

Abstract
Platelet-activating factor (PAF) is a phospholipid inter- and intracellular mediator implicated in intestinal injury primarily via induction of an inflammatory cascade. We find that PAF also has direct pathological effects on intestinal epithelial cells (IEC). PAF induces Cl− channel activation, which is associated with intracellular acidosis and apoptosis. Using the rat small IEC line IEC-6, electrophysiological experiments demonstrated that PAF induces Cl− channel activation. This PAF-activated Cl− current was inhibited by Ca2+ chelation and a calcium calmodulin kinase II inhibitor, suggesting PAF activation of a Ca2+-activated Cl− channel. To determine the pathological consequences of Cl− channel activation, microfluorimetry experiments were performed, which revealed PAF-induced intracellular acidosis, which is also inhibited by the Cl− channel inhibitor 4,4′diisothiocyanostilbene-2,2′disulfonic acid and Ca2+ chelation. PAF-induced intracellular acidosis is associated with caspase 3 activation and DNA fragmentation. PAF-induced caspase activation was abolished in cells transfected with a pH compensatory Na/H exchanger construct to enhance H+ extruding ability and prevent intracellular acidosis. As ClC-3 is a known intestinal Cl− channel dependent on both Ca2+ and calcium calmodulin kinase II phosphor-ylation, we generated ClC-3 knockdown cells using short hairpin RNA. PAF induced Cl− current; acidosis and apoptosis were all significantly decreased in ClC-3 knockdown cells. Our data suggest a novel mechanism of PAF-induced injury by which PAF induces intracellular acidosis via activation of the Ca2+-dependent Cl− channel ClC-3, resulting in apoptosis of IEC.
Keywords: intestinal injury, intestinal barrier, ion transport, Ca2+-activated Cl− channel
PLATELET-ACTIVATING FACTOR (PAF) is a phospholipid inter- and intracellular mediator that has been implicated in the pathology of inflammatory bowel disease (IBD). However, the mechanism of PAF-induced intestinal injury is incompletely understood. It is known that tissue and/or serum PAF levels are elevated in patients with Crohn's disease, ulcerative colitis, and neonatal necrotizing enterocolitis (NEC), and levels appear to correlate with disease severity (13, 26, 39, 45). In animal models of NEC, PAF receptor blockade or administration of the PAF-degrading enzyme PAF acetylhydrolase reduced the incidence of experimental NEC (7, 9). Moreover, in human studies of NEC, PAF has been shown to rise several days before the onset of clinical symptoms in some patients (38), suggesting that this is a critical and possibly initiating factor in the development of the disease.
Regulation begins at the receptor level where PAF acts through binding to a seven-transmembrane domain G protein-coupled receptor. Both high- and low-affinity PAF receptors have been identified (22). Multiple signaling cascades are then linked to the PAF receptor. Depending on the route of administration and the animal model used, PAF can induce a variety of effects including platelet aggregation, hypotension, increased vascular permeability, vasoconstriction, intestinal ischemia, neutrophil recruitment, and production of reactive oxygen species (18). In the intestine it is thought that PAF, once activated, initiates production of other inflammatory mediators such as tumor necrosis factor-α (TNF-α), prostaglandins, thromboxane, and complement that then lead to the clinical signs and symptoms of IBD (8, 19, 52).
Several lines of evidence suggest that the pathological consequences of elevated PAF levels in the intestine are not only the result of the initiation of an inflammatory cascade, but that PAF itself might have a direct effect on intestinal epithelial cells (IEC). Studies of PAF on tissue explants have shown that PAF alters intestinal ion transport, a primary function of the intestine. A study on isolated rat jejunum revealed that PAF induced Cl− secretion (15), whereas studies on rabbit colon have described that PAF stimulated Cl− and bicarbonate secretion (51). We have previously shown that this PAF-stimulated Cl− secretion occurs via the opening of a mucosal Cl− channel after activation of a mucosal PAF receptor; however, the pathological significance of this effect is unclear (12). Subsequently, it was shown that PAF induces apoptosis in a rat small IEC line (IEC-6), via a mechanism that involves mitochondrial depolarization (30). It is unknown whether there is any connection between the regulation of ion transport and apoptosis by PAF in epithelial cells.
Specific Cl− channels have been studied in the intestinal epithelium. Both the cystic fibrosis transmembrane conductance regulator (CFTR) in the duodenum and the outwardly rectifying chloride channel (ORCC) in the T84 human colonic adenocarcinoma cell line have been shown to conduct bicarbonate in addition to Cl− (11, 47). Bicarbonate secretion can lead to intracellular acidosis. Thus PAF-induced Cl− channel activation could alter the intracellular pH of IEC.
In other cell lines, intracellular acidosis has specifically been linked to apoptosis. Szabo et al. (46) showed that activation of ORCC in Jurkat T lymphocytes resulted in intracellular acidosis, leading to apoptosis, whereas Gottlieb and Dosanjh (14) studied mouse mammary epithelial cells and found that cells with mutated CFTR channels were unable to secrete bicarbonate, did not become acidotic, and did not undergo apoptosis.
A link between intracellular acidosis and apoptosis has not been previously demonstrated in IEC. To better understand the mechanism of PAF injury directly on IEC, we hypothesized that PAF-induced Cl− channel activation could lead to intra-cellular acidosis, resulting in accelerated apoptosis in IEC. In these studies we further define the previously described PAF-activated Cl− conductance and demonstrate that it represents activation of a Ca2+-dependent Cl− channel inhibited by 1,2-bis(oaminophenoxy)ethane-N,N,N′,N′-tetraacetic acid (BAPTA) and a calcium calmodulin kinase II inhibitor. This Cl− channel activation is associated with intracellular acidosis inhibited by the Cl− channel inhibitor 4,4′diisothiocyanostilbene-2,2′disulfonic acid (DIDS) and Ca2+ chelation. PAF-induced acidosis is associated with caspase 3 activation and DNA fragmentation characteristic of apoptosis. PAF-induced Cl− conductance and apoptosis is significantly decreased in ClC-3 knockdown cells, suggesting that this is that PAF-activated Cl− channel. Thus PAF directly damages IEC by activation of the Ca2+-dependent Cl− channel ClC-3, leading to intracellular acidosis and apoptosis.
MATERIALS AND METHODS
Epithelial Cell Culture
IEC-6 cells, a well-described immortalized nontransformed rat small IEC line, were used (36, 37). IEC-6 cells were grown in Dulbecco's Modified Eagle Media with 5% fetal bovine serum, 4 mM l-glutamine, 50 U/500 ml insulin, 50 U/ml penicillin, and 50 μg/ml streptomycin at 37°C in a 5% CO2 atmosphere. In addition, H4 cells, a human fetal nontransformed primary IEC line, was used (42). H4 cells were cultured in DMEM with 10% heat-inactivated fetal calf serum, 1% glutamine, 1% sodium pyruvate, 1% amino acids, 1% HEPES, penicillin (50 units/ml), streptomycin (50 μg/ml), and insulin (0.2 units/ml) under standard conditions.
Measurement of pH
IEC on coverslips were loaded for 1 h with the pH-sensitive fluores-cent dye BCECF-AM (Molecular Probes, Eugene, OR) in HEPES buffered solution (116 mM NaCl, 5.4 mM K acetate, .4 mM MgSO4, 5.5 mM Glucose, 1.8 mM CaSO4, 0.9 mM NaH2PO4, 1 mM HEPES hemisodium, and 4.9 mM Na pyruvate adjusted to pH 7.4). Using a spectrofluorometer, cells were exposed to a light source that alternated between excitation at the pH-sensitive wavelength 495 nm and the pH-insensitive wavelength 450 or 440 nm, depending on cell type. Ratio data was then recorded. At the completion of each experiment, pH values were calibrated by sequentially replacing the buffer with a series of high potassium buffers of known pH concentration, which contained the ionophore nigericin. Nigericin permeabilized the cell membrane allowing calibration of the intracellular pH with the known extracellular pH. Ratio data was then converted to pH data.
To measure pH chance in individual cells, cells were plated on glass coverslips at a density of 5,000 cells/coverslip. Two days after plating, cells were incubated for 1 h with the pH-sensitive dye 5-(and-6)-carboxy SNARF-1 acetoxymethyl ester (SNARF) (Molecular Probes). Intracellular pH was measured by microfluorimetry with a fluorescence digital imaging system excited at 480 nm with dual emission values recorded at 580 and 640 nm. Ratio data was converted to linear pH again using calibration data obtained by the K+/nigericin method, which allowed equilibration of intracellular pH with the known extracellular pH when cells were sequentially suspended in calibration buffer containing 75 mM K+ and 10 μM nigericin at pH values ranging from 6.2−8.5.
Measurement of [Ca2+]i
Intracellular calcium concentration ([Ca2+]i) was measured with Fura-2 (Kd 224 nm) loaded as AM esters for 1 h. After being loaded, cultures were washed in buffered saline for at least 15 min to ensure complete hydrolysis of the AM ester. The Fura dye was sequentially excited by using 10-nm bands of light centered on 340 and 380 nm, and a 40-nm-wide band of fluorescence centered on 535 nm was imaged using a fluorescence digital imaging system (31).
Electrophysiological Studies
Chloride channel activation was assessed by whole cell patch clamp. Chloride currents were elicited during a series of 200-ms test pulses delivered at 2-s intervals from −110 to +110 mV in 10-mV increments from a holding potential of −40 mV. The extracellular solution contained (in mM) 140 N-methyl d-glucamine (NMDG)-Cl, 1 MgCl2, 2 CaCl2, and 10 HEPES at a pH of 7.4. The intracellular recording solution contained (in mM) 40 NMDG-Cl, 100 NMDG-Glu, 2 MgCl2, 0.2 Ca (37 nM free), 5 Mg-ATP, 1 EGTA, and 10 HEPES (20).
Overexpression of NHE1
Na/H exchanger 1 (NHE1)-overexpressing IEC-6 cells were generated by transfecting IEC-6 cells with a pCN1/NHE1 plasmid-encoding full length NHE1 using Lipofectamine 2000 reagent. Selection in neomycin allowed establishment of a clone-expressing NHE1 at high levels maintained by selection in G418.
Measurement Of Caspase Activity
Caspase 3 activity was measured by quantifying cleavage of the fluorogenic peptide substrate Ac-DEVD-AFC (BioVision, Mountain-view, CA). Cells were treated as indicated and then harvested and lysed in cell lysis buffer (10 mM Tris·HCl, pH 7.5, 10 mM NaH2PO4, pH 7.5, 130 mM NaCl, 1% Triton X-100, and 10 mM Na pyrophosphate). After centrifugation at 10,000 g for 10 min, supernatants were collected and mixed with substrate (Ac-DEVD-AFC) or substrate plus inhibitor (Ac-DEVD-CHO) for 1 h, and fluorescence was measured at 400-nm excitation and 505-nm emission. Specific caspase activity was calculated by subtracting the fluorescence intensity measured in the presence of substrate plus inhibitor from the fluorescence observed by incubating with substrate alone. Sample fluorescence intensities were normalized to the intensity of untreated cells and are expressed as percentage of control (30).
shRNA Knockdown of ClC-3
RNAi
Two RNA interference (RNAi) target sequences (19 bp) in the rat ClC-3 cDNA (NM053363) were identified using an algorithm previously described (40). Oligonucleotides based on these target sequences, which incorporate the hairpin loop and restriction sites for cloning, were synthesized, phosphorylated, annealed, and cloned into a lentiviral transfer vector, pLentiLox3.7. This transfer vector contains self-inactivating long terminal repeat, a mouse U6 promoter for expression of the short hairpin RNA (shRNA), and green fluorescent protein (GFP) expressed from a cytomegalovirus (CMV) promoter (41). All constructs were verified by sequencing. A control shRNA construct was designed that has at least three base pair mismatches with rat genes in the GenBank database.
Lentivirus production
Replication-defective pseudotyped lentiviral particles were generated by transiently transfecting 3 × 106 HEK293T cells with the following combination of plasmids: 1) 20 μg of the transfer vector containing the cDNA or shRNA of interest; 2) 15 μg of a second generation packaging vector, psPax2 (a generous gift from Didier Trono); and 3) 6 μg of the VSV-G envelope protein vector, pHCMV-G (a generous gift from Garry Nolan, Stanford University) or an ecotropic envelope protein vector, pEnvEco (a generous gift from Toshio Kitamura, Tokyo, Japan) (34, 44), using standard CaPO4 precipitation or liposomal methods. The medium containing lentivirus was harvested 48 h posttransfection and filtered to remove cell debris. The viral supernatant was stored at −80°C.
Viral transduction
The titer of the resulting viral supernatant was determined using serial dilutions (1:10) to infect NIH 3T3 cells. Successfully transduced cells were identified by the expression of GFP detected by fluorescence microscopy. The percentage of GFP-positive cells was determined by manual counting and normalized to the total number of cells present in a low powered field. Titration experiments were performed on IEC-6 cells to determine the minimum multiplicity of infection to achieve >90% transduction on the basis of visual inspection using fluorescence microscopy. IEC-6 cells were incubated with the viral supernatant for4hat 33°C in 5% CO2. After this initial infection period, cultures were maintained at 37°C in 5% CO2. Transduced cells were identified 48 h postinfection by GFP expression with fluorescence microscopy. Medium from transduced IEC-6 cells was used to verify replication incompetence. Selection by flow cytometry for GFP fluorescence allowed establishment of a stable clone of ClC-3 knockdown IEC-6 cells.
RT-PCR Protocol
RNA isolation
Total RNA was isolated from the ClC-3-KD cells and control-KD cells using TRIzol method (Invitrogen, Carlsbad, CA). The concentration and purity of RNA was determined with a spectrophotometer (NanoDrop, Wilmington, DE). Samples were normalized to 1 μg of RNA per sample. Residual DNA was removed by ribonuclease-free deoxyribonuclease (DNase) I treatment (Invitrogen).
RT-PCR
1 μg of total RNA was then reverse transcribed to cDNA with a SuperScript II First-Strand cDNA Synthesis System (Invitrogen) in accordance with the manufacturer's protocol. RNA was preincubated with 0.5 μg (1 μl) oligo-dT12−18 (Invitrogen) and 10 mM (1 μl) dNTP mix (Invitrogen) at 65° for 5 min. After being chilled on ice, 4 μl of 5 × first strand buffer, 2 μl of 0.1 M DTT, and 40 units (1 μl) of RNaseOUT were added to each sample, and the samples were incubated for 2 min at 42°C, followed by the addition of 200 units (1 μl) of Superscript II reverse transcriptase (Invitrogen) and incubation for 50 min. Reverse transcriptase activity was terminated by incubation at 70°C for 15 min, and samples were stored at −80°C until use.
The first-strand cDNA was further amplified by PCR. The reaction was carried out in a DNA thermal cycler (PTC-100 Programmable thermal controller; MJ Research, Waltham, MA). PCR was started by a 3-min denaturation at 95°C, followed by 35 cycles of 30-s denaturation at 95°C, 45-s annealing at 46°C and 45-s extension at 72°C, with a final 10-min extension at 72°C.
In each PCR reaction, 0.5 μM primers of ClC-3, 5′-CCTCTTTCCAAAGTATAGCAC-3′ (sense), 5′-TTACTGGCATTCATGTCATTTC-3′ (antisense), were added. Primers (0.5 μM) of GAPDH were added as internal control: 5′-TAAAGGGCATCCTGGGCTACACT-3′ (sense); 5′-TTACTCCTTGGAGGCCATGTAGG-3′ (antisense).
Amplicons were electrophoresed through 1% agarose gels, stained with ethidium bromide, and visualized on a UV transilluminator.
Measurement of Apoptosis
20,000 cells/well were seeded on a 96-well plate and incubated overnight. Cells were then treated with or without 3 μM PAF. DNA fragmentation in all cell lines was analyzed using an ELISA kit (Roche, Indianapolis, IN) according to the manufacturer's instructions. Briefly, cell lysates were obtained and placed in a streptavidin-coated plate. Anti-histone-biotin-antibody was added to bind the histone component of nucleosomes to the plate. Additionally anti-DNA-peroxidase antibody was added to bind the DNA component of the nucleosomes and allow quantitative determination of the amount of nucleosomes photometrically with the addition of a peroxidase substrate. Absorbance at 405 nm was measured intermittently in 5-min intervals until absorbance values obtained from the sample representing cells were at least 0.2 optical density (OD) unit above the substrate blanks. Absorbance of blanks was subtracted, and OD values were normalized to the mean of OD values representing the samples of untreated cells and expressed as percentage of controls.
Pharmacological Agents
Carbamyl-PAF was obtained from Biomol (Plymouth Meeting, PA). For all experiments cells were treated with μM doses of PAF consistent with higher dosing previously shown to be necessary for effectiveness in epithelial cells (4, 48). BAPTA and DIDS were obtained from Sigma Chemical (St. Louis, MO). Autoinhibitory peptide (AIP) was obtained from Biomol Research. SNARF and Fura-2 were obtained from Molecular Probes.
Statistics
All data are presented as mean values ± SE. Differences were considered significant at P < 0.05 by Student's t-test or ANOVA for multiple variables (GraphPad Prism, San Diego, CA).
RESULTS
PAF induces Cl− channel-dependent intracellular acidification of IEC
Previous studies have demonstrated PAF induced Cl− secretion in tissue explants and colonic epithelial cell lines. The pathological consequences of this Cl− secretion are unknown; however, Cl− channel activation in other cell lines leads to nonspecific secretion of other anions such as , resulting in intracellular acidosis. To test the hypothesis that PAF-induced Cl− channel activation leads to intracellular acidosis, intracellular pH was measured in both the human fetal IEC line H4 and the rat small IEC line IEC-6, plated on coverslips using the pH-sensitive dye BCECF and pH measurement by spectrofluorometry. PAF decreased pH in H4 cells from 7.78 ± .08 to 7.37 ± .05 and in IEC-6 cells from 7.51 ± .08 to 7.29 ± .13 (Fig. 1, A and B).
Fig. 1.

Platelet-activating factor (PAF) induces dose-dependent intracellular acidification of intestinal epithelial cells (IEC). A: representative tracing of pH measurements in H4 cells by spectrofluorometry and the pH-sensitive dye BCECF. A stable baseline was obtained followed by the addition of PAF and calibration with buffers of known pH concentrations. Cells were transferred from standard media to HEPES buffered solutions before loading with BCECF, which accounts for the high baseline pH value. Data are representative of 3 separate experiments, all with similar results. B: summary bar graph of PAF-induced pH change in H4 and IEC-6 cells. Asterisk indicates significant PAF-induced pH change from baseline P < 0.05. C: summary curves representing data from >7 individual cells/dose for IEC-6 cells treated with 0, .75, 1, 3, or 6 μM PAF and pH measured by time-lapse imaging using the pH-sensitive fluorescent dye 5-(and-6)-carboxy SNARF-1 acetoxymethyl ester (SNARF). D: summary bar graph of PAF-induced change in pH. Single asterisk indicates statistically significant difference at P < 0.05 compared with 0.75 μM PAF dose. Double asterisk indicates statistical significance at P < 0.05 compared with 3 μM PAF dose.
Since spectrofluorometry represented an average pH across an entire population of cells, results were repeated using the pH-sensitive dye SNARF and microfluorimetry with a fluorescence digital imaging system, which allows pH measurement of individual cells. IEC-6 cells were treated with .75, 1, 3, or 6 μM PAF (n ≥ 3 separate experiments of minimum 10 cells/experiment for each dose). PAF induced a dose-dependent reduction in intracellular pH of 0.2 ± .02 for the lowest dose tested to maximal decrease in pH of 0.5 ± .03 when treated with PAF 6 μM (Fig. 1, C and D). A decrease in pH of 0.2−0.3 has previously been shown to be sufficient to induce apoptosis (3, 14). As our hypothesis linked PAF-induced intracellular acidosis and apoptosis, and 3 μM PAF induced a decrease in pH of 0.3 ± 0.1, this dose was used for further experiments.
To determine whether this PAF-induced acidification was mediated by Cl− channel activation, measurements were repeated in the presence of Cl− channel inhibitors. Cells treated with 3 μM PAF had a reduction in pH to a nadir of 7.07 ± .05 (Fig. 2A). PAF-induced acidosis was specifically blocked by the Cl− channel inhibitor DIDS (Fig. 2B), suggesting a link between Cl− channel activation and altered pH. Acidosis was not blocked by the two other tested Cl− channel inhibitors glybenclamide or 2,2′iminodibenzoic acid (DPC) (data not shown), suggesting that the activated channel is not the CFTR.
Fig. 2.

PAF-induced intracellular acidification of IEC-6 cells is inhibited by Cl− channel inhibition. A: representative tracing of pH measurements in individual IEC-6 cells by time-lapse imaging using the pH-sensitive fluores-cent dye SNARF. Data are representative of n > 3 separate experiments, each representing minimum 7 cells/experiment all with similar results. Based on previous calibration with buffers of known pH concentrations, 3 μM PAF induced a decrease in pH from a baseline of 7.39 ± .01 to 7.07 ± .05. B: summary data from separate experiments with measurements taken from >7 cells/experiment. Asterisk indicates statistical significance at P < 0.05 compared with baseline conditions. C: PAF-induced pH change was inhibited by the Cl− channel inhibitor 4,4′diisothiocyanostilbene-2,2′ disulfonic acid (DIDS). Data shown are representative of n > 3 separate experiments, each representing minimum 7 cells/experiment all with similar results. D: summary data from separate experiments with measurements taken from >7 cells/experiment indicating no PAF-induced change in pH in the presence of DIDS.
DIDS can inhibit both the exchanger and Cl− channels on IEC. As a Cl− channel inhibitor, DIDS generally blocks Ca2+-dependent anion conductances (17, 24). To determine an association between [Ca2+]i and intracellular pH, microfluorimetry experiments using the pH-sensitive dye SNARF were repeated with PAF in the presence of the Ca2+ chelator BAPTA. PAF-induced acidosis was blocked by BAPTA (Fig. 3) in addition to the previously demonstrated Cl− channel blocker DIDS, linking [Ca2+]i, Cl−, and pH.
Fig. 3.

PAF-induced intracellular acidification of IEC is inhibited by Ca2+ chelation. A: representative tracing of pH measurements in IEC-6 cells by microfluorimetry using the pH-sensitive fluorescence dye SNARF. Cells were loaded with BAPTA 5 μM before treatment with PAF 3 μM. In contrast to control cells (Fig. 1), no acidosis is seen in n > 3 separate experiments, each representing minimum 7 cells/experiment all with similar results. B: summary data from separate experiments with measurements taken from >7 cells/experiment indicating no PAF-induced acidosis in the presence of BAPTA. Asterisk indicates statistical significance at P < 0.05 compared with baseline conditions.
PAF increases [Ca2+]i in IEC
Since [Ca2+]i was potentially involved in PAF-induced Cl− channel activation and PAF has been shown to increase free [Ca2+]i in other cell lines (1, 28), the effect of PAF on free [Ca2+]i in IEC was investigated. The addition of 3 μM PAF induced a fourfold increase in [Ca2+]i as measured by Fura fluorescence (Fig. 4).
Fig. 4.

PAF increases intracellular calcium concentration ([Ca2+]i) in IEC-6 cells. A: representative tracing of [Ca2+]i measurements in IEC-6 cells by microfluorimetry using the Ca2+-sensitive fluorescence dye fura-2 acetoxymethyl ester (Fura-2). Cells were excited alternately between 340 and 380 with emission measured at 542. The addition of 3 μM PAF caused increased [Ca2+]i,in n > 3 separate experiments, each representing minimum 7 cells/experiment all with similar results. B: summary data from separate experiments with measurements taken from >7 cells/experiment. AU, arbitrary units. Asterisk indicates statistical significance at P < 0.05 compared with baseline conditions.
PAF activates a Ca2+-dependent Cl− channel
Our data suggest PAF induced intracellular acidosis associated with activation of a Ca2+-dependent Cl− channel. To better define the PAF-activated Cl− channel in IEC, whole-cell current recordings were obtained from IEC-6 cells. To clearly identify Cl− current activation, experiments were performed using solutions without permeant cations, where Cl− was the only permeant species. Basal current was determined following whole-cell configuration and was negligible (Fig. 5A). Test pulses of +110 mV resulted in a maximal current density of 47.8 ± 5.5 pA/pF (n = 13) following perfusion of IEC-6 cells with PAF (2 μM) containing media. The current-voltage (I-V) relationship of ICl,PAF was outwardly rectifying, with a reversal potential of approximately −8.4 mV (Fig. 5B).
Fig. 5.

PAF activates a Ca2+-dependent Cl− current in IEC. A: representative voltage-clamp recording of IEC-6 cells immediately following establishment of whole-cell configuration (basal) and following peak current activation of PAF 2 μM into the cell. Currents were elicited during a series of test pulses from −110 to +110 in 10-mV increments from a holding potential of −40 mV. B: averaged current-voltage (I-V) relationship shows significant outward rectification, with the reversal potential about −8.4. C: summary of pharmacology of the PAF-activated current. PAF elicited a maximal current density of 47.8 ± 5.5 pA/pF. BAPTA completely inhibited the PAF-induced current (n = 3 cells). Autoinhibitory peptide (AIP), a specific Ca2+/calmodulin-dependent protein kinase II (CaMKII) inhibitor significantly inhibited the PAF-induced current as well (n = 3 cells). Single asterisk indicates a statistically significant increase in current in response to PAF compared with basal conditions at P < 0.05. Double asterisk indicates a statistically significant inhibition of PAF-induced Cl− current by both BAPTA and AIP compared with PAF alone. Based on bathing solutions used, results are consistent with a Cl−-specific current. D: RT-PCR demonstrating significant ClC-3 mRNA (552 bp) knockdown (KD) in ClC-3 KD cell line (lane 3) compared with control KD cell line (lane 2). GAPDH was used as control with gene product expected length of 200 bp. E: summary of PAF-activated current in control and ClC-3 short hairpin RNA (shRNA)-transfected cells. There is significantly less PAF-activated current in ClC-3 knockdown cells even at higher PAF dosing (6 μM) compared with control cells. Number of cells examined as indicated above respective bar. Asterisk indicates statistical significance at P < 0.05.
To confirm that the induced current was activated by Ca2+, we added the Ca2+ chelator BAPTA (10 mM, n = 3) to the pipette solution along with PAF. The presence of BAPTA completely blocked the activation of PAF-induced currents. There are many Cl− channels on the intestinal epithelium. In addition to the well described CFTR and ORCC Cl− channels, other Cl− channels found in intestinal epithelium specifically require a rise in [Ca2+]i of 0.2−5 μM that can come from either [Ca2+]i influx or a release of [Ca2+]i from intracellular stores. These calcium-activated chloride channels are also known to be fairly nonselective in terms of ion conductance. The ClC family of Cl− channels includes ClC-3 an apical, Ca2+-dependent Cl− conductance described in many cell types including IEC (20). ClC-3 current activation is dependent on a Ca2+/calmodulin-dependent protein kinase II (CaMKII)-dependent phosphorylation step (20). To test the possibility that the PAF-induced Cl− channel requires CaMKII, we added AIP (2 μM, n = 3), a specific CaMKII inhibitor (23), to the pipette solution along with PAF. The presence of AIP significantly blocked PAF-activated currents (Fig. 5C).
To more definitively determine whether ClC-3 is the specific PAF-activated Ca2+-dependent Cl− channel, shRNA was used to create a clone of ClC-3 knockdown IEC-6 cells as shown in Fig. 5D. As shown in Fig. 5E, PAF-induced Cl− current was significantly decreased in these cells to 13.6 ± 10.7 compared with control transfected cells (67.4 ± 13.6) even with higher dose PAF (6 μM), demonstrating that ClC-3 is the PAF-activated Cl− channel.
PAF-induced caspase 3 activation is inhibited by NHE1 overexpression
Previous studies have demonstrated PAF-induced mitochondrial-dependent apoptosis leading to DNA fragmentation via caspase 3 activation (30). To determine the importance of PAF-induced intracellular acidosis on PAF-induced caspase 3 activation, it was necessary to inhibit the acidosis. In IEC, pH is regulated by an acid-extruding baso-lateral amiloride-sensitive Na+/H+ exchanger, a stilbene-sensitive exchanger, and a cotransporter (6, 53). Since there is no commercially available molecule to directly buffer intracellular pH, increasing the pH compensation capacity of cells via increased Na+/H+ exchanger activity by transfection has been used as a tool to prevent intracellular acidification in other studies (2). NHE1 was transfected into IEC-6 cells to increase the acid-extruding capacity of the cell and block the induced acidosis. Selection in neomycin and G418 allowed establishment of a clone expressing NHE1 at high levels as demonstrated by immunoblot in Fig. 6A. Caspase 3 activity was then measured by quantifying cleavage of the fluorogenic peptide substrate Ac-DEVD-AFC (BioVision) in response to PAF under baseline conditions and under conditions of inhibited acidosis by NHE1 transfection. PAF-induced caspase 3 activation was inhibited by inhibiting acidosis with NHE1 overexpression (Fig. 6B).
Fig. 6.

PAF-induced caspase 3 activity in IEC-6 cells is inhibited by Na/H exchanger 1 (NHE1) overexpression. A: immunoblot demonstrating overexpression of NHE1 in IEC-6 cells. B: caspase activity was determined in IEC-6 cells from cell lysates representing equal amounts of cellular protein using a fluorometric caspase 3 substrate in response to 1) basal conditions (no treatment), 2) PAF, 3) NHE1 transfection and 4) NHE1 transfection + PAF. Caspase activity was normalized to activity in untreated IEC-6 cells. The asterisk depicts statistical significance at P < 0.05 compared with basal conditions. C: representative tracing of pH measurements in ClC-3 knockdown IEC-6 cells by microfluorimetry using the pH-sensitive fluorescence dye SNARF and treatment with PAF 3 μM. In contrast to control cells (Figs. 1 and 2), no acidosis is seen in n > 3 separate experiments each representing minimum 7 cells/experiment, all with similar results. D: summary data from separate experiments with measurements taken from >7 cells/experiment indicating no PAF-induced acidosis in ClC-3 KD cells.
Since acidosis is important in PAF-induced caspase activation, and our hypothesis was that PAF-induced acidosis was associated with Cl− channel activation, which by electrophysiological studies appeared to be ClC-3, we wondered whether ClC-3 was associated with PAF-induced acidosis. ClC-3 knockdown cells were loaded with the pH-sensitive dye SNARF before treatment with 3 μM PAF and pH measurement by microfluorimetry. In contrast to the PAF-induced acidosis noted in control cells (Figs. 1 and 2), PAF did not induce acidosis in ClC-3 knockdown cells (Fig. 6C).
PAF induces DNA fragmentation. To next confirm a link between apoptosis and PAF activation of ClC-3-associated intracellular acidosis, a quantitative ELISA for histone-associated cytoplasmic DNA fragments was used to measure apoptosis in ClC-3 knockdown and control cells. As shown in Fig. 7, 3 μM PAF induced apoptosis by 171 ± 10% of control (0 PAF) in nontransfected cells and by 188 ± 19% of control in knockdown control cells. Consistent with our hypothesis, in ClC-3 knockdown cells there was significantly less induced apoptosis of only 112 ± 8% of control, suggesting that PAF-induced acidosis via ClC-3 is important for PAF-induced apoptosis.
Fig. 7.
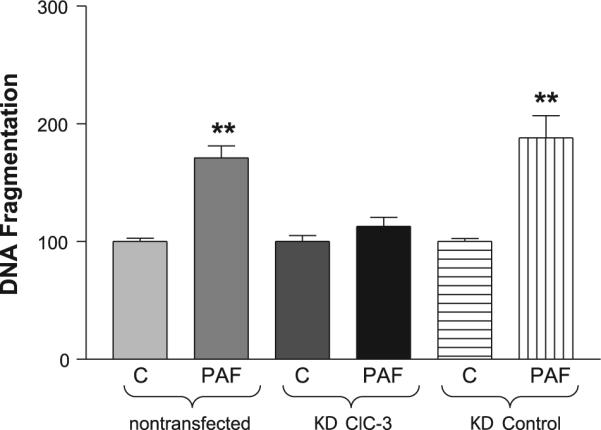
ClC-3 is important for PAF-induced apoptosis. A quantitative ELISA for histone-associated cytoplasmic DNA fragments was used to measure apoptosis in IEC-6 cells, ClC-3 shRNA-transfected IEC-6 cells, and control shRNA-transfected IEC-6 cells treated with 3 μM PAF or vehicle control (C). Each experiment was run in quadruplicate (4 wells/experiment, n = 3 separate experiments), and the mean of the 4 control values was used to standardize values within each experiment as percent of control with control being assigned a value of 100%. Asterisk denotes statistical significance at P < 0.05 compared with control. As shown, PAF induces significant apoptosis in both native and control shRNA-transfected IEC-6 cells but does not induce significant apoptosis in ClC-3 knockdown cells.
DISCUSSION
The central findings of this work are that PAF-induced Cl− channel activation leads to a decrease in pH of 0.2−0.3 units in IEC and that this acidification is associated with caspase activation and apoptosis. Cl− channel activation has not been previously associated with IEC apoptosis. This may be one mechanism behind the link between PAF and IBD.
It has been proposed that the injury in IBD results from an inappropriate immune response to commensal bacteria. A breach in the intestinal mucosal barrier, leading to bacterial translocation across the epithelium and exacerbation of an inflammatory cascade, can aggravate this response. A breach could be caused by disruption of the tight junctions between epithelial cells, or by destruction of the cells themselves. One means of cell destruction is apoptosis, or programmed cell death.
Apoptosis is a well-ordered process of removing damaged cells in which the cell membrane remains intact without induction of an inflammatory reaction. Unlike necrosis, which involves many cells, apoptosis can occur in a single cell surrounded by healthy cells. It is characterized by cell shrinkage, chromatin condensation, and DNA fragmentation (33). Caspase proteases are executors of apoptotic cell death. Caspase activation regulates endonuclease activation, resulting in cleavage of internucleosomal DNA and cell death (32, 50). Although apoptosis is a normal aspect of enterocyte turnover and renewal, accelerated apoptosis can lead to a breach in the intestinal mucosal barrier.
In isolated IEC lines and an animal model of the IBD necrotizing enterocolitis, PAF has been shown to induce apoptosis, but the mechanism of this effect is not fully known (25, 30). A caspase-dependent apoptosis pathway has been demonstrated in IEC in which PAF stimulation leads to Bax translocation to the mitochondria and mitochondrial membrane potential collapse after 1 h, followed by caspase 3 activation and DNA fragmentation (30). However, it is unknown what early PAF-induced signaling leads to mitochondrial initiated apoptosis.
Our data suggest a novel link between PAF-induced Cl− channel activation, intracellular acidosis, and apoptosis. Using whole-cell patch-clamp techniques, a PAF-induced Cl− current was confirmed in individual IEC-6 cells. To identify this anion conductance electrophysiologically, we used solutions that ensured that Cl− was the primary charge carrier. Glybenclamide and DPC did not inhibit PAF-induced acidosis, suggesting that the responsible channel is not CFTR. Instead PAF-induced acidosis was inhibited by DIDS, suggesting an effect on either the exchanger or a Ca2+-dependent Cl− channel. Since the exchanger is not Ca2+ sensitive and the Ca2+ chelator BAPTA also inhibited PAF-induced acidosis, our data suggest a link between a Ca2+-dependent Cl− channel and PAF-induced acidosis. Interestingly, measurements of pH in BAPTA-treated control IEC-6 cells not only did not become acidotic with PAF treatment, but also there was a small, transient, self-limited alkalosis. This is consistent with known effects of PAF in other cell types. PAF has been shown to induce intracellular alkalosis in neutrophils via activation of NHE1 (16, 43).
Electrophysiological studies also demonstrated that the PAF-activated Cl− current was inhibited by BAPTA, again indicating that the Cl− channel is Ca2+ dependent. There are only two known Ca2+-dependent DIDS-sensitive Cl− channels on intestinal epithelium, ClC-3 and bestrophins. The inhibition of the outwardly rectifying PAF-induced conductance by the specific CAMKII inhibitor AIP suggested activation of ClC-3, the only reported CaMKII-activated conductance (20). The current was not completely inhibited by AIP, suggesting incomplete inhibition of phosphorylation by AIP or perhaps an additional role for Ca2+-dependent Cl− channels such as bestrophins, which do not require a phosphorylation step. Studies using shRNA-generated ClC-3 knockdown cells confirm this data. PAF generates minimal current even at high doses in ClC-3 knockdown cells. The presence of some current may be due to incomplete knockdown of ClC-3 in the cells or, as noted above, the role of direct Ca2+-activated Cl− channels acting in compensation for the lack of ClC-3.
Many Cl− channels are relatively nonspecific and when activated can conduct bicarbonate in addition to Cl−, leading to intracellular acidosis. Our data demonstrate PAF-induced intracellular acidosis by 0.2−0.3 pH units. We overexpressed NHE1 in IEC to enhance recovery from an acidification insult. As caspase activation precedes DNA fragmentation and caspase 3 is one of the principal effector caspases of apoptosis, we measured the effect NHE1 overexpression on PAF-induced caspase 3 activity. NHE1 overexpression significantly decreased PAF-induced caspase 3 activity. Thus PAF-induced intracellular acidosis appears important for PAF-induced caspase activation and apoptosis in IEC. This is consistent with data from other cell lines in which intracellular acidosis has also been shown to trigger apoptosis both through enhanced activation of caspases and through activation of pH-sensitive endonucleases (3, 35).
To link our findings to PAF activation of ClC-3 we examined intracellular pH of PAF-treated ClC-3 knockdown cells. In contrast to control cells, PAF did not induce acidosis in ClC-3 knockdown cells. Correspondingly, there was a significant reduction in PAF-induced apoptosis in ClC-3 knockdown cells compared with control cells, linking PAF-induced ClC-3 activation, intracellular acidosis, and apoptosis. This validates our hypothesis that PAF-induced Cl− channel activation leads to intracellular acidosis, resulting in accelerated apoptosis in IEC.
Our data also demonstrate that PAF increases intracellular calcium levels in IEC. Since apoptosis can be triggered by a variety of physiological and pathological stimuli including sustained calcium increases, which act through activation of a Ca2+/Mg2+ endonuclease, it is possible that PAF-induced apoptosis is associated with calcium increase rather than induced intracellular acidosis. Although PAF-altered Ca2+ levels likely also affect cellular processes and would be an interesting separate line of investigation, again our data demonstrating inhibition of PAF-induced caspase 3 activation with NHE1 overexpression suggest a role for the specific effect of PAF-induced acidification in apoptosis.
Together our data suggest a pathological consequence of PAF-induced Cl− channel activation as a mechanism for initiating or enhancing signaling leading to apoptosis in IEC. This may lead to alterations in intestinal barrier function and contribute to the link between PAF and IBD. However, as our studies were performed in isolated cells, further studies in whole intestinal tissue are necessary to determine the clinical pathological consequences of PAF-induced acidosis. Interestingly, PAF has been shown to inhibit apoptosis in immune cells (49). The combined effects of IEC destruction and immune cell preservation may explain the potent role of PAF in intestinal injury.
The PAF doses used in these studies are higher than those commonly used in other studies of the effects of PAF on immune cells. Previously published reports on PAF-mediated changes in various cell types demonstrate two very distinct ranges of dose responses. Effective doses in the 1−100-μM range have been observed primarily in epithelial tissues, i.e., rat intestine and bronchial epithelial cells (4, 48), whereas endometrial cells, T and B lymphocytes, and Chinese hamster ovary cells transfected with the PAF receptor and macrophages exhibit several orders of magnitude higher sensitivity to PAF in the 10−500-nM range (5, 21, 29). Reasons for the lower affinity of PAF receptor in epithelial cells are not known; however, experimental evidence suggests that the affinity of PAF to its receptor can be modified by the interaction of PAF with G proteins, or with membrane lipids (10, 27). When the receptor is bound to heterotrimeric G proteins, it exhibits high PAF affinity, but this affinity is reduced when the G proteins dissociate (10). Also, the treatment of PAF receptor-expressing cells with polyunsaturated fatty acids that altered the lipid composition of extracted membranes also changed the binding affinity of PAF to PAF receptor (27). These findings suggest that the affinity of ligand to PAF receptor could be modified by G proteins or by the physico-chemical properties of the surrounding lipid domain. For the intestine, although the exact concentrations of PAF at the IEC surface are unknown, this lower sensitivity is likely appropriate for a cell potentially exposed to high levels of PAF at the interface between IEC and the intestinal microflora.
It is interesting that other calcium-dependent chloride secretagogues have not been shown to induce apoptosis in IEC and that activation of other Cl− channels, which similarly conduct bicarbonate, do not lead to acidosis. Ion transport is a complex process requiring simultaneous regulation not only of transporters, but also of basolateral loading mechanisms. Thus the difference between PAF and other secretagogues likely relates to simultaneous regulation of other transporters, or the lack of it. If the basolateral loading mechanisms are stimulated as well, the result is transepithelial transport, and if they are not, the result is net loss of Cl− and/or bicarbonate.
Clinical studies suggest that stool PAF levels can correlate with patients at risk for IBD and disease severity and that serum PAF levels can correlate with disease severity in NEC (38, 39, 45). If PAF is a critical molecule in the pathogenesis of IBD, and if an early effect is induced intracellular acidosis, inhibition of this effect may lead to a potential therapy for a subset of patients at risk for IBD due to elevated PAF levels. Further understanding of the direct effects of PAF on IEC separate from an induced inflammatory response will improve our understanding of the pathology of IBD. An improved understanding of factors relating intracellular pH to apoptosis in IEC has the potential to influence not only understanding of IBDs but also of intestinal malignancies.
ACKNOWLEDGMENTS
We thank Chan Bouribon, Anke Di, Sheng-Ru Shiou, and Yueyue Yu for technical assistance. E. Petrof's present affiliation is the Department of Medicine, Queen's University, Kingston, Ontario. Dr. Jun Sun's present affiliation is University of Rochester, Rochester, New York, Department of Medicine.
GRANTS
This study was supported by National Institutes of Health Grant K08-HD043839 (to E. Claud). The DK-42086 grant supported Pilot and feasibility award (to E. Claud) and maintenance of core facilities used.
REFERENCES
- 1.Bankers-Fulbright JL, Kephart GM, Bartemes KR, Kita H, O'Grady SM. Platelet-activating factor stimulates cytoplasmic alkalinization and granule acidification in human eosinophils. J Cell Sci. 2004;117:5749–5757. doi: 10.1242/jcs.01498. [DOI] [PubMed] [Google Scholar]
- 2.Barriere H, Poujeol C, Tauc M, Blasi JM, Counillon L, Poujeol P. CFTR modulates programmed cell death by decreasing intracellular pH in Chinese hamster lung fibroblasts. Am J Physiol Cell Physiol. 2001;281:C810–C824. doi: 10.1152/ajpcell.2001.281.3.C810. [DOI] [PubMed] [Google Scholar]
- 3.Barry MA, Eastman A. Endonuclease activation during apoptosis: the role of cytosolic Ca2+ and pH. Biochem Biophys Res Commun. 1992;186:782–789. doi: 10.1016/0006-291x(92)90814-2. [DOI] [PubMed] [Google Scholar]
- 4.Bern MJ, Sturbaum CW, Karayalcin SS, Berschneider HM, Wachsman JT, Powell DW. Immune system control of rat and rabbit colonic electrolyte transport. Role of prostaglandins and enteric nervous system. J Clin Invest. 1989;83:1810–1820. doi: 10.1172/JCI114086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bonaccorsi L, Luconi M, Maggi M, Muratori M, Forti G, Serio M, Baldi E. Protein tyrosine kinase, mitogen-activated protein kinase and protein kinase C are involved in the mitogenic signaling of platelet-activating factor (PAF) in HEC-1A cells. Biochim Biophys Acta. 1997;1355:155–166. doi: 10.1016/s0167-4889(96)00125-5. [DOI] [PubMed] [Google Scholar]
- 6.Busche R, Jeromin A, von Engelhardt W, Rechkemmer G. Basolateral mechanisms of intracellular pH regulation in the colonic epithelial cell line HT29 clone 19A. Pflügers Arch. 1993;425:219–224. doi: 10.1007/BF00374170. [DOI] [PubMed] [Google Scholar]
- 7.Caplan MS, Hedlund E, Adler L, Lickerman M, Hsueh W. The platelet-activating factor receptor antagonist WEB 2170 prevents neonatal necrotizing enterocolitis in rats. J Pediatr Gastroenterol Nutr. 1997;24:296–301. doi: 10.1097/00005176-199703000-00012. [DOI] [PubMed] [Google Scholar]
- 8.Caplan MS, Kelly A, Hsueh W. Endotoxin and hypoxia-induced intestinal necrosis in rats: the role of platelet activating factor. Pediatr Res. 1992;31:428–434. doi: 10.1203/00006450-199205000-00002. [DOI] [PubMed] [Google Scholar]
- 9.Caplan MS, Lickerman M, Adler L, Dietsch GN, Yu A. The role of recombinant platelet-activating factor acetylhydrolase in a neonatal rat model of necrotizing enterocolitis. Pediatr Res. 1997;42:779–783. doi: 10.1203/00006450-199712000-00010. [DOI] [PubMed] [Google Scholar]
- 10.Carlson SA, Chatterjee TK, Murphy KP, Fisher RA. Mutation of a putative amphipathic alpha-helix in the third intracellular domain of the platelet-activating factor receptor disrupts receptor/G protein coupling and signaling. Mol Pharmacol. 1998;53:451–458. doi: 10.1124/mol.53.3.451. [DOI] [PubMed] [Google Scholar]
- 11.Clarke LL, Harline MC. Dual role of CFTR in cAMP-stimulated HCO3− secretion across murine duodenum. Am J Physiol Gastrointest Liver Physiol. 1998;274:G718–G726. doi: 10.1152/ajpgi.1998.274.4.G718. [DOI] [PubMed] [Google Scholar]
- 12.Claud EC, Li D, Xiao Y, Caplan MS, Jilling T. Platelet-activating factor regulates chloride transport in colonic epithelial cell monolayers. Pediatr Res. 2002;52:155–162. doi: 10.1203/00006450-200208000-00005. [DOI] [PubMed] [Google Scholar]
- 13.Denizot Y, Chaussade S, Nathan N, Colombel JF, Bossant MJ, Cherouki N, Benveniste J, Couturier D. PAF-acether and acetylhydrolase in stool of patients with Crohn's disease. Dig Dis Sci. 1992;37:432–437. doi: 10.1007/BF01307739. [DOI] [PubMed] [Google Scholar]
- 14.Gottlieb RA, Dosanjh A. Mutant cystic fibrosis transmembrane conductance regulator inhibits acidification and apoptosis in C127 cells: possible relevance to cystic fibrosis. Proc Natl Acad Sci USA. 1996;93:3587–3591. doi: 10.1073/pnas.93.8.3587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hanglow AC, Bienenstock J, Perdue MH. Effects of platelet-activating factor on ion transport in isolated rat jejunum. Am J Physiol Gastrointest Liver Physiol. 1989;257:G845–G850. doi: 10.1152/ajpgi.1989.257.5.G845. [DOI] [PubMed] [Google Scholar]
- 16.Hidalgo MA, Ojeda F, Eyre P, LaBranche TP, Smith C, Hancke JL, Burgos RA. Platelet-activating factor increases pH(i) in bovine neutrophils through the PI3K-ERK1/2 pathway. Br J Pharmacol. 2004;141:311–321. doi: 10.1038/sj.bjp.0705590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hogg RC, Wang Q, Large WA. Effects of Cl channel blockers on Ca-activated chloride and potassium currents in smooth muscle cells from rabbit portal vein. Br J Pharmacol. 1994;111:1333–1341. doi: 10.1111/j.1476-5381.1994.tb14891.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hsueh W, Gonzalez-Crussi F. Ischemic bowel necrosis induced by platelet-activating factor: an experimental model. Methods Achiev Exp Pathol. 1988;13:208–239. [PubMed] [Google Scholar]
- 19.Hsueh W, Gonzalez-Crussi F, Arroyave JL. Platelet-activating factor-induced ischemic bowel necrosis. An investigation of secondary mediators in its pathogenesis. Am J Pathol. 1986;122:231–239. [PMC free article] [PubMed] [Google Scholar]
- 20.Huang P, Liu J, Di A, Robinson NC, Musch MW, Kaetzel MA, Nelson DJ. Regulation of human CLC-3 channels by multifunctional Ca2+/calmodulin-dependent protein kinase. J Biol Chem. 2001;276:20093–20100. doi: 10.1074/jbc.M009376200. [DOI] [PubMed] [Google Scholar]
- 21.Huang YH, Schafer-Elinder L, Owman H, Lorentzen JC, Ronnelid J, Frostegard J. Induction of IL-4 by platelet-activating factor. Clin Exp Immunol. 1996;106:143–148. doi: 10.1046/j.1365-2249.1996.d01-813.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hwang SB. Specific receptors of platelet-activating factor, receptor heterogeneity, and signal transduction mechanisms. J Lipid Mediat. 1990;2:123–158. [PubMed] [Google Scholar]
- 23.Ishida A, Kameshita I, Okuno S, Kitani T, Fujisawa H. A novel highly specific and potent inhibitor of calmodulin-dependent protein kinase II. Biochem Biophys Res Commun. 1995;212:806–812. doi: 10.1006/bbrc.1995.2040. [DOI] [PubMed] [Google Scholar]
- 24.Jensen BL, Skott O. Blockade of chloride channels by DIDS stimulates renin release and inhibits contraction of afferent arterioles. Am J Physiol Renal Fluid Electrolyte Physiol. 1996;270:F718–F727. doi: 10.1152/ajprenal.1996.270.5.F718. [DOI] [PubMed] [Google Scholar]
- 25.Jilling T, Lu J, Jackson M, Caplan MS. Intestinal epithelial apoptosis initiates gross bowel necrosis in an experimental rat model of neonatal necrotizing enterocolitis. Pediatr Res. 2004;55:622–629. doi: 10.1203/01.PDR.0000113463.70435.74. [DOI] [PubMed] [Google Scholar]
- 26.Kald B, Olaison G, Sjodahl R, Tagesson C. Novel aspect of Crohn's disease: increased content of platelet-activating factor in ileal and colonic mucosa. Digestion. 1990;46:199–204. doi: 10.1159/000200346. [DOI] [PubMed] [Google Scholar]
- 27.Kang JX, Man SF, Hirsh AJ, Clandinin MT. Characterization of platelet-activating factor binding to human airway epithelial cells: modulation by fatty acids and ion-channel blockers. Biochem J. 1994;303:795–802. doi: 10.1042/bj3030795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Khoo C, Helm J, Choi HB, Kim SU, McLarnon JG. Inhibition of store-operated Ca(2+) influx by acidic extracellular pH in cultured human microglia. Glia. 2001;36:22–30. doi: 10.1002/glia.1092. [DOI] [PubMed] [Google Scholar]
- 29.Kravchenko VV, Pan Z, Han J, Herbert JM, Ulevitch RJ, Ye RD. Platelet-activating factor induces NF-kappa B activation through a G protein-coupled pathway. J Biol Chem. 1995;270:14928–14934. doi: 10.1074/jbc.270.25.14928. [DOI] [PubMed] [Google Scholar]
- 30.Lu J, Caplan MS, Saraf AP, Li D, Adler L, Liu X, Jilling T. Platelet-activating factor-induced apoptosis is blocked by Bcl-2 in rat intestinal epithelial cells. Am J Physiol Gastrointest Liver Physiol. 2004;286:G340–G350. doi: 10.1152/ajpgi.00182.2003. [DOI] [PubMed] [Google Scholar]
- 31.Marks JD, Boriboun C, Wang J. Mitochondrial nitric oxide mediates decreased vulnerability of hippocampal neurons from immature animals to NMDA. J Neurosci. 2005;25:6561–6575. doi: 10.1523/JNEUROSCI.1450-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McConkey DJ, Orrenius S. Signal transduction pathways in apoptosis. Stem Cells. 1996;14:619–631. doi: 10.1002/stem.140619. [DOI] [PubMed] [Google Scholar]
- 33.Metcalfe A, Streuli C. Epithelial apoptosis. Bioessays. 1997;19:711–720. doi: 10.1002/bies.950190812. [DOI] [PubMed] [Google Scholar]
- 34.Morita S, Kojima T, Kitamura T. Plat-E: an efficient and stable system for transient packaging of retroviruses. Gene Ther. 2000;7:1063–1066. doi: 10.1038/sj.gt.3301206. [DOI] [PubMed] [Google Scholar]
- 35.Park HJ, Lyons JC, Ohtsubo T, Song CW. Acidic environment causes apoptosis by increasing caspase activity. Br J Cancer. 1999;80:1892–1897. doi: 10.1038/sj.bjc.6690617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Quaroni A, Isselbacher KJ, Ruoslahti E. Fibronectin synthesis by epithelial crypt cells of rat small intestine. Proc Natl Acad Sci USA. 1978;75:5548–5552. doi: 10.1073/pnas.75.11.5548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Quaroni A, Wands J, Trelstad RL, Isselbacher KJ. Epithelioid cell cultures from rat small intestine. Characterization by morphologic and immunologic criteria. J Cell Biol. 1979;80:248–265. doi: 10.1083/jcb.80.2.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rabinowitz SS, Dzakpasu P, Piecuch S, Leblanc P, Valencia G, Kornecki E. Platelet-activating factor in infants at risk for necrotizing enterocolitis. J Pediatr. 2001;138:81–86. doi: 10.1067/mpd.2001.110132. [DOI] [PubMed] [Google Scholar]
- 39.Rachmilewitz D, Eliakim R, Simon P, Ligumsky M, Karmeli F. Cytokines and platelet-activating factor in human inflamed colonic mucosa. Agents Actions. 1992:C32–C36. Spec No. [PubMed] [Google Scholar]
- 40.Reynolds A, Leake D, Boese Q, Scaringe S, Marshall WS, Khvorova A. Rational siRNA design for RNA interference. Nat Biotechnol. 2004;22:326–330. doi: 10.1038/nbt936. [DOI] [PubMed] [Google Scholar]
- 41.Rubinson DA, Dillon CP, Kwiatkowski AV, Sievers C, Yang L, Kopinja J, Rooney DL, Ihrig MM, McManus MT, Gertler FB, Scott ML, Van Parijs L. A lentivirus-based system to functionally silence genes in primary mammalian cells, stem cells and transgenic mice by RNA interference. Nat Genet. 2003;33:401–406. doi: 10.1038/ng1117. [DOI] [PubMed] [Google Scholar]
- 42.Sanderson IR, Ezzell RM, Kedinger M, Erlanger M, Xu ZX, Pringault E, Leon-Robine S, Louvard D, Walker WA. Human fetal enterocytes in vitro: modulation of the phenotype by extracellular matrix. Proc Natl Acad Sci USA. 1996;93:7717–7722. doi: 10.1073/pnas.93.15.7717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sandoval AJ, Riquelme JP, Carretta MD, Hancke JL, Hidalgo MA, Burgos RA. Store-operated calcium entry mediates intracellular alkalinization, ERK1/2, and Akt/PKB phosphorylation in bovine neutrophils. J Leukoc Biol. 2007;82:1266–1277. doi: 10.1189/jlb.0307196. [DOI] [PubMed] [Google Scholar]
- 44.Schambach A, Galla M, Modlich U, Will E, Chandra S, Reeves L, Colbert M, Williams DA, von Kalle C, Baum C. Lentiviral vectors pseudotyped with murine ecotropic envelope: increased biosafety and convenience in preclinical research. Exp Hematol. 2006;34:588–592. doi: 10.1016/j.exphem.2006.02.005. [DOI] [PubMed] [Google Scholar]
- 45.Sobhani I, Hochlaf S, Denizot Y, Vissuzaine C, Rene E, Benveniste J, Lewin MM, Mignon M. Raised concentrations of platelet activating factor in colonic mucosa of Crohn's disease patients. Gut. 1992;33:1220–1225. doi: 10.1136/gut.33.9.1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Szabo I, Lepple-Wienhues A, Kaba KN, Zoratti M, Gulbins E, Lang F. Tyrosine kinase-dependent activation of a chloride channel in CD95-induced apoptosis in T lymphocytes. Proc Natl Acad Sci USA. 1998;95:6169–6174. doi: 10.1073/pnas.95.11.6169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tabcharani JA, Jensen TJ, Riordan JR, Hanrahan JW. Bicarbonate permeability of the outwardly rectifying anion channel. J Membr Biol. 1989;112:109–122. doi: 10.1007/BF01871272. [DOI] [PubMed] [Google Scholar]
- 48.Tamaoki J, Sakai N, Isono K, Kanemura T, Yamawaki I, Takizawa T. Effects of platelet-activating factor on bioelectric properties of cultured tracheal and bronchial epithelia. J Allergy Clin Immunol. 1991;87:1042–1049. doi: 10.1016/0091-6749(91)92148-t. [DOI] [PubMed] [Google Scholar]
- 49.Toledano BJ, Bastien Y, Noya F, Baruchel S, Mazer B. Platelet-activating factor abrogates apoptosis induced by cross-linking of the surface IgM receptor in a human B lymphoblastoid cell line. J Immunol. 1997;158:3705–3715. [PubMed] [Google Scholar]
- 50.Torriglia A, Perani P, Brossas JY, Altairac S, Zeggai S, Martin E, Treton J, Courtois Y, Counis MF. A caspase-independent cell clearance program. The LEI/L-DNase II pathway. Ann NY Acad Sci. 2000;926:192–203. doi: 10.1111/j.1749-6632.2000.tb05612.x. [DOI] [PubMed] [Google Scholar]
- 51.Travis SP, Jewell DP. Regional differences in the response to platelet-activating factor in rabbit colon. Clin Sci (Colch) 1992;82:673–680. doi: 10.1042/cs0820673. [DOI] [PubMed] [Google Scholar]
- 52.Wallace JL, Steel G, Whittle BJ, Lagente V, Vargaftig B. Evidence for platelet-activating factor as a mediator of endotoxin-induced gastrointestinal damage in the rat. Effects of three platelet-activating factor antagonists. Gastroenterology. 1987;93:765–773. doi: 10.1016/0016-5085(87)90438-0. [DOI] [PubMed] [Google Scholar]
- 53.Wenzl E, Sjaastad MD, Weintraub WH, Machen TE. Intracellular pH regulation in IEC-6 cells, a cryptlike intestinal cell line. Am J Physiol Gastrointest Liver Physiol. 1989;257:G732–G740. doi: 10.1152/ajpgi.1989.257.5.G732. [DOI] [PubMed] [Google Scholar]