

Abstract
Neurodegenerative diseases have become a global issue due to the aging population. These disorders affect a vast patient population and represent a huge area of unmet therapeutic need. Axon degeneration is a common pathological character of those neurodegenerative diseases. It results in the loss of communication between neurons. Two decades ago, the Wallerian degeneration slow (Wlds) mouse strain was identified, in which the degeneration of transected axons is delayed. The phenotype is attributed to the overexpression of a chimeric protein Wlds which contains a short fragment of the ubiquitin assembly protein UFD2 and the full-length nicotinamide adenine dinucleotide (NAD) synthetic enzyme Nicotinamide mononucleotide adenylyl-transferase-1 (Nmnat-1). However, the underlying molecular mechanism remains largely unknown. Recently, it's reported by independent researchers that the full length coding sequence of mouse Nmnat-1 could mimic the axonal protective effect of the Wlds gene when overexpressed in primary neural cultures. Together with a significant number of subsequential reports, this finding highlighted the substantial role of nicotinamide adenine dinucleotide (NAD) in the process of axon degeneration. Here we reviewed the history of axon degeneration research from a neurochemical standpoint and discuss the potential involvement of NAD synthesis, NAD consumption and NAD-dependent proteins and small molecules in axon degeneration.
Key words: axon degeneration, Wallerian degeneration, Wlds, NAD, UPS, neurodegenerative diseases
Introduction
Axon degeneration is a common phenomenon which occurs under both physiological conditions during normal development, such as axon remodeling, as well as pathological conditions, such as injury or neurodegenerative disorders. During development, neurons use the strategy of overshooting to establish the connection with their targets. The exuberant axons are eliminated or pruned by degeneration without damaging the cell body or desired branches.1,2 In adults, damaged axons are removed by a similar process to maintain the normal neuronal function of neighboring circuits. In case of trauma or injury, the degeneration of the distal portion of the severed axon was first described by Augustus Waller in 1850 and termed Wallerian Degeneration.3 In motor degenerative diseases and peripheral nerve diseases caused by toxic insults, the axons of the unhealthy neurons develop a ‘dying-back’ phenomenon, which starts from the distal terminal and progressively spreads toward the cell body, before death of the cell body.4,5 Axonal degeneration also represents a major pathological feature in epilepsy, ischemia and aging as reported in the following publications.6,7 The common morphological changes that occur during axon degeneration include axonal beading or focal swelling followed by axon fragmentation. However, the underlying molecular mechanism of axon degeneration is still largely unknown. Understanding the pathways which control this process will provide a critical input into the unmet therapeutic needs of these disorders.
Axon Degeneration in Neurodegenerative Diseases
Neurodegenerative diseases, such as Alzheimer disease, Parkinson disease, amyotrophic lateral sclerosis and multiple sclerosis, affect a vast patient population and represent a huge area of unmet therapeutic need. Pathological features of neurodegenerative diseases include neuron loss and axonal degeneration.6,7 Previously, it has been proposed that entire neuron loss is the major causative feature which contributes to the development of clinical signs in those disorders. However, there is growing evidence suggesting that axonal defects are important in the expression of those diseases. During the onset of neurodegenerative diseases, axon degeneration occurs before the neuronal cell body undergoes apoptosis.5 The degeneration of axons separates the neurons from their targets, therefore inducing the loss of neuronal function. For example, axonal swelling, an early sign of axon degeneration, has been identified in mouse models of Alzheimer disease and at the early stages of Alzheimer disease in human.8 These swelling axons accumulate abnormal amounts of microtubule-associated proteins and molecular motor proteins, organelles and vesicles. Strategies used to rescue neurons from programmed cell death have been largely unsuccessful in attenuating the progression of disease phenotypes. For example in the Wobbler mouse, an early post-natal motoneuron degeneration model, overexpression of bcl-2, a key regulator of cell death, reduced neuron loss but had no detectable effect on axonal degeneration.9 More importantly, the clinical signs as well as biochemical and histological parameters of the disease were not improved. These observations indicate that only preventing neuron death may not be sufficient to treat neurodegenerative diseases. Efficient therapeutic strategies, which could protect axons, are also essential for curing such disorders. Therefore, understanding the molecular mechanism of axon degeneration is very important.
Axon Degeneration and Cell Death
The axon is a special structure specific to neurons. The distal potion of an axon can be as far as one meter away from the cell body. This raises the question of whether axon degeneration may be under the control of the same machinery which commands the process of neuronal cell death. Possible mechanisms of neuronal cell death include apoptosis, autophagy and necrosis.10 It has been reported that the process of Wallerian degeneration occurs in an apoptosis-independent manner.11 Finn and colleagues found that the activation of caspases was neither involved in Wallerian degeneration nor axon degeneration induced by local neurotrophin deprivation.
Autophagy is a lysosome-mediated catabolic mechanism, which is responsible for the bulk degradation and recycling of damaged or dysfunctional intracellular organelles and cytoplasmic components.12 Autophagy has been well studied as a recycling mechanism used by lower eukaryotic organism to survive nutrient starvation conditions. Increased lysosomal activity has been observed during neuronal cell death associated with neurodegenerative diseases.13 It has also been reported that NGF deprivation-induced sympathetic neuronal cell death exhibits autophagic features, and could be delayed by the autophagy inhibitor 3-methyladenine (3-MA), also known as a PI-3K inhibitor.14 These observations suggest that autophagy might be involved in the cell death process of neurodegenerative diseases. However, there is no definitive evidence for the involvement of autophagy in axonal degeneration. Unpublished results from our lab showed that 3-MA can significantly delay Wallerian degeneration. However, 3-MA must be used at high concentration, where its specificity is questionable. Thus more genetic evidence may be obligatory before any conclusions can be drawn.
Previous ultrastructural studies showed that the death of developing motor neurons exhibits typical necrotic features, such as dilation of the endoplasmic reticulum (ER), Golgi and nuclear membrane.10 In C. elegans, Ca2+ released from the ER is believed to be the main candidate for initiating the downstream necrotic cell death events.15 When the intracellular Ca2+ level exceeds the physiological condition, it will trigger the activation of calpains, the Ca2+-activated cysteine proteases, to degrade cytoplasmic proteins. Calpains have been reported to play a major role in ischemic neuronal death.16 In the case of injury-induced axon degeneration, Ca2+ influx and activation of calpains appear to be both necessary and sufficient to induce axonal degeneration.17 During degeneration, calpain activation induces the breakdown of neurofilaments, an axonal cytoskeletal component. At the same time, calpain-dependent neurofilament breakdown is also detected in anoxic and ischemic axons.18 These findings suggest that the process of axonal degeneration shares more features with that of necrosis than it does with apoptosis and autophagy.
From a bioenergetics point of view, one significant difference between necrosis and the energy-dependent cell death mechanisms concerns ATP levels.19,20 Defects in ATP synthesis, which might be due to failure in energy homeostasis, occurs at an early stage of necrosis, while a certain level of ATP synthesis can still be maintained until the late stages of apoptosis and autophagy.21–24 Results from our lab revealed that axonal ATP levels start to decrease while the AMP level increases at early stages of Wallerian degeneration.25 These observations further support the idea that mechanisms of axon degeneration might share more similarities with the mechanisms of necrosis than with those of apoptosis or autophagic cell death.
Wlds Mice and Wallerian Degeneration
Wallerian degeneration is a commonly used model to study axonal degeneration. It is observed at the distal portion of transected axons upon injury. It occurs in both central and peripheral nervous systems.26 In rodents, within 24–26 hours after axotomy, distal axons and synaptic terminals will degenerate.3 The debris and fragments of degenerated axons and their surrounding myelin sheaths will then be cleared by macrophages and microglia. The earliest observable cellular events are local swelling and disruption of neurofilament, which depends on Ca2+ influx and activation of the Ca2+-dependent protease calpain.17 At the mammalian neuromuscular junction (NMJ), degeneration of nerve terminals occurs before the degeneration of the motor axons, which depends on the length of the remaining distal nerve.27 The most recent in vivo imaging observation shows that an acute form of axonal degeneration occurs within 30 min after trauma, which removes the distal axon hundreds of micrometers from the lesion site, while the further fragmentation of the distal axon starts near the lesion site and spread in a proximal to distal direction.28
It has long been thought that Wallerian degeneration is caused solely by the lack of protein synthesized in the cell body. This view was challenged by the discovery of a spontaneous mutant mouse strain, the slow Wallerian degeneration mice (C57/Wlds).29 Wallerian degeneration is significantly delayed in the Wlds mice. After nerve injury, the distal potion of the transected axon remains alive and functional for about 3–4 weeks in vivo. This neuronal-protective effect is genetically dominant and intrinsic to the axon. Programmed cell death is not affected in Wlds mice.30 providing additional support for the idea that axon degeneration and programmed cell death are regulated by different molecular mechanisms.
It has been demonstrated that the Wlds phenotype is caused by the overexpression of a chimeric Wlds gene, which contains the coding sequence of the N-terminal 70-amino acid fragment from the E4 ubiquitin ligase Ube4b (homologous to yeast Ufd2) fused to the entire coding region of D4Colele gene (homologous to human Nmnat-1) (Fig. 1).31,32 The Wlds mouse contain thee copies of this chimeric gene in an 85 KB tandem triplication unit on the distal part of mouse chromosome 4.33 This fusion protein, which protects both sensory and motor axons from Wallerian degeneration, is abundantly expressed in both the CNS and PNS of Wlds mice. The Wlds gene also protects axons from vincristine-induced neuronal toxicity.34 The protective effect of this gene has already been confirmed by transgenic mice overexpressing the fusion protein,35 in which motor nerve conduction, synaptic transmission, vesicle recycling and motor nerve terminal morphology were preserved after lesion to an extent that depends on the expressing level of the transgene. On the contrary, recent studies showed the Wlds gene cannot protect the neuronal cell body from cell death induced by axonal injury, supporting the idea that axon degeneration and the death of its cell body are controlled by different mechanism.36,37
Figure 1.

The components of the Wlds chimeric gene. The Wlds gene contains the N-terminal 70-amino acid short fragment of the ubiquitin assembly protein UFD2 and the full-length nicotinamide adenine dinucleotide (NAD) synthetic enzyme Nicotinamide mononucleotide adenylyl-transferase-1 (Nmnat-1).
The discovery of the Wlds mice indicates that lack of protein synthesized at the cell body may not be the only cause of Wallerian degeneration. Rather, it is more likely caused by events intrinsic to the axon. The two components of Wlds gene imply that the ubiquitin-proteasome system and NAD+ synthesis might be involved in the process of axon degeneration.
Axon Degeneration and Proteolysis
The Wlds protein contains the N-terminal 70-amino acid fragment of Ube4b protein. This discovery suggested that the Ubiquitin Proteasome System (UPS) might be involved in the process of axon degeneration. UPS contributes to several neuronal processes, such as axon path-finding, synapse formation and the pathophysiology of neurodegenerative diseases.38,39 In the Gracile axonal dystrophy (Gad) mutant mouse, inactivation of the ubiquitin carboxy-terminal hydrolase, UCH-L1, is the major reason for the degeneration of the gracile tract of the spinal cord.40 However, based on the dual activities of UCH-L1 as both a ubiquitin ligase and a de-ubiquitination enzyme,41 the exact role of the UPS in Gad mice is still unknown.
We and others demonstrated that inhibition of UPS by pharmacological and genetic approaches could prolong the survival of transected axons both in vivo and in vitro.42,43 At the same time, studies in Drosophila showed that the UPS is involved in the axon degeneration during pruning.44 These results indicate that the UPS might be involved as a common mechanism of axon degeneration. However, the response of the neuronal cell body to UPS inhibition is quite different from that in the axon. In particular proteasome inhibitors can induce apoptosis in cultured cortical neurons.24 This cellular toxicity might come from the general shutdown of the clearance of wasted or damaged proteins or many other possibilities. Cell bodies might have a higher demand for the normal function of UPS than axons, in accordance with the different complexity of these two structures. More specific inhibition of the UPS is required for developing therapeutic strategies against axon degeneration.
Both the Drosophila study44 and our results42 identified the loss of microtubule integrity as the earliest event during axon degeneration. However, our biochemistry data showed that tubulin is not the immediate target of UPS. Unpublished data from our lab obtained by electronic microscopic analysis of sympathetic ganglions cultured in vitro showed that at early stages of Wallerian degeneration, dozens of single layer-membrane vesicles, sometimes accompanied by a normally-shaped mitochondria, accumulate at the microtubule fragmentation site. These sites are evenly distributed along the axon, with an approximate 2 to 3 um distance from each other. One possible explanation is that some microtubule associated proteins, which might exist in a huge complex and attach to microtubules with a certain spatial frequency, might be the target of UPS during early the stage of Wallerian degeneration. Degradation of these proteins might induce vesicles to fall off from microtubules during axonal transport. Thus it is critical to identify relevant E2/E3 and their targets in axon degeneration. Wallerian degeneration models in a lower animal system would be a big advantage for this purpose. Such a model system has already been established recently in Drosophila.45
Axon Degeneration and NAD Synthesis
Although the UPS is involved in early stages of axon degeneration, structure-function analysis of the Wlds protein showed that the N-terminal 70 amino acids of Ube4b alone has no neural protective effect on axon degeneration.25,46 While the other large segment of the Wlds protein, the full length coding sequence of mouse Nmnat-1, could mimic the axonal protective effect of the Wlds gene when overexpressed in primary neural cultures mediated by Lentivirus46 or Herpes simplex virus (HSV).25
Nmnat-1 is an indispensable enzyme catalyzing the last step in both the de novo and salvage pathways for NAD+ biosynthesis in mammal (Fig. 2).47 The combination of these two pathways contributes to the biosynthesis of NAD+.48 De novo biosynthesis refers to NAD+ synthesis from tryptophan, an essential aromatic amino acid. The intermediate product from tryptophan to NAD+ is quinolinic acid, which is excitotoxic to the brain.49 Quinolinic acid phosphoribosyltransferase (QPRTase) is the enzyme catalyzing the first step from quinolinic acid to NAD+. This enzyme is found primarily in glial cells and sporadically in neurons in the brain,50 indicating that tryptophan is not the major NAD+ precursor in neurons. The salvage pathway refers to NAD+ synthesis from niacin (or vitamin B3), which is the generic name used to describe both nicotinic acid and nicotinamide. Similar to quinolinic acid, nicotinic acid has also been reported to have excitotoxic effects when injected into rat brain.49 On the other hand, nicotinamide represents the main source of NAD+ for most cells in mammals. Its concentration in human plasma could be five-fold higher than nicotinic acid levels in the case of imperfect dietary supplementation.51 Moreover, nicotinamide is also the product of NAD+ hydrolysis catalyzed by NAD+-consuming enzymes, such as the PARP-PARG system. The nicotinamide phosphoribosyltransferase (NAmPRTase) activity, which catalyzes the first step converting nicotinamide to NAD+, has been found in diverse mammalian tissues including brain.52 Results from our lab showed that exogenous nicotinamide supplementation has a very strong axonal protective effect.25 It profoundly delayed axon degeneration induced by axotomy or the toxicity of vincristine, and appeared to act by increasing local NAD+ levels. In case of Wallerian degeneration, the protective effect of nicotinamide is rapid and does not require the present of cell body. Up to 2 or 3 hours of post-treatment (applying nicotinamide to soma free axon 2 or 3 hours after the axotomy) is as efficient as 24 hours pretreatment, indicating its potential as a therapeutic strategy. On the contrary, neither quinolinic acid nor nicotinic acid have any protective effect, but instead induced neural toxicity, consistent with the fact that nicotinamide is the major NAD+ precursor in neurons.
Figure 2.

The mammalian NAD synthesis pathways. In mammalian cells, NAD could be synthesized from three different precursors, such as from tryptophan (the de novo synthesis), as while as from nicotinamide or nicotinic acid (the salvage synthesis). Nmnat-1 is a indispensable enzyme controlling the last step of both de novo and salvage NAD biosynthesis pathways. Nicotinamide represents the main source of NAD in most cells in mammals, including neurons, and it is also the product of NAD+ hydrolysis catalyzed by NAD+ consuming proteins.
Three distinct human Nmnat genes have been recently identified. Each of these adenylyltransferases can participate in both the de novo and salvage NAD+ biosynthetic routes.47,53–55 These three isoforms have distinct cellular localizations. Nmnat-1 is a nuclear protein, while Nmnat-2 and Nmnat-3 are present in Golgi complexes and mitochondria, respectively.56 It has been demonstrated that the protective effect of Nmnat-1 depends on its enzymatic activity.25,46 In Wlds mice, the Nmnat enzymatic activity of the whole brain homogenate is three-fold higher than that of wild type,35 probably due to the three extra copies of this gene in the genome.32 However, the whole brain total NAD+ level of the Wlds mice is not significantly altered.35 These observations raise the question of whether and how NAD+ is involved in axon degeneration. NAD has been well known as a coenzyme in oxidation-reduction reactions, in which it could accept and donate electrons, and participate in ATP generation. The oxidized form of NAD, NAD+, is the major form. The NAD+/NADH ratio of cortical slices is about 3 to 4,57 while the ratio in neuronal cell lines is as high as 10 to 30,58 indicating that neurons might have much higher NAD+/NADH ratio than non-neuronal cells in the brain. HPLC (Fig. 3) results from our lab demonstrated that axonal NAD+ decreases more than 50 percent before the axon shows any morphological sign of degeneration.25 Delaying the axonal NAD+ level decrease by either Nmnat-1 overexpression or exogenous supplementation of nicotinamide could delay axon degeneration, indicating that maintaining axonal NAD+ levels is very important for the integrity of the axonal structure. Exogenous NAD+ supplementation could also delay Wallerian degeneration without the requirement of pretreatment, while NADH has no protective effect and is neurotoxic, suggesting that NAD+ is the major form of NAD which is required to maintain axonal integrity. Moreover, a recent study reported that nicotinamide phosphoribosyl transferase and nicotinic acid phosphoribosyl transferase showed moderated protective activity in the presence of their substrate, indicating the stimulation of NAD biosynthesis delays Wallerian degeneration.59 The same study also showed that overexpressed Nmnat3 located at mitochondria and mutant Nmnat1 relocated to cytoplasm both could provide strong axonal protection. Together with the finding that a small portion of the overexpressed Nmnat-1 localized in the axon,25 these results suggest that the protective effect of Nmnat-1 is mediated by a gain-of-function local NAD+ synthesis.
Figure 3.

The method used to measure axonal NAD+ level. Axons from 12 culture DRG explants were washed with PBS and extracted with perchloric acid. The precipitated protein was separated by centrifuge. The amount of the precipitated protein was measured by the modified BCA protein assay. The protein-free extract was neutralized and applied to HPLC. The axonal NAD+ level is normalized against the protein contents in the same sample and expressed as nmol/mg.
Recent studies from Bellen and colleagues showed in Drosophila that Nmnat functions as a chaperone in protecting neuronal degeneration independent of it NAD synthesis activity.60,61 The author demonstrated that the mutant Drosophila Nmnat, remains only 1% of enzymatic activity, could provide neuroprotection as good as the wild type and both the wild type and mutant Drosophila Nmnat have chaperone activity. This important result leads to the question if mammalian Nmnats also has chaperone activity and if it is required for its axon protection. As mentioned previously, mammalian Nmnat has three distinct isoforms located in nucleus, Golgi and mitochondria, while Drosophila only has one located in cytoplasm.48 In this study, the authors showed that human Nmnat3 has similar chaperone activity to the Drosophila Nmnat. It will be very interesting to find out if the human Nmnat1 and Nmnat2 have chaperone activity, and if the chaperone activity is required to their axon protective function. It is possible that the mammalian Nmnat isoforms have more divided biological functions and only some of them, such as Nmnat3, still keep the chaperone activity as in Drosophila. The NAD synthesis pathways between mammal and Drosophila are also very different. As mentioned previously, the mammalian Nmnats control both the de novo and the salvage NAD synthesis, while the Drosophila Nmnat only controls one of the salvage NAD synthesis pathways. Taking all these difference in consideration, it is possible that the Drosophila Nmnat does not share the exact functions as the mammalian Nmnat1 in neuronal degeneration.
The HPLC results from our lab revealed a depletion of axonal ATP levels during Wallerian degeneration, parallel to, but slightly delayed, when compared to the NAD+ depletion.25 At the same time, AMP levels increased, accompanied by an increase in the AMP/ATP ratio, which activated AMPKK (Fig. 5A). NAD+ is a major electron acceptor in both cytoplasmic glycolysis and the mitochondrial Kreb cycle (Fig. 4). Because it is impossible to bypass the NAD+ requirement for ATP synthesis in the neuron, we supplied the glycolysis end product pyruvate to degenerating axons to partially bypass the requirement for cytoplasmic NAD+, and found that pyruvate also had the ability to delay Wallerian degeneration, suggesting that cellular bioenergetics might be an important downstream event of affected by NAD+ depletion (Fig. 5C). It would be very informative to monitor the change of the NAD+/NADH ratio during axon degeneration; however the NADH level is not easily detectable in axonal samples, probably due to the high neuronal NAD+/NADH ratio and the limited amount of the starting material. On the other hand, ATP is required in the last reaction (NMN + ATP = NAD + PPi) of NAD+ synthesis, catalyzed by Nmnats, indicating that ATP depletions might have a positive feedback on NAD+ depletion (Fig. 5B and C).
Figure 5.

The interaction between NAD+ and ATP during Wallerian degeneration. (A) Increase of the AMP/ATP ration indicates low cellular energy level, and would induce the activation of AMPKK, which would further catalyze the phosphorylation of AMPK. (B) NAD+ is required for ATP generation, while ATP is one substrate of NAD synthesis from nicotinamide. (C) the proposed molecular pathway participated in axon degeneration.
Figure 4.
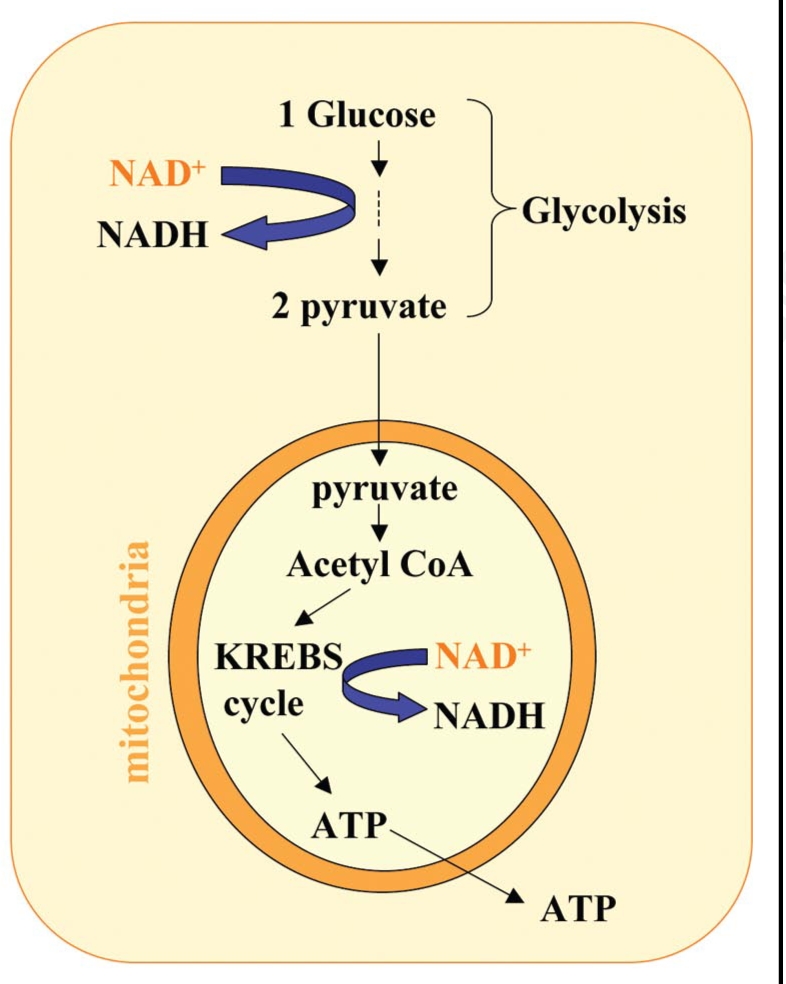
NAD+ is essential for ATP-synthesizing redox reactions. Both cytosolic glycolysis and mitochondrial oxidative phosphorylation required the participation of NAD+. Cytosolic NAD+ is required in the glycolytic pathway for the conversion of glyceraldehyde 3-phosphate to 1,3-bisphosphoglycerate. Thus, upon cytosolic NAD+ depletion, glucose can no longer be converted to the pyruvate needed to fuel the oxidative phosphorylation in mitochondria.
Axon Degeneration and NAD Consumption
During energy metabolism, NAD is rapidly inter-converted between its oxidized and reduced forms without net consumption. On the other hand, the oxidized form of NAD, NAD+, also serves as substrate for covalent protein modification and precursor for biologically active compounds.62 In these processes, NAD+ functions as a donor of ADP-ribose, and releases nicotinamide as a side product (Fig. 6). Therefore NAD+ synthesis is required for maintaining the intracellular NAD+ pool from being consumed by these reactions.
Figure 6.

The three families of NAD+ consuming proteins. All the reactions catalyzed by these NAD consuming proteins use NAD+ as a donor of ADPR, while generating nicotinamide (NAm) as a side product. Nicotinamide has the product inhibition effect on the enzymatic activities of most of the NAD+ consuming proteins, probably by competing for the NAD+ binding pocket.
ADP-ribosyl cyclases are one group of NAD+ consuming proteins which cleave NAD+ to generate cyclic ADP-ribose (cADPR) and nicotinamide.63 cADPR has been indicated as an important signaling molecule and second messenger, participating in cytosolic calcium regulation by releasing calcium from intracellular stores.64 In neurons, there are both membrane-bound and cytosolic forms of ADP-ribosyl cyclases65 and the activity of ADP-ribosyl cyclases is regulated by nitric oxide65 or glutamate stimulation.66 The other product of this enzymatic reaction nicotinamide, which is also an inhibitor of ADP-ribosyl cyclases,67,68 has strong axonal protective effect,25 suggesting the possibility that ADP-ribosyl cyclases may participate in the calcium signaling during early stages of Wallerian degeneration.
NAD+-dependent protein modification is catalyzed by ADP-ribosyl transferases (ADPRTs), which attach the ADP-ribose moiety of NAD+ to target proteins and release nicotinamide at the same time.69 One well known example of ADP-ribosyl transferase is poly (ADP-ribose) polymerase (PARP) protein family, which has been implicated in several important processes. For example, PARP-1 catalyses the transfer of the ADP-ribose moiety from NAD+ to other nuclear proteins and itself, in response to DNA damage.70 Excessive activation of PARP-1 could deplete the cellular NAD+ and ATP pools. Unpublished data from our lab showed that 3-aminobenzamide (3-AB), a well known PARPs inhibitor,71 could modestly delay Wallerian degeneration. At the same time, nicotinamide has also been reported as a potent PARPs inhibitor a long time ago,72 indicating that PARPs activation might be one cause of axonal NAD+ depletion during Wallerian degeneration. However, the poor specificity of 3-AB limits the strength of such a hypothesis.
Sir2 family proteins (or sirtuins) are another NAD+-dependent protein family.73,74 These enzymes catalyze a unique reaction in which the cleavage of NAD+ and the deacetylation of substrate are coupled, leading to the formation of O-acetyl-ADP-ribose. The sir2 gene was first identified as a gene silencing factor in yeast.75 It has been demonstrated that it may play a very important role in calorie restriction-related life span elongation in lower organisms, such as yeast and Drosophila. Sirt1 is one of the seven mammalian homologues of sir2.74 Unlike yeast sir2, Sirt1 targets not only histones, but also some other proteins, such as p53,76,77 and forkhead (Foxo) proteins.78,79 Sirt1 could deacetylate and downregulate p53 and forkhead proteins, therefore negatively regulating cellular damage or stress-induced apoptosis. Study from Araki et al. reported that Nmnat1/NAD+-mediated axonal protection occurs by upregulating the activity of sirt1.46 They found that RNA interference of Sirt1 expression attenuated the protective effects of the Wlds gene or exogenous NAD+ supplementation in vitro, at the same time a Sirt1 activator, Resveratrol, could also protected axon from Wallerian degeneration. In this study, the protective effect of NAD+ requires NAD+ (0.1 to 1 mM) to be supplied to the culture 24 hours before triggering Wallerian degeneration by axotomy, indicating the protective of NAD+ requires nuclear events. However, results from our lab showed that at such low concentration (less than 1 mM) NAD+ has no significant protective effect, and the protective effect of NAD+ at high concentrations (1 to 20 mM) does not require any pretreatment, arguing against the need for nuclear effects.25 Exogenous NAD+ supplementation, up to 5 hours after axotomy and removal of cell bodies, still has the protective function. These results suggest that the protective effect of NAD+ doesn't require the participation of the nucleus. Furthermore, Sirt1 knockout background does not attenuate the protective effect of Wlds/Nmnat or NAD+, suggesting that Sirt1 may not be the downstream effector of the Wlds protein, though our study cannot rule out the involvement of nuclear events directly regulated by the Wlds protein. Another possibility is that other Sirt family members might be upregulated by the Wlds gene. However, it has been shown that the NAD+ levels of the Wlds brain is not altered, raising the question of how Nmnat1 can enhance Sirt activity without changing NAD+ levels. On the contrary, nicotinamide has been shown to be a potent inhibitor of the deacetylation activity of Sirts, including at least mammalian sirt1, 2, 3 and yeast Sir2,80–82 suggesting that Sirts activation might be a reason for NAD+ depletion during Wallerian degeneration.
Results from our lab demonstrated that axonal NAD+ levels are very important in maintaining the axon structure during Wallerian degeneration.25 However, one important remaining question is what triggers the NAD+ depletion during degeneration. As discussed above, excessive activation of any of the NAD+ consuming proteins has the potential to deplete NAD+, which makes them candidate factors upstream of NAD+ depletion. Interestingly, a recent study showed that Resveratrol, the Sirt1 activator, diminished the axonal protection of the Wlds gene against colchicines toxicity, a microtubule de-polymerizing drug, by activating the enzymatic activity of Sirt2, an NAD-dependent tubulin deacetylase.83 One possibility is that the activation of Sirt2 depleted the axonal NAD level, thus over-counted the local NAD synthesis medicated axonal protection provided by the Wlds gene. It would be even more interesting to see if Sirt2 is activated during Wallerian degeneration in wild type mice, in effort to provide more insight into what triggered NAD depletion during this process.
Moreover, nicotinamide is the major NAD+ precursor in neurons as well as a common product of all NAD+ consuming reactions. It functions as a product inhibitor on most of the NAD+ consuming enzymes, probable by competing with NAD+ for the binding pocket.81 These functions of nicotinamide, an endogenous small molecule, make it a perfect candidate to enhance NAD+ levels in therapeutic strategies for pathological axonal degeneration. Study from our lab has demonstrated that increase NAD level by nicotinamide profoundly prevents the degeneration of demyelinated axon and improved the behavioral deficits in EAE models.84
Wallerian Degeneration: Positive Self-Disruption or Starvation?
Axons, the unusual structure of neurons, could be as far as one meter away from the cell body, where most of the structural and functional proteins are synthesized. Initially, it was thought that Wallerian degeneration was due to starvation from proteins synthesized in the cell body. However, the prolonged survival of the functional injured distal axon in Wlds mice eliminates the possibility that only somatic protein starvation is responsible for axonal degeneration. So it was hypothesized that a positive self-disruption process is triggered during Wallerian degeneration. Based on the results from our study,25 we hypothesized that maintaining axonal NAD+ level by local synthesis could prolong the survival of transected axons. At least two models might be possible to explain the NAD+ depletion during axon degeneration. One possibility is that axotomy triggers the over activation of one or more of the NAD+ consuming proteins, which deplete the axonal NAD+ pool. For example as discussed previously, the NAD+-consuming protein ADP-ribosyl cyclases, which catalyze the synthesis of cyclic ADP-ribose, a second messenger for the release of endogenous calcium,64 could be the responsible candidate. This model favors the idea of positive self-disruption and implicates an axonal protective strategy by inhibiting the activity of these NAD+-dependent proteins. On the other hand, it is also possible that axons rely on a constant NAD+ supply from the cell body to complement the constant NAD+ consumption by NAD+-dependent proteins which might be essential for maintaining normal axonal functions. This model is similar to the idea of starvation, but instead of lacking in protein synthesis, NAD+ is the missing molecule. As mentioned earlier, the three mammalian Nmnat proteins are localized in nuclear, Golgi complex and mitochondria respectively,56 which are all mainly located in the cell body rather than in the axon, supporting the idea that the cell body is the major area producing NAD+. To test these theories, the dynamic metabolism of the axonal NAD+ pool needs to be studied under both physiological and pathological conditions.
Wlds vs Nmnat1: Stronger?
In vitro data suggested that Nmnat1 overexpression could provide axonal protection as the Wlds gene.25,45,46 However, the Nmnat1-overexpressing transgenic mice failed to delay Wallerian degeneration in transected sciatic nerves.85 At the same time, recent result from Drosophila showed that the Drosophila Nmnat also protected severed axons from Wallerian degeneration, but the efficacy of protection was slightly reduced when compared to Wlds.45 These conflicting results raised the question whether Nmnat1 could fully mimic the protective effect of the Wlds gene. To answer this question, a well controlled experiments need to be perform to compare the protective effects of both genes, as well as the protein level and Nmnat1 enzymatic activity at different time points after transaction, since the expression level and half life of these proteins might be alternated by the N-terminal 70 amino acids (N70) of Wlds. It has been reported that the UFD2 protein was abundantly expressed in neurons of the cerebrum and cerebellum,86 suggesting the N70 might increase the expression level of the Wlds gene.
The same study also demonstrated that in the cerebrum and cerebellum UFD2 protein is localized predominantly in the cytoplasm of neurons including pyramidal cells in the cerebral cortex and Purkinje cells in the cerebellum,86 which indicated the possibility that the N70 might change the sub-cellular distribution of Nmnat1 and therefore provide more local protection in the axons. Interestingly, this study also showed that UFD2 interacted with calosin-containing protein (VCP), a member of the large family of AAA-type ATPases that are thought to possess chaperone activity, indicating that the mammalian UFD2a/VCP complex might function as an E3/chaperon complex in the response of neurons to stress. Recently, it has been reported the N70 of the Wlds protein bind to VCP directly,87 indicating that the N70 might increase the expression level and protein stability of Wlds especially under stress conditions such as axonal injury. On the other hand, virus mediated in vitro overexpression may generate much more Wlds or Nmnat1 protein and make the potential slight difference in expression levels and protein stability less important. It is also possible that the N70 might recruit other binder partners to Nmnat1, but the fact that virus overexpressed Nmnat1 could fully mimic the protective effect of Wlds in vitro indicated that the Nmnat1 and it enzymatic activity is critical to the axonal protection of Wlds and N70 might serve as a enhancer or stabilizer for the chimeric protein.
NAD+ Decline, Neuronal Atrophy and Autophagy
Unpublished results from our lab showed that NAD+ level decrease could induce neuronal atrophy in intact neurons, indicating the importance of maintaining the cellular NAD+ pool. We identified several genetic or pharmacological reagents which are very useful in manipulating neuronal NAD+ levels. It has been reported that a single point mutation in the (T/H)XGH of Nmnat-1 could completely abolish the enzymatic activity of this proteins without inducing detectable conformational change.88 The mutant Nmnat-1 could form a symmetric hexamer together with the wild type ones. In the catalytic site of this mutant Nmnat-1, the substrate nicotinamide mononucleotide (NMN) was trapped, suggesting that this mutant Nmnat-1 might have a dominant-negative effect on the wild type ones by either abolishing the potential synergetic effect in the Nmnat-1 hexamer or sequestering the substrate NMN. As expected, we found that overexpression of this mutant Nmnat1, gradually decreased neuronal NAD+ levels. As a result, hippocampal neurons overexpressing this mutant Nmnat1 developed morphological changes such as synaptic loss and neuronal atrophy. However, the caveat of dominant-negative Nmnat-1 overexpression is that it might also interrupt other cellular events, such as protein-protein interactions between Nmnat-1 and others.
Because nicotinamide is the major source of NAD+ synthesis, another possible way to decrease NAD+ level is by interfering with other enzymes participating in NAD+ synthesis from nicotinamide. In mammalian cells, nicotinamide is first converted to nicotinamide mononucleotide (NMN) by nicotinamide phosphoribosyltransferase (PBEF).89 NMN is further converted to NAD+ by Nmnats. FK866, a highly specific noncompetitive inhibitor of nicotinamide phosphoribosyltransferase, has been reported to deplete NAD+ levels in mammalian cells.90 We found that FK866 could deplete NAD+ levels quite efficiently in mature hippocampal neuron culture, while it only slightly decreasing ATP levels (Fig. 7). This treatment induced the neuronal atrophy phenotype, similar to the one detected in neurons overexpressing the mutant Nmnat-1 protein. Interfering with PBEF expression by Lentivirus mediated RNAi also decreased hippocampal NAD+ levels, but not as potently as FK866, probably due to the incomplete knock done of PBEF. These results suggest the importance of NAD+ in maintaining the neuron morphology. These results supported the idea that the neuronal NAD+ level is primarily important to maintain neuronal morphology, and ATP synthesis may not be the only downstream events affected by NAD+ decline.
Figure 7.

PBEF inhibitor FK866 induces neuronal atrophy in cultured hippocampal neurons (A and B) The effects of FK866 on cellular NAD and ATP levels. Mature hippocampal neurons (DIV = 22) were treated with FK866 at different concentration, and collected at indicated time-points post-KF866 treatment for HPLC analysis. The NAD and ATP levels were expressed as percentages of the control groups without treatment. (C) Representative confocal images showing that FK866 treatment induces decreases of dendritic spines and dendritic branching in hippocampal neurons. DIV 25 neurons were treated with FK866 (10 nM) alone, or FK866 (10 nM) together with NAD (5 mM) for three days. To visualized individual neurons, the same cultures were transfected with enhanced GFP expressing plasmids by the calcium phosphate transfection method. Hippocampal neurons were cultured at a high density (about 1,000 neurons per mm2). Transfection efficiency is less than 0.1%. The top panel shows the representative higher magnification views of dendritic segments (Scale bar: 5 µm). The bottom panel is the representative images of whole the entire neuron showing dendritic branching patterns (Scale bar: 50 µm).
When NAD+ is decreased, the neuronal processes undergo gradual atrophy, which starts from the terminals of those processes such as spines and growth cones. These structures are important for function, but not for the survival of the neuron. The underlying principle of this phenomenon might be similar to the principle of autophagy during nutrient or trophic factor deprivation. It was thought that mammalian cells activate pathways to survive through the temporary hardship through recycling intracellular components, rather than a death mechanism. It is possible that neurons activate autophagy to recycle its distal processes to provide energy, when cellular energy levels are low. It would be very interesting to find out whether NAD+ decline induced atrophy is through autophagy and whether this process is reversible.
Age, NAD and Neural Protection
Although one study showed that the prolonged survival of axons in transected nerves of Wlds mice is independent of age,91 most other studies found that the Wlds phenotype is age-dependent. One study showed that although the expression of Wlds protein was independent of age in these mice, loss of ability of the sciatic nerve to conduct action potentials after transection is much more rapid in mice one year of age when compared to one month old animals.92 Another study showed that in Wlds mice over seven months of age, axotomy-induced synapse withdrawal from motor endplates is no longer protected, and reverts to a wild-type pattern.93,94 These findings indicate that the molecular pathways involved in the NAD+/Wlds mediated neural protection might be regulated by age-related changes. The age factor is probably working downstream of the Wlds gene. At the same time, these factors might also regulate NAD+ levels in wild type animals. Moreover, the neural morphological changes induced by NAD+ decline are very similar to those exhibited by the neuronal atrophy in aging animals. We measured the NAD+ levels of several brain areas, an age-dependent decrease of NAD+ levels was observed in the hippocampus and cerebellum, which could be detected as early as 7–10 months of age, suggesting that NAD metabolism might be involved in normal aging (Fig. 8).
Figure 8.

Age-related NAD decrease in mouse tissues (A–D) Hippocampus (A), cerebellum (B), cortex (C) or liver (D) tissues from mice of indicated age were subjected to HPLC analysis. NAD levels were normalized against the total weight of the tissue and expressed as nmol/mg. Quantification was performed on results from duplicate experiments from at least three animals at each age. Statistical analysis was done by student t-test. **p < 0.01, ***p < 0.001.
UPS and NAD Synthesis
At the early stage of Wallerian degeneration, loss of microtubule integrity and decrease of NAD+ level occur at similar time. The loss of microtubule integrity could be prevented by nicotinamide and Nmnat-1 overexpression, indicating that NAD+ level change might be upstream of the cytoskeletal disruption. On the other hand, it is very difficult to link the UPS activity to NAD+ depletion, except the fact that the UPS activity consumes ATP. It is possible that UPS activation might contribute to the axonal energy depletion during degeneration, or it is also possible that the UPS activation and NAD+ depletion are involved in two parallel pathways. However, it is hard to demonstrate the relationship between these two processes, because the protective effect of NAD+ is much stronger than UPS inhibition. Recent study showed that wlds has no effect on naturally occurring developmental axon degeneration in flies or mice, while the UPS is intrinsically required for both developmental and injury-induced axon degeneration,1 suggesting that NAD might control early steps in injury-induced degeneration while UPS may be involved in a common execution pathway. As a conclusion, to identify the upstream factor which induced NAD depletion during axon degeneration will provide more meaningful insight into the cue of axonal degeneration in neurodegenerative diseases.
Acknowledgements
We thank Drs. Tim Mitchison, John Flanagan, Marc Freeman, Joshua Kaplan and Glenn Yiu for critical comments on our manuscript.
Footnotes
Previously published online as a Cell Adhesion & Migration E-publication: http://www.landesbioscience.com/journals/celladhesion/article/7483
References
- 1.Hoopfer ED, McLaughlin T, Watts RJ, Schuldiner O, O'Leary DD, Luo L. Wlds protection distinguishes axon degeneration following injury from naturally occurring developmental pruning. Neuron. 2006;50:883–895. doi: 10.1016/j.neuron.2006.05.013. [DOI] [PubMed] [Google Scholar]
- 2.Luo L, O'Leary DD. Axon retraction and degeneration in development and disease. Annu Rev Neurosci. 2005;28:127–156. doi: 10.1146/annurev.neuro.28.061604.135632. [DOI] [PubMed] [Google Scholar]
- 3.Waller AV. Experiments on the section of the glossopharyngeal and hypoglossal nerves of the frog, and observations on the alterations produced thereby in the structure of their primitive fibres. Philosophical Transactions of the Royal Society of London. 1850;140:423–429. [Google Scholar]
- 4.Schmalbruch H, Jensen HJ, Bjaerg M, Kamieniecka Z, Kurland L. A new mouse mutant with progressive motor neuronopathy. J Neuropathol Exp Neurol. 1991;50:192–204. doi: 10.1097/00005072-199105000-00002. [DOI] [PubMed] [Google Scholar]
- 5.Raff MC, Whitmore AV, Finn JT. Axonal self-destruction and neurodegeneration. Science. 2002;296:868–871. doi: 10.1126/science.1068613. [DOI] [PubMed] [Google Scholar]
- 6.Jellinger K, Jirasek A. Neuroaxonal dystrophy in man: character and natural history. Acta Neuropathol (Berl) 1971;5:3–16. doi: 10.1007/978-3-642-47449-1_2. [DOI] [PubMed] [Google Scholar]
- 7.Gold BG. The pathophysiology of proximal neurofilamentous giant axonal swellings: implications for the pathogenesis of amyotrophic lateral sclerosis. Toxicology. 1987;46:125–139. doi: 10.1016/0300-483x(87)90123-5. [DOI] [PubMed] [Google Scholar]
- 8.Stokin GB, Lillo C, Falzone TL, Brusch RG, Rockenstein E, Mount SL, et al. Axonopathy and transport deficits early in the pathogenesis of Alzheimer's disease. Science. 2005;307:1282–1288. doi: 10.1126/science.1105681. [DOI] [PubMed] [Google Scholar]
- 9.Ait-Ikhlef A, Murawsky M, Blondet B, Hantaz-Ambroise D, Martinou JC, Rieger F. The motoneuron degeneration in the wobbler mouse is independent of the overexpression of a Bcl2 transgene in neurons. Neurosci Lett. 1995;199:163–166. doi: 10.1016/0304-3940(95)12061-8. [DOI] [PubMed] [Google Scholar]
- 10.Yuan J, Lipinski M, Degterev A. Diversity in the mechanisms of neuronal cell death. Neuron. 2003;40:401–413. doi: 10.1016/s0896-6273(03)00601-9. [DOI] [PubMed] [Google Scholar]
- 11.Finn JT, Weil M, Archer F, Siman R, Srinivasan A, Raff MC. Evidence that Wallerian degeneration and localized axon degeneration induced by local neurotrophin deprivation do not involve caspases. J Neurosci. 2000;20:1333–1341. doi: 10.1523/JNEUROSCI.20-04-01333.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science. 2000;290:1717–1721. doi: 10.1126/science.290.5497.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nixon RA, Cataldo AM. The endosomal-lysosomal system of neurons: new roles. Trends Neurosci. 1995;18:489–496. doi: 10.1016/0166-2236(95)92772-i. [DOI] [PubMed] [Google Scholar]
- 14.Xue L, Fletcher GC, Tolkovsky AM. Autophagy is activated by apoptotic signalling in sympathetic neurons: an alternative mechanism of death execution. Mol Cell Neurosci. 1999;14:180–198. doi: 10.1006/mcne.1999.0780. [DOI] [PubMed] [Google Scholar]
- 15.Xu K, Tavernarakis N, Driscoll M. Necrotic cell death in C. elegans requires the function of calreticulin and regulators of Ca(2+) release from the endoplasmic reticulum. Neuron. 2001;31:957–971. doi: 10.1016/s0896-6273(01)00432-9. [DOI] [PubMed] [Google Scholar]
- 16.Yamashima T. Implication of cysteine proteases calpain, cathepsin and caspase in ischemic neuronal death of primates. Prog Neurobiol. 2000;62:273–295. doi: 10.1016/s0301-0082(00)00006-x. [DOI] [PubMed] [Google Scholar]
- 17.George EB, Glass JD, Griffin JW. Axotomy-induced axonal degeneration is mediated by calcium influx through ion-specific channels. J Neurosci. 1995;15:6445–6452. doi: 10.1523/JNEUROSCI.15-10-06445.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stys PK, Jiang Q. Calpain-dependent neurofilament breakdown in anoxic and ischemic rat central axons. Neurosci Lett. 2002;328:150–154. doi: 10.1016/s0304-3940(02)00469-x. [DOI] [PubMed] [Google Scholar]
- 19.Leist M, Single B, Naumann H, Fava E, Simon B, Kuhnle S, et al. Inhibition of mitochondrial ATP generation by nitric oxide switches apoptosis to necrosis. Exp Cell Res. 1999;249:396–403. doi: 10.1006/excr.1999.4514. [DOI] [PubMed] [Google Scholar]
- 20.Lelli JL, Jr, Becks LL, Dabrowska MI, Hinshaw DB. ATP converts necrosis to apoptosis in oxidant-injured endothelial cells. Free Radic Biol Med. 1998;25:694–702. doi: 10.1016/s0891-5849(98)00107-5. [DOI] [PubMed] [Google Scholar]
- 21.Stefanelli C, Bonavita F, Stanic I, Farruggia G, Falcieri E, Robuffo I, et al. ATP depletion inhibits glucocorticoid-induced thymocyte apoptosis. Biochem J. 1997;322:909–917. doi: 10.1042/bj3220909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Plomp PJ, Wolvetang EJ, Groen AK, Meijer AJ, Gordon PB, Seglen PO. Energy dependence of autophagic protein degradation in isolated rat hepatocytes. Eur J Biochem. 1987;164:197–203. doi: 10.1111/j.1432-1033.1987.tb11011.x. [DOI] [PubMed] [Google Scholar]
- 23.Schellens JP, Vreeling-Sindelarova H, Plomp PJ, Meijer AJ. Hepatic autophagy and intracellular ATP. A morphometric study. Exp Cell Res. 1988;177:103–108. doi: 10.1016/0014-4827(88)90028-6. [DOI] [PubMed] [Google Scholar]
- 24.Mizushima N, Noda T, Yoshimori T, Tanaka Y, Ishii T, George MD, et al. A protein conjugation system essential for autophagy. Nature. 1998;395:395–398. doi: 10.1038/26506. [DOI] [PubMed] [Google Scholar]
- 25.Wang J, Zhai Q, Chen Y, Lin E, Gu W, McBurney MW, et al. A local mechanism mediates NAD-dependent protection of axon degeneration. J Cell Biol. 2005;170:349–355. doi: 10.1083/jcb.200504028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gillingwater TH, Ribchester RR. Compartmental neurodegeneration and synaptic plasticity in the Wld(s) mutant mouse. J Physiol. 2001;534:627–639. doi: 10.1111/j.1469-7793.2001.00627.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Birks R, Katz B, Miledi R. Physiological and structural changes at the amphibian myoneural junction, in the course of nerve degeneration. J Physiol. 1960;150:145–168. doi: 10.1113/jphysiol.1960.sp006379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kerschensteiner M, Schwab ME, Lichtman JW, Misgeld T. In vivo imaging of axonal degeneration and regeneration in the injured spinal cord. Nat Med. 2005;11:572–577. doi: 10.1038/nm1229. [DOI] [PubMed] [Google Scholar]
- 29.Lunn ER, Perry VH, Brown MC, Rosen H, Gordon S. Absence of Wallerian Degeneration does not Hinder Regeneration in Peripheral Nerve. Eur J Neurosci. 1989;1:27–33. doi: 10.1111/j.1460-9568.1989.tb00771.x. [DOI] [PubMed] [Google Scholar]
- 30.Deckwerth TL, Johnson EM., Jr Neurites can remain viable after destruction of the neuronal soma by programmed cell death (apoptosis) Dev Biol. 1994;165:63–72. doi: 10.1006/dbio.1994.1234. [DOI] [PubMed] [Google Scholar]
- 31.Fernando FS, Conforti L, Tosi S, Smith AD, Coleman MP. Human homologue of a gene mutated in the slow Wallerian degeneration (C57BL/Wld(s)) mouse. Gene. 2002;284:23–29. doi: 10.1016/s0378-1119(02)00394-3. [DOI] [PubMed] [Google Scholar]
- 32.Conforti L, Tarlton A, Mack TG, Mi W, Buckmaster EA, Wagner D, et al. A Ufd2/D4Cole1e chimeric protein and overexpression of Rbp7 in the slow Wallerian degeneration (Wlds) mouse. Proc Natl Acad Sci USA. 2000;97:11377–11382. doi: 10.1073/pnas.97.21.11377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lyon MF, Ogunkolade BW, Brown MC, Atherton DJ, Perry VH. A gene affecting Wallerian nerve degeneration maps distally on mouse chromosome 4. Proc Natl Acad Sci USA. 1993;90:9717–9720. doi: 10.1073/pnas.90.20.9717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang MS, Fang G, Culver DG, Davis AA, Rich MM, Glass JD. The Wlds protein protects against axonal degeneration: a model of gene therapy for peripheral neuropathy. Ann Neurol. 2001;50:773–779. doi: 10.1002/ana.10039. [DOI] [PubMed] [Google Scholar]
- 35.Mack TG, Reiner M, Beirowski B, Mi W, Emanuelli M, Wagner D, et al. Wallerian degeneration of injured axons and synapses is delayed by a Ube4b/Nmnat chimeric gene. Nat Neurosci. 2001;4:1199–1206. doi: 10.1038/nn770. [DOI] [PubMed] [Google Scholar]
- 36.Adalbert R, Nogradi A, Szabo A, Coleman MP. The slow Wallerian degeneration gene in vivo protects motor axons but not their cell bodies after avulsion and neonatal axotomy. Eur J Neurosci. 2006;24:2163–2168. doi: 10.1111/j.1460-9568.2006.05103.x. [DOI] [PubMed] [Google Scholar]
- 37.Wang AL, Yuan M, Neufeld AH. Degeneration of neuronal cell bodies following axonal injury in Wld(S) mice. J Neurosci Res. 2006;84:1799–1807. doi: 10.1002/jnr.21075. [DOI] [PubMed] [Google Scholar]
- 38.Campbell DS, Holt CE. Chemotropic responses of retinal growth cones mediated by rapid local protein synthesis and degradation. Neuron. 2001;32:1013–1026. doi: 10.1016/s0896-6273(01)00551-7. [DOI] [PubMed] [Google Scholar]
- 39.DiAntonio A, Haghighi AP, Portman SL, Lee JD, Amaranto AM, Goodman CS. Ubiquitination-dependent mechanisms regulate synaptic growth and function. Nature. 2001;412:449–452. doi: 10.1038/35086595. [DOI] [PubMed] [Google Scholar]
- 40.Saigoh K, Wang YL, Suh JG, Yamanishi T, Sakai Y, Kiyosawa H, et al. Intragenic deletion in the gene encoding ubiquitin carboxy-terminal hydrolase in gad mice. Nat Genet. 1999;23:47–51. doi: 10.1038/12647. [DOI] [PubMed] [Google Scholar]
- 41.Liu Y, Fallon L, Lashuel HA, Liu Z, Lansbury PT., Jr The UCH-L1 gene encodes two opposing enzymatic activities that affect alpha-synuclein degradation and Parkinson's disease susceptibility. Cell. 2002;111:209–218. doi: 10.1016/s0092-8674(02)01012-7. [DOI] [PubMed] [Google Scholar]
- 42.Zhai Q, Wang J, Kim A, Liu Q, Watts R, Hoopfer E, et al. Involvement of the ubiquitin-proteasome system in the early stages of wallerian degeneration. Neuron. 2003;39:217–225. doi: 10.1016/s0896-6273(03)00429-x. [DOI] [PubMed] [Google Scholar]
- 43.MacInnis BL, Campenot RB. Regulation of Wallerian degeneration and nerve growth factor withdrawal-induced pruning of axons of sympathetic neurons by the proteasome and the MEK/Erk pathway. Mol Cell Neurosci. 2005;28:430–439. doi: 10.1016/j.mcn.2004.10.003. [DOI] [PubMed] [Google Scholar]
- 44.Watts RJ, Hoopfer ED, Luo L. Axon pruning during Drosophila metamorphosis: evidence for local degeneration and requirement of the ubiquitin-proteasome system. Neuron. 2003;38:871–885. doi: 10.1016/s0896-6273(03)00295-2. [DOI] [PubMed] [Google Scholar]
- 45.MacDonald JM, Beach MG, Porpiglia E, Sheehan AE, Watts RJ, Freeman MR. The Drosophila cell corpse engulfment receptor Draper mediates glial clearance of severed axons. Neuron. 2006;50:869–881. doi: 10.1016/j.neuron.2006.04.028. [DOI] [PubMed] [Google Scholar]
- 46.Araki T, Sasaki Y, Milbrandt J. Increased nuclear NAD biosynthesis and SIRT1 activation prevent axonal degeneration. Science. 2004;305:1010–1013. doi: 10.1126/science.1098014. [DOI] [PubMed] [Google Scholar]
- 47.Emanuelli M, Carnevali F, Saccucci F, Pierella F, Amici A, Raffaelli N, et al. Molecular cloning, chromosomal localization, tissue mRNA levels, bacterial expression, and enzymatic properties of human NMN adenylyltransferase. J Biol Chem. 2001;276:406–412. doi: 10.1074/jbc.M008700200. [DOI] [PubMed] [Google Scholar]
- 48.Rongvaux A, Andris F, Van Gool F, Leo O. Reconstructing eukaryotic NAD metabolism. Bioessays. 2003;25:683–690. doi: 10.1002/bies.10297. [DOI] [PubMed] [Google Scholar]
- 49.Lapin IP. Stimulant and convulsive effects of kynurenines injected into brain ventricles in mice. J Neural Transm. 1978;42:37–43. doi: 10.1007/BF01262727. [DOI] [PubMed] [Google Scholar]
- 50.Kohler C, Okuno E, Flood PR, Schwarcz R. Quinolinic acid phosphoribosyltransferase: preferential glial localization in the rat brain visualized by immunocytochemistry. Proc Natl Acad Sci USA. 1987;84:3491–3495. doi: 10.1073/pnas.84.10.3491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jacobson EL, Dame AJ, Pyrek JS, Jacobson MK. Evaluating the role of niacin in human carcinogenesis. Biochimie. 1995;77:394–398. doi: 10.1016/0300-9084(96)88152-1. [DOI] [PubMed] [Google Scholar]
- 52.Dietrich LS, Fuller L, Yero IL, Martinez L. Nicotinamide mononucleotide pyrophosphorylase activity in animal tissues. J Biol Chem. 1966;241:188–191. [PubMed] [Google Scholar]
- 53.Schweiger M, Hennig K, Lerner F, Niere M, Hirsch-Kauffmann M, Specht T, et al. Characterization of recombinant human nicotinamide mononucleotide adenylyl transferase (NMNAT), a nuclear enzyme essential for NAD synthesis. FEBS Lett. 2001;492:95–100. doi: 10.1016/s0014-5793(01)02180-9. [DOI] [PubMed] [Google Scholar]
- 54.Raffaelli N, Sorci L, Amici A, Emanuelli M, Mazzola F, Magni G. Identification of a novel human nicotinamide mononucleotide adenylyltransferase. Biochem Biophys Res Commun. 2002;297:835–840. doi: 10.1016/s0006-291x(02)02285-4. [DOI] [PubMed] [Google Scholar]
- 55.Zhang X, Kurnasov OV, Karthikeyan S, Grishin NV, Osterman AL, Zhang H. Structural characterization of a human cytosolic NMN/NaMN adenylyltransferase and implication in human NAD biosynthesis. J Biol Chem. 2003;278:13503–13511. doi: 10.1074/jbc.M300073200. [DOI] [PubMed] [Google Scholar]
- 56.Berger F, Lau C, Dahlmann M, Ziegler M. Subcellular compartmentation and differential catalytic properties of the three human nicotinamide mononucleotide adenylyltransferase isoforms. J Biol Chem. 2005 doi: 10.1074/jbc.M508660200. [DOI] [PubMed] [Google Scholar]
- 57.Kannurpatti SS, Sanganahalli BG, Mishra S, Joshi PG, Joshi NB. Glutamate-induced differential mitochondrial response in young and adult rats. Neurochem Int. 2004;44:361–369. doi: 10.1016/s0197-0186(03)00164-5. [DOI] [PubMed] [Google Scholar]
- 58.Ellerby LM, Ellerby HM, Park SM, Holleran AL, Murphy AN, Fiskum G, et al. Shift of the cellular oxidation-reduction potential in neural cells expressing Bcl-2. J Neurochem. 1996;67:1259–1267. doi: 10.1046/j.1471-4159.1996.67031259.x. [DOI] [PubMed] [Google Scholar]
- 59.Sasaki Y, Araki T, Milbrandt J. Stimulation of nicotinamide adenine dinucleotide biosynthetic pathways delays axonal degeneration after axotomy. J Neurosci. 2006;26:8484–8491. doi: 10.1523/JNEUROSCI.2320-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhai RG, Cao Y, Hiesinger PR, Zhou Y, Mehta SQ, Schulze KL, et al. Drosophila NMNAT maintains neural integrity independent of its NAD synthesis activity. PLoS Biol. 2006;4:416. doi: 10.1371/journal.pbio.0040416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhai RG, Zhang F, Hiesinger PR, Cao Y, Haueter CM, Bellen HJ. NAD synthase NMNAT acts as a chaperone to protect against neurodegeneration. Nature. 2008;452:887–891. doi: 10.1038/nature06721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ziegler M. New functions of a long-known molecule. Emerging roles of NAD in cellular signaling. Eur J Biochem. 2000;267:1550–1564. doi: 10.1046/j.1432-1327.2000.01187.x. [DOI] [PubMed] [Google Scholar]
- 63.Lee HC, Walseth TF, Bratt GT, Hayes RN, Clapper DL. Structural determination of a cyclic metabolite of NAD+ with intracellular Ca2+-mobilizing activity. J Biol Chem. 1989;264:1608–1615. [PubMed] [Google Scholar]
- 64.Galione A. Cyclic ADP-ribose, the ADP-ribosyl cyclase pathway and calcium signalling. Mol Cell Endocrinol. 1994;98:125–131. doi: 10.1016/0303-7207(94)90130-9. [DOI] [PubMed] [Google Scholar]
- 65.Higashida H, Hashii M, Yokoyama S, Hoshi N, Asai K, Kato T. Cyclic ADP-ribose as a potential second messenger for neuronal Ca2+ signaling. J Neurochem. 2001;76:321–331. doi: 10.1046/j.1471-4159.2001.00082.x. [DOI] [PubMed] [Google Scholar]
- 66.Higashida H, Zhang JS, Mochida S, Chen XL, Shin Y, Noda M, et al. Subtype-specific coupling with ADP-ribosyl cyclase of metabotropic glutamate receptors in retina, cervical superior ganglion and NG108-15 cells. J Neurochem. 2003;85:1148–1158. doi: 10.1046/j.1471-4159.2003.01751.x. [DOI] [PubMed] [Google Scholar]
- 67.Willmott N, Sethi JK, Walseth TF, Lee HC, White AM, Galione A. Nitric oxide-induced mobilization of intracellular calcium via the cyclic ADP-ribose signaling pathway. J Biol Chem. 1996;271:3699–3705. doi: 10.1074/jbc.271.7.3699. [DOI] [PubMed] [Google Scholar]
- 68.Sethi JK, Empson RM, Galione A. Nicotinamide inhibits cyclic ADP-ribose-mediated calcium signalling in sea urchin eggs. Biochem J. 1996;319:613–617. doi: 10.1042/bj3190613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Koch-Nolte F, Reche P, Haag F, Bazan F. ADP-ribosyltransferases: plastic tools for inactivating protein and small molecular weight targets. J Biotechnol. 2001;92:81–87. doi: 10.1016/s0168-1656(01)00356-x. [DOI] [PubMed] [Google Scholar]
- 70.Ziegler M, Oei SL. A cellular survival switch: poly(ADP-ribosyl)ation stimulates DNA repair and silences transcription. Bioessays. 2001;23:543–548. doi: 10.1002/bies.1074. [DOI] [PubMed] [Google Scholar]
- 71.Pivazyan AD, Birks EM, Wood TG, Lin TS, Prusoff WH. Inhibition of poly(ADP-ribose)polymerase activity by nucleoside analogs of thymidine. Biochem Pharmacol. 1992;44:947–953. doi: 10.1016/0006-2952(92)90127-5. [DOI] [PubMed] [Google Scholar]
- 72.Clark JB, Ferris GM, Pinder S. Inhibition of nuclear NAD nucleosidase and poly ADP-ribose polymerase activity from rat liver by nicotinamide and 5′-methyl nicotinamide. Biochim Biophys Acta. 1971;238:82–85. doi: 10.1016/0005-2787(71)90012-8. [DOI] [PubMed] [Google Scholar]
- 73.Frye RA. Characterization of five human cDNAs with homology to the yeast SIR2 gene: Sir2-like proteins (sirtuins) metabolize NAD and may have protein ADP-ribosyltransferase activity. Biochem Biophys Res Commun. 1999;260:273–279. doi: 10.1006/bbrc.1999.0897. [DOI] [PubMed] [Google Scholar]
- 74.Imai S, Armstrong CM, Kaeberlein M, Guarente L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature. 2000;403:795–800. doi: 10.1038/35001622. [DOI] [PubMed] [Google Scholar]
- 75.Rine J, Herskowitz I. Four genes responsible for a position effect on expression from HML and HMR in Saccharomyces cerevisiae. Genetics. 1987;116:9–22. doi: 10.1093/genetics/116.1.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Luo J, Nikolaev AY, Imai S, Chen D, Su F, Shiloh A, et al. Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell. 2001;107:137–148. doi: 10.1016/s0092-8674(01)00524-4. [DOI] [PubMed] [Google Scholar]
- 77.Vaziri H, Dessain SK, Ng Eaton E, Imai SI, Frye RA, Pandita TK, et al. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell. 2001;107:149–159. doi: 10.1016/s0092-8674(01)00527-x. [DOI] [PubMed] [Google Scholar]
- 78.Brunet A, Sweeney LB, Sturgill JF, Chua KF, Greer PL, Lin Y, et al. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science. 2004;303:2011–2015. doi: 10.1126/science.1094637. [DOI] [PubMed] [Google Scholar]
- 79.Motta MC, Divecha N, Lemieux M, Kamel C, Chen D, Gu W, et al. Mammalian SIRT1 represses forkhead transcription factors. Cell. 2004;116:551–563. doi: 10.1016/s0092-8674(04)00126-6. [DOI] [PubMed] [Google Scholar]
- 80.Bitterman KJ, Anderson RM, Cohen HY, Latorre-Esteves M, Sinclair DA. Inhibition of silencing and accelerated aging by nicotinamide, a putative negative regulator of yeast sir2 and human SIRT1. J Biol Chem. 2002;277:45099–45107. doi: 10.1074/jbc.M205670200. [DOI] [PubMed] [Google Scholar]
- 81.Marcotte PA, Richardson PR, Guo J, Barrett LW, Xu N, Gunasekera A, et al. Fluorescence assay of SIRT protein deacetylases using an acetylated peptide substrate and a secondary trypsin reaction. Anal Biochem. 2004;332:90–99. doi: 10.1016/j.ab.2004.05.039. [DOI] [PubMed] [Google Scholar]
- 82.Schwer B, North BJ, Frye RA, Ott M, Verdin E. The human silent information regulator (Sir)2 homologue hSIRT3 is a mitochondrial nicotinamide adenine dinucleotide-dependent deacetylase. J Cell Biol. 2002;158:647–657. doi: 10.1083/jcb.200205057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Suzuki K, Koike T. Resveratrol abolishes resistance to axonal degeneration in slow Wallerian degeneration (Wlds) mice: activation of SIRT2, an NAD-dependent tubulin deacetylase. Biochem Biophys Res Commun. 2007;359:665–671. doi: 10.1016/j.bbrc.2007.05.164. [DOI] [PubMed] [Google Scholar]
- 84.Kaneko S, Wang J, Kaneko M, Yiu G, Hurrell JM, Chitnis T, et al. Protecting axonal degeneration by increasing nicotinamide adenine dinucleotide levels in experimental autoimmune encephalomyelitis models. J Neurosci. 2006;26:9794–9804. doi: 10.1523/JNEUROSCI.2116-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Conforti L, Fang G, Beirowski B, Wang MS, Sorci L, Asress S, et al. NAD(+) and axon degeneration revisited: Nmnat1 cannot substitute for Wld(S) to delay Wallerian degeneration. Cell Death Differ. 2007;14:116–127. doi: 10.1038/sj.cdd.4401944. [DOI] [PubMed] [Google Scholar]
- 86.Kaneko C, Hatakeyama S, Matsumoto M, Yada M, Nakayama K, Nakayama KI. Characterization of the mouse gene for the U-box-type ubiquitin ligase UFD2a. Biochem Biophys Res Commun. 2003;300:297–304. doi: 10.1016/s0006-291x(02)02834-6. [DOI] [PubMed] [Google Scholar]
- 87.Laser H, Conforti L, Morreale G, Mack TG, Heyer M, Haley JE, et al. The slow Wallerian degeneration protein, WldS, binds directly to VCP/p97 and partially redistributes it within the nucleus. Mol Biol Cell. 2006;17:1075–1084. doi: 10.1091/mbc.E05-04-0375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Saridakis V, Christendat D, Kimber MS, Dharamsi A, Edwards AM, Pai EF. Insights into ligand binding and catalysis of a central step in NAD+ synthesis: structures of Methanobacterium thermoautotrophicum NMN adenylyltransferase complexes. J Biol Chem. 2001;276:7225–7232. doi: 10.1074/jbc.M008810200. [DOI] [PubMed] [Google Scholar]
- 89.Rongvaux A, Shea RJ, Mulks MH, Gigot D, Urbain J, Leo O, et al. Pre-B-cell colony-enhancing factor, whose expression is upregulated in activated lymphocytes, is a nicotinamide phosphoribosyltransferase, a cytosolic enzyme involved in NAD biosynthesis. Eur J Immunol. 2002;32:3225–3234. doi: 10.1002/1521-4141(200211)32:11<3225::AID-IMMU3225>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 90.Hasmann M, Schemainda I. FK866, a highly specific noncompetitive inhibitor of nicotinamide phosphoribosyltransferase, represents a novel mechanism for induction of tumor cell apoptosis. Cancer Res. 2003;63:7436–7442. [PubMed] [Google Scholar]
- 91.Crawford TO, Hsieh ST, Schryer BL, Glass JD. Prolonged axonal survival in transected nerves of C57BL/Ola mice is independent of age. J Neurocytol. 1995;24:333–340. doi: 10.1007/BF01189060. [DOI] [PubMed] [Google Scholar]
- 92.Perry VH, Brown MC, Tsao JW. The Effectiveness of the Gene Which Slows the Rate of Wallerian Degeneration in C57BL/Ola Mice Declines With Age. Eur J Neurosci. 1992;4:1000–1002. doi: 10.1111/j.1460-9568.1992.tb00126.x. [DOI] [PubMed] [Google Scholar]
- 93.Gillingwater TH, Thomson D, Mack TG, Soffin EM, Mattison RJ, Coleman MP, et al. Age-dependent synapse withdrawal at axotomised neuromuscular junctions in Wld(s) mutant and Ube4b/Nmnat transgenic mice. J Physiol. 2002;543:739–755. doi: 10.1113/jphysiol.2002.022343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Tsao JW, Brown MC, Carden MJ, McLean WG, Perry VH. Loss of the compound action potential: an electrophysiological, biochemical and morphological study of early events in axonal degeneration in the C57BL/Ola mouse. Eur J Neurosci. 1994;6:516–524. doi: 10.1111/j.1460-9568.1994.tb00295.x. [DOI] [PubMed] [Google Scholar]