a regularization parameter, plays a central role: as it approaches zero, the solution to (2) tends to ordinary least squares, while large values of 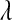 penalize the L1 norm of
penalize the L1 norm of 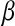 ,
, 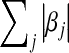 , highly. In the Bayesian view of LASSO,
, highly. In the Bayesian view of LASSO, 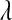 controls the prior on
controls the prior on 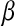 , with large values of this parameter associated with more informative (sharper) priors.
, with large values of this parameter associated with more informative (sharper) priors.
By construction, the non-Bayesian LASSO solution admits at most n − 1 nonzero regression coefficients (e.g., Park and Casella 2008). This is not desirable in models with dense marker-based regressions since, a priori, there is no reason why the number of markers with effectively nonzero effects should be smaller than the number of observations. This problem does not arise in BL, which is discussed next.
A computationally convenient hierarchical formulation of a DE distribution is obtained by exploiting the fact that the DE density can be represented as a mixture of scaled Gaussian densities (e.g., Andrews and Mallows 1974; Rosa 1999), where the mixing process of the variances is an exponential distribution. Following Park and Casella (2008),
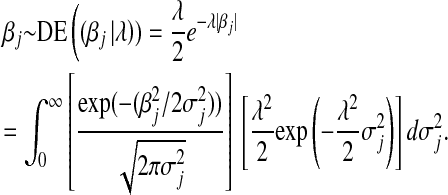 |
Above, 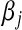 is the unknown effect of the jth marker and
is the unknown effect of the jth marker and 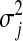 is a variance parameter (measuring prior uncertainty) associated with
is a variance parameter (measuring prior uncertainty) associated with 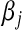 . Using this, Park and Casella (2008) suggested the following hierarchical model (BL),
. Using this, Park and Casella (2008) suggested the following hierarchical model (BL),
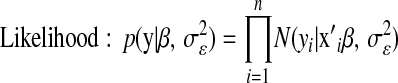 |
(3) |
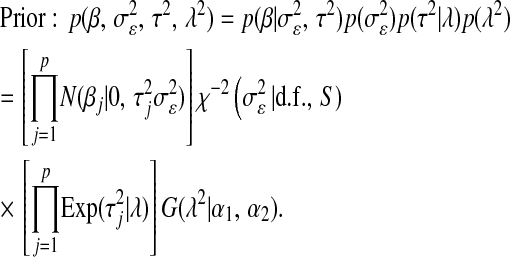 |
(4) |
Above, 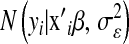 and
and 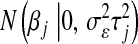 are normal densities centered at
are normal densities centered at 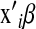 and 0, with variances
and 0, with variances 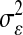 and
and 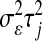 , respectively;
, respectively; 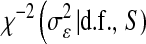 is a scaled-inverted chi-square density, with degrees of freedom
is a scaled-inverted chi-square density, with degrees of freedom 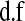 . and scale
. and scale 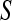 , in this parameterization,
, in this parameterization, 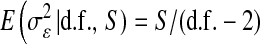 ;
; 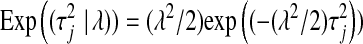 is an exponential density indexed by a single parameter,
is an exponential density indexed by a single parameter, 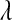 , and
, and 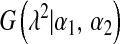 is a Gamma distribution, with shape parameter
is a Gamma distribution, with shape parameter 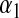 and rate parameter
and rate parameter 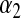 .
.
The role of the 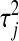 's becomes more clear by changing variables in (3) and (4) from
's becomes more clear by changing variables in (3) and (4) from 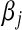 to
to 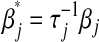 . After this change of variables, the product of the likelihood function and of the joint prior for the regression coefficients,
. After this change of variables, the product of the likelihood function and of the joint prior for the regression coefficients, 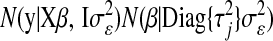 , becomes
, becomes 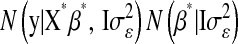 , where
, where 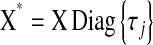 and
and 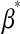 is a vector of regression coefficients with homogeneous variance. Thus, one way of viewing this class of regression models is as a standard BR model with additional unknowns,
is a vector of regression coefficients with homogeneous variance. Thus, one way of viewing this class of regression models is as a standard BR model with additional unknowns, 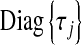 , which assign different weights to the columns of X, with
, which assign different weights to the columns of X, with 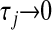 being equivalent to removing the jth covariate from the model.
being equivalent to removing the jth covariate from the model.
Park and Casella (2008) presented a set of fully conditional distributions that allows fitting the BL model via the Gibbs sampler. Some of these distributions are discussed next, to illustrate main features of the algorithm.
Location parameters:
In the Gibbs sampler of Park and Casella (2008), the fully conditional distribution of the regression coefficients is multivariate normal with mean (covariance matrix) equal to the solution (inverse of the coefficient matrix) of the system of equations,
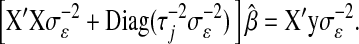 |
(5) |
Recall that ordinary least-squares estimates are obtained by solving 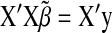 and that the counterpart of (5) in BR is
and that the counterpart of (5) in BR is 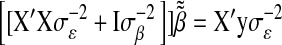 . A key aspect of BL is that it produces a shrinkage that is marker specific, contrary to BR. Since
. A key aspect of BL is that it produces a shrinkage that is marker specific, contrary to BR. Since  is a scaling factor common to all regression coefficients, the differential shrinkage is due to the
is a scaling factor common to all regression coefficients, the differential shrinkage is due to the 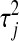 's. If
's. If 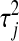 is large, i.e., a large variance is associated with the effect of the jth marker, the quantity added to the diagonal will be small. Conversely, if a small variance is associated with the effect of the jth coefficient,
is large, i.e., a large variance is associated with the effect of the jth marker, the quantity added to the diagonal will be small. Conversely, if a small variance is associated with the effect of the jth coefficient, 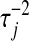 will be large. Adding a large constant to the jth diagonal element shrinks the least-squares estimates toward zero and reduces the variance of its fully conditional distribution.
will be large. Adding a large constant to the jth diagonal element shrinks the least-squares estimates toward zero and reduces the variance of its fully conditional distribution.
Variances of the regression coefficients:
An important aspect of the algorithm is how samples of the regression coefficients affect realizations of the variances of marker effects. In BL, the fully conditional posterior distributions of the  's can be shown to be inverse Gaussian (e.g., Chhikara and Folks 1989), with mean
's can be shown to be inverse Gaussian (e.g., Chhikara and Folks 1989), with mean 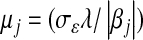 and scale parameter
and scale parameter 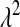 . For a given
. For a given 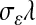 , a small absolute value of
, a small absolute value of 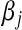 will lead to a fully conditional distribution of
will lead to a fully conditional distribution of 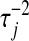 with a large mean, which in turn will generate relatively small values of
with a large mean, which in turn will generate relatively small values of 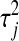 .
.
The λ parameter of the exponential prior:
In the standard LASSO, 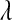 controls the trade-off between goodness of fit and model complexity, and this may be crucial in defining the ability of a model to uncover signal. Small values of
controls the trade-off between goodness of fit and model complexity, and this may be crucial in defining the ability of a model to uncover signal. Small values of 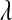 produce better fit, in the sense of the residual sum of squares (
produce better fit, in the sense of the residual sum of squares (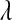 = 0 gives ordinary least squares); as
= 0 gives ordinary least squares); as 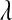 increases, the penalty on model complexity increases (in optimization problem (1) the feasible set is smaller). On the other hand, in BL,
increases, the penalty on model complexity increases (in optimization problem (1) the feasible set is smaller). On the other hand, in BL, 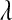 controls the shape of the prior distribution assigned to the
controls the shape of the prior distribution assigned to the 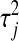 's. In general, the exponential prior assigns more density to small values of the
's. In general, the exponential prior assigns more density to small values of the 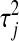 's than to large ones, and this may be reasonable for most SNPs under the expectation that most of their effects are nil.
's than to large ones, and this may be reasonable for most SNPs under the expectation that most of their effects are nil.
In BL 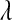 can be treated as any other unknown. If, as in (4), a Gamma prior is assigned to
can be treated as any other unknown. If, as in (4), a Gamma prior is assigned to 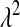 , the fully conditional posterior distribution of
, the fully conditional posterior distribution of 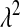 is also Gamma, with shape and rate parameters equal to
is also Gamma, with shape and rate parameters equal to 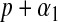 and
and 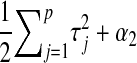 , respectively. The expectation of this Gamma distribution is
, respectively. The expectation of this Gamma distribution is  , so a large value of
, so a large value of 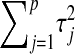 will lead to a relatively small
will lead to a relatively small  , and the opposite will occur if the sum of the variances of the regression coefficients is small.
, and the opposite will occur if the sum of the variances of the regression coefficients is small.
Relationship between LASSO and other regression models used in genomic selection:
Standard BR may not be suitable for regressing phenotypes on a large number of markers because shrinkage of regression coefficients is homogeneous across markers (Fernando et al. 2007). In contrast, in BL the variance is marker specific, producing shrinkage whose extent is related to the absolute value of the estimated regression coefficient.
Meuwissen et al. (2001) recognized that marker-specific variances may be needed and suggested regression models based on marginal priors that are also mixtures of scaled-Gaussian distributions (“Bayes A”) or mixtures of scaled-Gaussian distributions and of a point mass at zero (“Bayes B”). In these models, the likelihood is as in (3) and, in Bayes A, the prior is
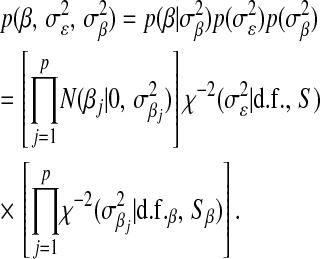 |
(6) |
The first two components of (6), 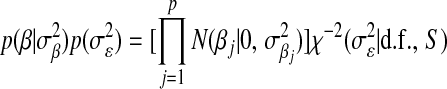 , are the counterparts of the first two components of (4),
, are the counterparts of the first two components of (4), 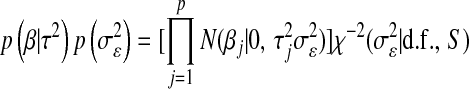 , with
, with 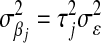 . The difference between BL and Bayes A (or Bayes B) is how the priors of the variances of the marker-specific regression coefficients (
. The difference between BL and Bayes A (or Bayes B) is how the priors of the variances of the marker-specific regression coefficients (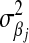 in Bayes A and
in Bayes A and 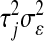 in BL) are specified. At this level, Bayes A and BL differ in two respects:
in BL) are specified. At this level, Bayes A and BL differ in two respects:
-
In Bayes A, the prior assumption is that the marker-specific variances are independent random variables following the same scaled-inverted chi-square distribution with known prior degree of belief
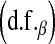 and scale
and scale 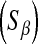 . In BL, the assumption is that these variances are independent as well, but each following the same exponential distribution with unknown parameter
. In BL, the assumption is that these variances are independent as well, but each following the same exponential distribution with unknown parameter 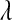 . The conditional (given
. The conditional (given 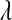 ) marginal prior in BL
) marginal prior in BL 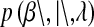 is DE, while in Bayes A
is DE, while in Bayes A 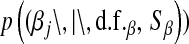 is a t-distribution. Although a t-distribution may place more density at zero than the Gaussian prior of BR, the density at zero is larger in the DE. This issue was recognized by Meuwissen et al. (2001), leading to the development of Bayes B.
is a t-distribution. Although a t-distribution may place more density at zero than the Gaussian prior of BR, the density at zero is larger in the DE. This issue was recognized by Meuwissen et al. (2001), leading to the development of Bayes B.Xu (2003) employed an improper prior for the marker-specific variances; if
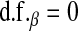 and
and 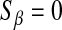 , then
, then 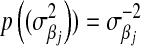 . Similar to the exponential prior, this density decreases monotonically with
. Similar to the exponential prior, this density decreases monotonically with 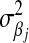 . However, unlike the exponential distribution, where
. However, unlike the exponential distribution, where 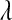 can be used to “tune” the shape of the distribution, this prior does not have parameters to allow any control. In addition, as noted by Ter Braak et al. (2005),
can be used to “tune” the shape of the distribution, this prior does not have parameters to allow any control. In addition, as noted by Ter Braak et al. (2005), 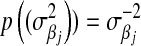 yields an improper posterior. As an alternative, these authors suggested to use
yields an improper posterior. As an alternative, these authors suggested to use 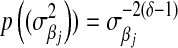 with
with 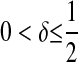 ; although this prior is improper, it does not yield an improper posterior. As with the exponential prior,
; although this prior is improper, it does not yield an improper posterior. As with the exponential prior, 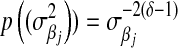 is decreasing with respect to
is decreasing with respect to 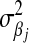 . Ter Braak (2006) furthered discussed the role of
. Ter Braak (2006) furthered discussed the role of 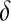 , which, as
, which, as 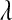 in the BL, controls the shape of the prior density on the variance of the regression coefficients.
in the BL, controls the shape of the prior density on the variance of the regression coefficients. A second difference is that, in Bayes A, values of parameters
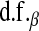 and
and 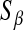 are specified as known a priori. On the other hand, in BL there is an extra level in the model:
are specified as known a priori. On the other hand, in BL there is an extra level in the model: 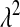 is assigned a Gamma distribution, and information from all regression coefficients is pooled. This difference is illustrated in Figure 2: in Bayes A,
is assigned a Gamma distribution, and information from all regression coefficients is pooled. This difference is illustrated in Figure 2: in Bayes A, 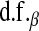 and
and 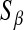 control, as
control, as 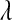 does in BL, the trade-offs between goodness of fit and model complexity.
does in BL, the trade-offs between goodness of fit and model complexity.
Figure 2.—

Graphical representation of the hierarchical structure of the Bayesian LASSO (top) and Bayes A (bottom). In the Bayesian LASSO, the variances of the marker effects are 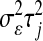 ,
, 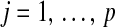 , with counterparts
, with counterparts 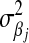 in Bayes A.
in Bayes A.
Yi and Xu (2008) discuss an extension of Bayes A where a prior is assigned to 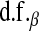 and
and 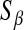 , and these quantities are treated as nuisances, as
, and these quantities are treated as nuisances, as 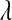 is in BL. However, as argued earlier, the DE seems to be a better choice, if the assumption is that most markers have no effect on the trait of interest.
is in BL. However, as argued earlier, the DE seems to be a better choice, if the assumption is that most markers have no effect on the trait of interest.
MONTE CARLO STUDY
Although in the BL 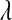 can be treated as unknown, it is not clear how sensitive results might be with respect to the choice of hyperparameters
can be treated as unknown, it is not clear how sensitive results might be with respect to the choice of hyperparameters 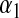 and
and 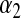 . Park and Casella (2008, p. 683) recognized that this may be an issue: “The prior density for
. Park and Casella (2008, p. 683) recognized that this may be an issue: “The prior density for 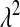 should approach 0 sufficiently fast as
should approach 0 sufficiently fast as 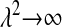 (to avoid mixing problems) but should be relatively flat and place high probability near the maximum likelihood estimate.” The main problem of applying this recommendation is that one does not know in advance what the maximum-likelihood estimate is.
(to avoid mixing problems) but should be relatively flat and place high probability near the maximum likelihood estimate.” The main problem of applying this recommendation is that one does not know in advance what the maximum-likelihood estimate is.
The sensitivity of BL with respect to the choice of the prior distribution of 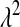 was investigated here by fitting the model under different priors to simulated data. In addition to the conjugate Gamma prior, we also considered (see File S1 and File S2)
was investigated here by fitting the model under different priors to simulated data. In addition to the conjugate Gamma prior, we also considered (see File S1 and File S2)
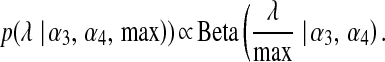 |
(7) |
The above distribution gives great flexibility for specifying a relatively flat prior over a wide range of values. The uniform prior appears as a special case when 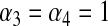 . When the Beta prior is used, the fully conditional distribution of
. When the Beta prior is used, the fully conditional distribution of 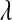 does not have closed form; however, draws from the distribution can be obtained using the Metropolis–Hastings algorithm (see File S1 and File S2).
does not have closed form; however, draws from the distribution can be obtained using the Metropolis–Hastings algorithm (see File S1 and File S2).
Data-generating process:
Data were simulated in a simple setting, such that problems could be identified easily, while the phenotypic and genotypic structure attempted to resemble those encountered in real data sets.
Data were generated under the additive model,
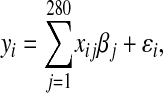 |
where 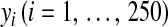 is the phenotype for individual i,
is the phenotype for individual i, 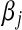 is the effect of allele substitution at marker j
is the effect of allele substitution at marker j 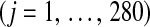 , and
, and 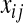 is the code for the genotype of subject i at locus j,
is the code for the genotype of subject i at locus j, 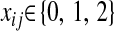 . Residuals were independently sampled from a standard normal distribution; that is,
. Residuals were independently sampled from a standard normal distribution; that is, 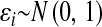 .
.
Two scenarios regarding the genotypic distribution were considered. In scenario X0, markers were in low linkage disequilibrium (LD), with almost no correlation between adjacent markers (Table 1). In scenario X1 a relatively high LD was considered (Table 1).
TABLE 1.
Absolute values of the correlation between marker genotypes (average across markers and 100 Monte Carlo simulations) by scenario (X0, low linkage disequilibrium; X1, high linkage disequilibrium)
| Adjacency between markers
|
||||
|---|---|---|---|---|
| Scenario | 1 | 2 | 3 | 4 |
| X0 | 0.091 | 0.089 | 0.090 | 0.001 |
| X1 | 0.754 | 0.602 | 0.479 | 0.381 |
The effects of allele substitutions 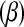 were kept constant across simulations and set to zero for all markers except for 10 (Figure 3). The locations of markers with nonnull effects were chosen such that different situations regarding effects of linked markers were represented.
were kept constant across simulations and set to zero for all markers except for 10 (Figure 3). The locations of markers with nonnull effects were chosen such that different situations regarding effects of linked markers were represented.
Figure 3.—

Positions (chromosome and marker number) and effects of markers (there were 280 markers, with 270 with no effect).
Choice of prior distribution of λ:
For each Monte Carlo (MC) replicate, five variations of BL were fitted, and four involved a Gamma prior 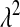 with the following values of parameters:
with the following values of parameters: 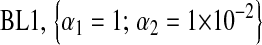 ;
; 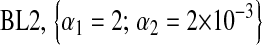 ;
; 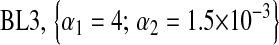 ;
; 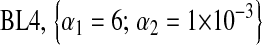 . In BL5, the prior on
. In BL5, the prior on 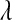 was
was 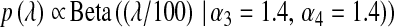 (Figure 4).
(Figure 4).
Figure 4.—

Unnormalized density of the five priors evaluated in the MC study (BL1–BL4 use Gamma priors on 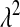 , and BL5 uses a prior for
, and BL5 uses a prior for 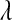 based on a Beta distribution; the densities in this figure are the corresponding densities for
based on a Beta distribution; the densities in this figure are the corresponding densities for 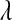 ).
).
Results:
Table 2 shows the average (across 100 MC replicates) of posterior means of the residual variance, the regularization parameter, and the correlation between the true and the estimated quantity of several features (phenotypes, genomic values, and marker effects). 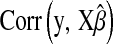 is a goodness-of-fit measure,
is a goodness-of-fit measure, 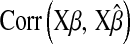 measures how well the model estimates genomic values, and
measures how well the model estimates genomic values, and 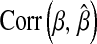 evaluates how well the model estimates marker effects.
evaluates how well the model estimates marker effects.
TABLE 2.
Posterior mean of residual variance, 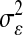 , regularization parameter,
, regularization parameter, 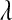 , and correlation between the true and estimated value for several items (
, and correlation between the true and estimated value for several items (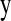 , phenotypes;
, phenotypes; 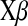 , true genomic value;
, true genomic value; 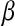 , marker effects; all quantities averaged over 100 MC replicates)
, marker effects; all quantities averaged over 100 MC replicates)
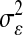
|
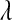
|
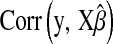
|
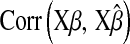
|
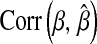
|
||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Meana | SDb | Meana | SDb | Meanc | SDb | Meanc | SDb | Meanc | SDb | |
| Low linkage disequilibrium between markers (X0) | ||||||||||
| BL1 | 0.909 | 0.088 | 24.110 | 1.883 | 0.640 | 0.039 | 0.657 | 0.070 | 0.328 | 0.062 |
| BL2 | 0.990 | 0.095 | 35.986 | 4.944 | 0.615 | 0.044 | 0.661 | 0.073 | 0.332 | 0.061 |
| BL3 | 1.035 | 0.096 | 45.506 | 6.755 | 0.601 | 0.045 | 0.660 | 0.074 | 0.329 | 0.059 |
| BL4 | 1.093 | 0.098 | 63.748 | 9.200 | 0.584 | 0.044 | 0.657 | 0.077 | 0.323 | 0.056 |
| BL5 | 1.006 | 0.104 | 41.920 | 11.570 | 0.610 | 0.050 | 0.660 | 0.073 | 0.330 | 0.059 |
| High linkage disequilibrium between markers (X1) | ||||||||||
| BL1 | 0.917 | 0.088 | 23.770 | 1.621 | 0.607 | 0.039 | 0.699 | 0.071 | 0.294 | 0.050 |
| BL2 | 0.991 | 0.093 | 36.168 | 4.163 | 0.569 | 0.044 | 0.705 | 0.074 | 0.300 | 0.046 |
| BL3 | 1.030 | 0.095 | 45.572 | 5.735 | 0.551 | 0.045 | 0.704 | 0.076 | 0.296 | 0.044 |
| BL4 | 1.078 | 0.097 | 62.373 | 8.435 | 0.529 | 0.046 | 0.700 | 0.079 | 0.291 | 0.043 |
| BL5 | 1.003 | 0.099 | 41.072 | 9.724 | 0.564 | 0.051 | 0.704 | 0.074 | 0.299 | 0.046 |
Mean (across 100 MC replicates) of the posterior mean.
Between-replicate standard deviation of the estimate.
Mean (across MC replicates) of the correlation evaluated at the posterior mean of 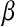 . The priors on
. The priors on 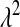 were as follows: BL1,
were as follows: BL1, 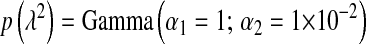 ; BL2,
; BL2, 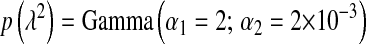 ; BL3,
; BL3, 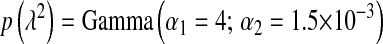 ; and BL4,
; and BL4, 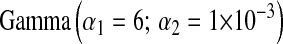 . In BL5,
. In BL5, 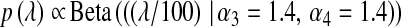 .
.
The posterior mean and standard deviation of 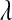 were influenced by the prior (Table 2). The posterior mean was shrunk toward the prior mode, and the posterior standard deviation was larger for more dispersed priors (see Table 2 and Figure 4). These results suggest that there is not much information about
were influenced by the prior (Table 2). The posterior mean was shrunk toward the prior mode, and the posterior standard deviation was larger for more dispersed priors (see Table 2 and Figure 4). These results suggest that there is not much information about 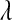 in the type of samples evaluated. On the other hand, model goodness of fit and the ability of the model to uncover signal were not affected markedly by the choice of prior. This suggest that, while it may be difficult to learn about
in the type of samples evaluated. On the other hand, model goodness of fit and the ability of the model to uncover signal were not affected markedly by the choice of prior. This suggest that, while it may be difficult to learn about 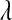 from data, inferences on quantities of interest (e.g., genetic values) may be robust with respect to values of
from data, inferences on quantities of interest (e.g., genetic values) may be robust with respect to values of 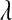 over a fairly wide range. For example, differences in
over a fairly wide range. For example, differences in 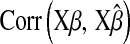 or in
or in 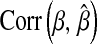 were small when the prior was changed.
were small when the prior was changed.
A relatively flat prior based on a Beta distribution (BL5) produced a more dispersed posterior distribution of 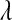 , and mixing was not as good as when the sharper Gamma priors (BL1–BL4) were used. For example, the average (across MC replicates) effective sample sizes (e.g., Plummer et al. 2008) for the residual variance were 1468, 1155, 1091, 1138, and 578 for BL1–BL5, respectively.
, and mixing was not as good as when the sharper Gamma priors (BL1–BL4) were used. For example, the average (across MC replicates) effective sample sizes (e.g., Plummer et al. 2008) for the residual variance were 1468, 1155, 1091, 1138, and 578 for BL1–BL5, respectively.
BAYESIAN REGRESSION COUPLED WITH LASSO
In practice, the information set available for prediction of genomic values may include components other than genetic markers. For example, data may cluster into known contemporary groups (e.g., individuals may be measured under different experimental conditions), or a pedigree may be available in addition to genetic markers. It is natural to treat the various classes of predictors in a different way. From a penalized-likelihood point of view, this amounts to using penalty functions that are specific to each class of predictors. From a Bayesian standpoint, treating predictors differently may be achieved by assigning different priors. A straightforward extension of the BL is described next.
The data structure is denoted as 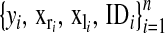 , where
, where 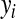 is the phenotype of subject i,
is the phenotype of subject i, 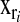 is a vector of covariates that is treated as in a standard BR with a normal prior and variance common to all regressions,
is a vector of covariates that is treated as in a standard BR with a normal prior and variance common to all regressions, 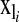 is a set of covariates whose effects are assigned a double-exponential prior as in BL, and
is a set of covariates whose effects are assigned a double-exponential prior as in BL, and 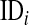 is a label that allows tracking subjects in a pedigree. The equation for the data is
is a label that allows tracking subjects in a pedigree. The equation for the data is
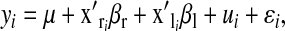 |
where 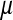 is an intercept,
is an intercept, 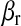 and
and 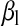 are regressions of
are regressions of 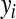 on
on 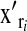 and
and 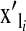 , respectively,
, respectively, 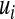 is an infinitesimal genetic effect pertaining to individual i for which the prior (co)variance structure is determined by a pedigree, and
is an infinitesimal genetic effect pertaining to individual i for which the prior (co)variance structure is determined by a pedigree, and 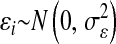 is a model residual, assumed to be identically and independently distributed of other residuals. The likelihood function is
is a model residual, assumed to be identically and independently distributed of other residuals. The likelihood function is
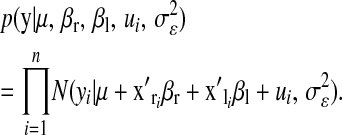 |
(8) |
Prior specification (4) is modified as
 |
(9) |
where 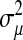 ,
, 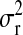 , and
, and 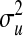 are the variances of
are the variances of 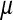 ,
, 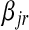 , and
, and 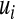 , respectively;
, respectively; 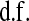 and
and 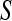 are prior degrees of freedom and scale parameter of the corresponding distributions;
are prior degrees of freedom and scale parameter of the corresponding distributions; 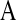 is a (co)variance structure computed from the genealogy (for example, a numerator-relationship matrix); and,
is a (co)variance structure computed from the genealogy (for example, a numerator-relationship matrix); and, 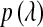 is the prior on
is the prior on 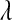 that may be as in (4) or (7).
that may be as in (4) or (7).
In the model defined by (8) and (9) all fully conditional distributions (except that of 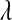 if a nonconjugate prior is chosen for
if a nonconjugate prior is chosen for 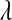 ) have closed form, so a Gibbs sampler (with a Metropolis–Hastings step) can be used to draw samples from the joint posterior distribution (see File S1 and File S2). To distinguish the above model from the standard BL we refer to it as Bayesian regression coupled with LASSO (BRL).
) have closed form, so a Gibbs sampler (with a Metropolis–Hastings step) can be used to draw samples from the joint posterior distribution (see File S1 and File S2). To distinguish the above model from the standard BL we refer to it as Bayesian regression coupled with LASSO (BRL).
DATA ANALYSIS
Two data sets were analyzed with the BRL model. The first set pertains to a collection of wheat lines (see File S1 and File S2); the second set contains information from a population of mice (publicly available at http://gscan.well.ox.ac.uk).
The wheat data set is from the Global Wheat program of the International Maize and Wheat Improvement Center (CIMMYT). This program conducted several international trials across a wide variety of environments. For this study, we took a subset of 599 wheat lines derived from 25 years of Elite Spring Wheat Yield Trials (ESWYT) conducted from 1979 through 2005. The environments represented in these trials were grouped into four macroenvironments. The phenotype considered here was average grain yield performance of the 599 wheat lines evaluated in one of the macroenvironments. An association mapping study based on a reduced number of these ESWYT trials is presented in Crossa et al. (2007).
The Browse application of the International Crop Information System (ICIS), as described in http://cropwiki.irri.org/icis/index.php/TDM_GMS_Browse (McLaren et al. 2005), was used for deriving the relationship matrix A between the 599 lines, and it accounts for selection and inbreeding.
A total of 1447 Diversity Array Technology (DArT) markers were generated by Triticarte (Canberra, Australia; http://www.triticarte.com.au). The DArT markers may take on two values, denoted by their presence or their absence.
The mouse data come from an experiment carried out to detect and locate QTL for complex traits in a mouse population (Valdar et al. 2006a,b). These data have already been analyzed for comparing genome-assisted genetic evaluation methods (Legarra et al. 2008). The data file consists of 1884 individuals (168 full-sib families), each genotyped for 10,946 polymorphic markers. The trait analyzed here was body mass index (BMI), precorrected by body weight, season, month, and day. Mice were housed in 359 cages; on average, each litter was allocated into 2.84 cages.
Three models were fitted to each of the data sets: P (standing for pedigree) is a pedigree-based model where markers were not included; M is a model where the only genetic component is the regression on markers; P&M (standing for pedigree and markers) includes regressions on markers and an additive effect with (co)variance structure computed from the pedigree. For both data sets, phenotypes were standardized to have a sample variance equal to one, so that results are easily compared across data sets.
In the mouse data set, 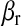 were the effects of cages where groups of mice were reared. In the wheat data set, the component
were the effects of cages where groups of mice were reared. In the wheat data set, the component 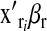 was omitted because there was no such set of regressors.
was omitted because there was no such set of regressors.
Models were first fitted to the entire data set. Subsequently, a fivefold cross-validation (CV) was carried out with assignment of individuals into folds at random. The CV yields prediction of phenotypes 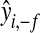 (f = 1, … , 5) obtained from a model in which all observations in the fth fold were excluded. The ability of each model to predict out-of-sample data was evaluated via the correlation between phenotypes and predictions from CV. Inferences for each fit were based on 70,000 samples (after 5000 were discarded as burn-in). Convergence was checked by inspection of trace plots and with estimates of effective sample size for (co)variance components computed using the coda package of R (Plummer et al. 2008). Parameters of the prior distributions were
(f = 1, … , 5) obtained from a model in which all observations in the fth fold were excluded. The ability of each model to predict out-of-sample data was evaluated via the correlation between phenotypes and predictions from CV. Inferences for each fit were based on 70,000 samples (after 5000 were discarded as burn-in). Convergence was checked by inspection of trace plots and with estimates of effective sample size for (co)variance components computed using the coda package of R (Plummer et al. 2008). Parameters of the prior distributions were 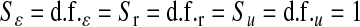 and
and 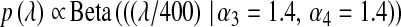 . This latter prior is flat over a wide range of values of
. This latter prior is flat over a wide range of values of 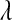 .
.
RESULTS AND DISCUSSION
Table 3 shows summaries of the posterior distributions of the variance components and of λ by model and data set. In both populations, a moderate reduction in the posterior mean of the residual variance was observed when the P&M model was fitted, relative to P. Using model P in the wheat population gave a posterior mean of heritability of grain yield of 0.34, while in the mouse population the posterior mean of h2 of body-mass index was 0.11. These results are in agreement with previous reports (Valdar et al. 2006b and Legarra et al. 2008 for the mouse data and Crossa et al. 2007 for the wheat data) for these traits and populations. The inclusion of markers (P&M) reduced the estimate of the variance of the infinitesimal additive effect, relative to P. This happens because, in P&M, part of the infinitesimal additive effect is captured by the regression on markers (e.g., Habier et al. 2007; Bink et al. 2008). In the model for body-mass index in M, the variance between cages 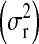 was reduced only slightly when the effects of the markers were fitted.
was reduced only slightly when the effects of the markers were fitted.
TABLE 3.
Posterior means (standard deviations) of variance components for yield in wheat and body-mass index in mice, and of 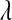 for each of the models, by data set
for each of the models, by data set
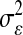 |
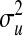 |
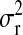 |
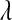 |
||
|---|---|---|---|---|---|
| Wheat | P | 0.561 (0.058) | 0.294 (0.056) | — | — |
| M | 0.546 (0.046) | — | — | 19.33 (2.43) | |
| P&M | 0.410 (0.055) | 0.139 (0.046) | — | 18.32 (2.86) | |
| Mice | P | 0.754 (0.038) | 0.092 (0.041) | 0.156 (0.030) | — |
| M | 0.741 (0.037) | — | 0.153 (0.026) | 223.84 (25.78) | |
| P&M | 0.723 (0.032) | 0.021 (0.009) | 0.153 (0.026) | 217.90 (26.26) |
P, infinitesimal models using pedigree; M, model including regressions on markers, but not pedigree information; P&M, model including an infinitesimal additive effect and regressions on markers; 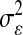 ,
, 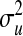 ,
, 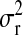 , and
, and 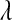 , residual variance, variance of the infinitesimal additive effect, variance of cage-effects, and smoothing parameter of the BL regression, respectively.
, residual variance, variance of the infinitesimal additive effect, variance of cage-effects, and smoothing parameter of the BL regression, respectively.
Figure 5 gives absolute values of the posterior means of marker effects. In the mouse data there are several regions showing groups of markers with relatively large estimated effects. This is not evident in the wheat data set where fewer markers were available.
Figure 5.—

Absolute values of the posterior means of effects of allele substitution in a model including markers and pedigree information (P&M), by data set.
From a breeder's perspective, a relevant question is whether or not the P, M, and P&M models lead to different ranking of individuals on the basis of the estimated genetic values. Table 4 shows the rank (Spearman) correlation of estimated genetic values. As expected, these correlations were high, but not perfect. The correlation between predicted genetic values from M and P&M was larger than that of the estimates from P and P&M, suggesting that the inclusion of markers in the model is probably critical. This was clearer in the mouse data set, where (a) the extent of additive relationships was not as strong as in the wheat population and (b) a much larger number of markers were available.
TABLE 4.
Rank correlation (Spearman) between genetic values estimated from models including different sources of genetic information (pedigree, markers, and pedigree and markers), by data set (mouse data set above diagonal, wheat data set below diagonal)
| Source of genetic information
|
|||
|---|---|---|---|
| Source of genetic information | Pedigree | Marker | Pedigree and markers |
| Pedigree | — | 0.715 | 0.729 |
| Marker | 0.802 | — | 0.986 |
| Pedigree and markers | 0.927 | 0.944 | — |
Figure 6 shows scatter plots of predicted genomic values in P and P&M for both data sets. Although the correlation between genetic values estimated from different models was high, using P and P&M would lead to different sets of selected individuals. The difference was more marked in the mouse data set, illustrating that the impact of considering markers in breeding decisions depends on the data structure and on how informative the pedigree and markers are. Also, the dispersion of predicted genetic values was larger when markers were fitted, and this is consistent with the smaller posterior mean of the residual variance observed for P&M (Table 3). An interpretation of this result is that, in certain contexts, markers may help to uncover genetic variance that would not be captured if only pedigree-based predictions were used.
Figure 6.—

Predicted genetic value using markers and pedigree (P&M) vs. using pedigree only (P), by data set.
The aforementioned results indicate that incorporation of markers into a genetic model can influence inferences and breeding decisions. In contrast, cross-validation allows comparing models from the standpoint of their ability to predict future outcomes. Table 5 shows the correlation between phenotypic records and predictions from cross-validation. Two CV correlations were considered:
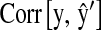 is the correlation between phenotypic records and their prediction from CV. That is,
is the correlation between phenotypic records and their prediction from CV. That is, 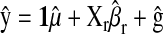 , where
, where 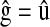 in P,
in P, 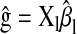 in M, and
in M, and 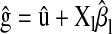 in P&M.
in P&M.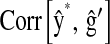 is the correlation between the CV estimate of the genetic value and phenotypic records adjusted with CV estimates of nongenetic effects. That is,
is the correlation between the CV estimate of the genetic value and phenotypic records adjusted with CV estimates of nongenetic effects. That is, 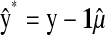 in the wheat population, and
in the wheat population, and  in the mouse data set.
in the mouse data set.
TABLE 5.
Rank correlation (Spearman) between phenotypic values or corrected phenotypic records and predictions from cross-validation, by population and model (P, pedigree-based model; P&M, pedigree and marker information)
| Wheat
|
Mice
|
|||||
|---|---|---|---|---|---|---|
| P | M | P&M | P | M | P&M | |
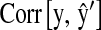 a a
|
0.408 | 0.423 | 0.462 | 0.263 | 0.306 | 0.300 |
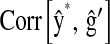 b b
|
0.423 | 0.594 | 0.602 | 0.109 | 0.211 | 0.225 |
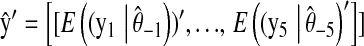 , where
, where 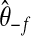 is the posterior mean from a model with the observations in the fth fold excluded.
is the posterior mean from a model with the observations in the fth fold excluded.
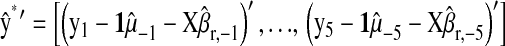 , where
, where 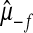 ,
, 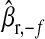 are the posterior mean of the intercept and of the regression coefficients of systematic effects from a model with the observations of the fth fold excluded; and
are the posterior mean of the intercept and of the regression coefficients of systematic effects from a model with the observations of the fth fold excluded; and 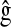 is the posterior mean, from CV, of genetic effects (sum of additive value and genomic value in P&M).
is the posterior mean, from CV, of genetic effects (sum of additive value and genomic value in P&M).
Overall, P&M models had better predictive ability than models based on pedigrees or markers only. In the wheat data set, the increases in the correlation observed when markers were included in the model were 13% for 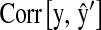 and 42% for
and 42% for  . In the mouse data set the relative increases in correlation were 14 and 100% for
. In the mouse data set the relative increases in correlation were 14 and 100% for 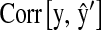 and
and 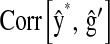 , respectively. We conclude that there are sizable benefits from using markers for breeding decisions and that the relative impact of the contribution depends upon data structure and on how informative the pedigree and the set of markers are.
, respectively. We conclude that there are sizable benefits from using markers for breeding decisions and that the relative impact of the contribution depends upon data structure and on how informative the pedigree and the set of markers are.
CONCLUDING REMARKS
Additive models with infinitesimal effects are ubiquitous in animal and plant breeding. For many decades, predictions of genetic values have been made using phenotypic records and pedigrees, i.e., some sort of family-based evaluation. Markers capture Mendelian segregation and may enhance prediction of genomic values, independently of the mode of gene action.
With highly dense markers, marker-specific shrinkage may be needed. Priors on marker effects based on mixtures of scaled-Gaussian distributions allow this type of shrinkage and constitute a promising tool for genomic-based additive models. This family of models includes, among others, the t or DE distributions. Models based on marginal priors that belong to the t family have been proposed for marker-based regressions (e.g., Meuwissen et al. 2001).
If the hypothesis that most markers do not have any effect holds, a DE prior may be a better choice than the t. For this reason, the Bayesian LASSO appears to be an interesting alternative for performing regressions on markers, at least under an additive model.
Our results indicate that in the type of samples that are relevant for genomic selection (i.e., p ≫ n) the choice of prior for 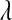 matters in terms of inferences about this unknown. However, estimates of genetic values and of marker effects may be robust with respect to the choice of prior, over a wide range. To circumvent the potential influence of the prior, we proposed an alternative formulation of the BL where the prior on
matters in terms of inferences about this unknown. However, estimates of genetic values and of marker effects may be robust with respect to the choice of prior, over a wide range. To circumvent the potential influence of the prior, we proposed an alternative formulation of the BL where the prior on 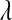 is formulated using a Beta distribution. Unlike the Gamma prior, this prior allows expressing vague prior preferences over a wide range of values of
is formulated using a Beta distribution. Unlike the Gamma prior, this prior allows expressing vague prior preferences over a wide range of values of 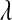 .
.
Two data analyses carried out with the proposed model showed that (a) markers may allow capturing fractions of additive variance that would be lost if pedigrees are the only source of genetic information used, (b) considering markers has a sizable impact on selection decisions, and (c) models including marker and pedigree information had better predictive ability than pedigree-based or marker-based models.
Acknowledgments
We greatly appreciate suggestions of two anonymous reviewers and of the Associate Editor. The Wellcome Trust Center for Human Genetics, Oxford, is gratefully acknowledged for making the mouse data available at http://gscan.well.ox.ac.uk. Vivi Arief from the School of Land Crop and Food Sciences of the University of Queensland, Australia, is thanked for assembling the historical wheat phenotypic and molecular marker data and for computing additive relationships between wheat lines. Financial support by the Wisconsin Agriculture Experiment Station, grant DMS-NSF DMS-044371, and by the Chaire D'Excellence Pierre de Fermat programme of the Midi-Pyrennées Region, France, is acknowledged.
Supporting information is available online at http://www.genetics.org/cgi/content/full/genetics.109.101501/DC1.
References
- Andrews, D. F., and C. L. Mallows, 1974. Scale mixtures of normal distributions. J. R. Stat. Soc. Ser. B 36 99–102. [Google Scholar]
- Bink, M. C. A. M., P. Uimari, M. J. Sillanpää, L. L. G. Janss and R. C. Jansen, 2002. Multiple QTL mapping in related plant populations via a pedigree-analysis approach. Theor. Appl. Genet. 104 751–762. [DOI] [PubMed] [Google Scholar]
- Bink, M. C. A. M., M. P. Boer, C. J. F. Ter Braak, J. Jansen, R. E. Voorrips et al., 2008. Bayesian analysis of complex traits in pedigreed populations. Euphytica 161 85–96. [Google Scholar]
- Chhikara, R. S, and J. L. Folks, 1989. The Inverse Gaussian Distribution: Theory, Methodology and Applications. Marcel Dekker, NY.
- Crossa, J., J. Burgueño, S. Dreisigacker, M. Vargas, S. A. Herrera-Foessel et al., 2007. Association analysis of historical bread wheat germplasm using additive genetic covariance of relatives and population structure. Genetics 177 1889–1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De los Campos, G., D. Gianola and G. J. M. Rosa, 2009. Reproducing kernel Hilbert spaces regression: a general framework for genetic evaluation. J. Anim. Sci.(in press). [DOI] [PubMed]
- Fernando, R. L., and M. Grossman, 1989. Marker assisted selection using best linear unbiased prediction. Genet. Sel. Evol. 21 467–477. [Google Scholar]
- Fernando, R. L., D. Habier, C. Stricker, J. C. M. Dekkers and L. R. Totir, 2007. Genomic selection. Acta Agric. Scand. Sect. A 57 192–195. [Google Scholar]
- Gianola, D., and J. B. van Kaam, 2008. Reproducing kernel Hilbert spaces regression methods for genomic assisted prediction of quantitative traits. Genetics 178 2289–2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gianola, D., M. Perez-Enciso and M. A. Toro, 2003. On marker-assisted prediction of genetic value: beyond the ridge. Genetics 163 347–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gianola, D., R. Fernando and A. Stella, 2006. Genomic-assisted prediction of genetic value with semiparametric procedures. Genetics 173 1761–1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habier, D., R. L. Fernando and J. C. M. Dekkers, 2007. The impact of genetic relationship information on genome-assisted breeding values. Genetics 177 2389–2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hans, C., 2008. Bayesian LASSO regression. Technical Report No. 810. Department of Statistics, Ohio State University, Columbus, OH. (http://www.stat.osu.edu/∼hans/Papers/blasso.pdf).
- Legarra, A., C. Robert-Granié, E. Manfredi and J. M. Elsen, 2008. Performance of genomic selection in mice. Genetics 180 611–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaren, C. G., R. Bruskiewich, A. M. Portugal and A. B. Cosico, 2005. The international rice information system. A platform for meta-analysis of rice crop data. Plant Physiol. 139 637–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meuwissen, T. H. E., B. J. Hayes and M. E. Goddard, 2001. Prediction of total genetic value using genome-wide dense marker maps. Genetics 157 1819–1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park, T., and G. Casella, 2008. The Bayesian LASSO. J. Am. Stat. Assoc. 103 681–686. [Google Scholar]
- Plummer, M., N. Best, K. Cowles and K. Vines, 2008. coda: output analysis and diagnostics for MCMC. http://cran.r-project.org/web/packages/coda/index.html
- R Development Core Team, 2008. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna. http://www.R-project.org.
- Rosa, G. J. M., 1999. Robust mixed linear models in quantitative genetics: Bayesian analysis via Gibbs sampling. International Symposium on Animal Breeding and Genetics, September 21–24, Viçosa, Minas Gerais, Brazil, pp. 133–159.
- Ter Braak, C. J. F., 2006. Bayesian sigmoid shrinkage with improper variance priors and an application to wavelet denoising. Comput. Stat. Data Anal. 51 1232–1242. [Google Scholar]
- Ter Braak, C. J. F, M. P. Boer and M. C. A. M. Bink, 2005. Extending Xu's Bayesian model for estimating polygenic effects using markers of the entire genome. Genetics 170 1435–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tibshirani, R., 1996. Regression shrinkage and selection via the LASSO. J. R. Stat. Soc. B 58 267–288. [Google Scholar]
- Valdar, W., L. C. Solberg, D. Gauguier, S. Burnett, P. Klenerman et al., 2006. a Genome-wide genetic association of complex traits in heterogeneous stock mice. Nat. Genet. 38 879–887. [DOI] [PubMed] [Google Scholar]
- Valdar, W., L. C. Solberg, D. Gauguier, W. O. Cookson, J. N. P. Rawlins et al., 2006. b Genetic and environmental effects on complex traits in mice. Genetics 174 959–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, W. Y., B. J. Barratt, D. G. Clayton and J. A. Todd, 2005. Genome-wide association studies: theoretical and practical concerns. Nat. Rev. Genet. 6 109–118. [DOI] [PubMed] [Google Scholar]
- Xu, S., 2003. Estimating polygenic effects using markers of the entire genome. Genetics 163 789–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi, N., and S. Xu, 2008. Bayesian LASSO for quantitative trait loci mapping. Genetics 179 1045–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]