

Abstract
Native mammalian prions exist in self-propagating strains that exhibit distinctive clinical, pathological and biochemical characteristics. Prion strain diversity is associated with variations in PrPSc conformation, but it remains unknown precisely which physical properties of the PrPSc molecules are required to encipher mammalian prion strain phenotypes. In this study, we subjected prion-infected brain homogenates derived from three different hamster scrapie strains to either (i) proteinase K digestion or (ii) sonication, and inoculated the modified samples into normal hamsters. The results show that the strain-specific clinical features and neuropathological profiles of inoculated animals were not affected by either treatment. Similarly, the strain-dependent biochemical characteristics of the PrPSc molecules (including electrophoretic mobility, glycoform composition, conformational stability and susceptibility to protease digestion) in infected animals were unaffected by either proteolysis or sonication of the original inocula. These results indicate that the infectious strain properties of native prions do not appear to be altered by PrPSc disaggregation, and that maintenance of such properties does not require the N-domain (approximately residues 23–90) of the protease-resistant PrPSc molecules or protease-sensitive PrPSc molecules.
INTRODUCTION
Prion diseases are fatal neurodegenerative illnesses that occur in genetic, sporadic and infectious forms (Glatzel et al., 2005). From a public health perspective, prion diseases are challenging to control because infectious prions are highly resistant to environmental degradation (Brown & Gajdusek, 1991) and can potentially be transmitted by several different routes (Holada et al., 2000; Ligios et al., 2005; Mathiason et al., 2006; Seeger et al., 2005). The critical molecular event in the pathogenesis of prion diseases is the misfolding of the host-encoded prion protein (PrPC) into an infectious isoform (PrPSc), but the mechanism of this conformational change remains unknown (Prusiner, 1982). Mature PrP molecules contain 208 aa (residues 23–231), two N-linked glycosylation sites, an intramolecular disulfide bond and a C-terminal glycophosphatidylinositol anchor (Endo et al., 1989; Locht et al., 1986; Stahl et al., 1987; Turk et al., 1988). Recently, purified native PrPC molecules containing only prion protein and co-purified lipids have been converted into infectious PrPSc molecules de novo, through an in vitro reaction requiring accessory polyanions (Deleault et al., 2007).
Interestingly, mammalian prions occur in a variety of different ‘strains’. Strains are defined as natural isolates of infectious prions characterized by distinctive clinical and neuropathological features, which are faithfully recapitulated upon serial passage within the same animal species (Bruce, 1993; Carlson, 1996). Strain diversity is associated with variations in PrPSc conformation (Bessen & Marsh, 1992; Collinge et al., 1996; Peretz et al., 2001; Safar et al., 1998; Telling et al., 1996), but it remains unknown precisely which PrPSc conformers or domains are required to encode mammalian prion strain phenotypes.
Studies using a conformation-dependent immunoassay (CDI) technique have shown that prion-infected brains contain both protease-sensitive and protease-resistant PrPSc molecules (denoted sPrPSc and rPrPSc, respectively), and the ratio of sPrPSc : rPrPSc molecules generated in infected animals is characteristic for each prion strain (Safar et al., 1998). These observations have raised the possibility that sPrPSc molecules may be essential for encoding the strain properties of mammalian prions. In addition, other studies have demonstrated that a variety of physical and chemical treatments alter the strain-dependent relationship between infectious titre and incubation time (Dickinson & Fraser, 1969; Somerville & Carp, 1983; Taylor & Fernie, 1996), raising the possibility that such treatments might also modify other strain properties, possibly by disrupting PrPSc aggregates.
Currently, the molecular structure of PrPSc molecules is unknown. Protease digestion experiments indicate that approximately 70 N-terminal amino acids of the rPrPSc molecules are accessible to proteolytic enzymes, and that the precise length of the protease-sensitive domain varies between different prion strains (Bessen & Marsh, 1992). It is not known whether this externally accessible domain might play a critical role in encoding strain properties.
Here, we performed a series of experiments with native mammalian prions to investigate the physical basis of prion strain variation in greater depth. Specifically, we studied the effects of (i) proteinase K (PK) digestion (which degrades the sPrPSc conformer and removes the N terminus from the rPrPSc conformer to form PrP27–30 molecules) and (ii) sonication (which physically disrupts PrPSc aggregates) upon the clinical, pathological and biochemical properties of hamster scrapie prions. Although prior studies have established that both protease-digested PrP27–30 molecules and sonication-disaggregated prions are infectious (Gabizon et al., 1988; Prusiner et al., 1983; Supattapone et al., 2001), the in vivo properties of different prion strains subjected to these treatments have not previously been analysed systematically.
METHODS
Preparation of brain homogenates
All procedures were done at 4 °C. To avoid cross-contamination of various prion strains, all work was done in laminar flow biosafety cabinets, using disposable surface liners and aerosol barrier tips. Between each brain homogenate and inoculum preparation, safety cabinets were disinfected using 10 % SDS/2 % acetic acid (Peretz et al., 2006).
Normal and scrapie-infected brains from female Syrian golden hamsters were homogenized in 10 vols (w/v) of ice-cold PBS without calcium or magnesium by using Precision disposable tissue grinders. Homogenates were then centrifuged at 200 g for 30 s and the supernatant was removed. This step was repeated twice for each homogenate before use.
Preparation of inocula
Three strains of hamster scrapie [drowsy (DY), 139H and Sc237] were kindly provided by Dr Stanley Prusiner (University of California, San Francisco, USA). Prior to experimental treatments, homogenates were subjected to further disruption by five passages each through 26G and 28G hypodermic needles. In order to normalize the amount of PrPSc present in each homogenate (based on Western blot signals), 4 ml DY brain homogenate and 1 ml each of 139H and Sc237 were centrifuged for 1 h at 100 000 g at 4 °C. Supernatants were removed and discarded. Pellets of 139H and Sc237 were resuspended in 500 μl PBS, 1 % Triton X-100 and the DY pellet was resuspended in 750 μl PBS, 1 % Triton X-100. Homogenates were then subjected to another round of homogenization by five passages each through 26G and 28G hypodermic needles.
To prepare control and experimental inocula for each strain, separate 90 μl aliquots were treated as follows. For ‘control’ samples, 10 μl deionized water was added to each homogenate and the samples were incubated at 37 °C for 1 h with shaking at 800 r.p.m. in an Eppendorf Thermomixer (Fisher Scientific). For PK digested samples (denoted as ‘PrP27–30’ in figures), 10 μl PK (30 U mg−1; Roche Applied Science) was added to each homogenate to achieve a final concentration of 10 μg ml−1, and the sample was incubated as described above. ‘Sonicated’ samples were mixed with 10 μl deionized water, and were incubated at 37 °C in a Misonix 3000-MPD programmable sonicator equipped with a deep well microplate horn containing 350 ml water (Misonix). The samples were subjected to three 30 s bursts of sonication separated by two 30 min incubations. After incubation, each treatment then received 20 μl Pefabloc SC (Roche Applied Sciences), to a final concentration of 0.9–1.0 mM. Inocula were prepared by adding 50 μl each sample (adjusted so that each group had similar concentrations of PK-resistant PrP as determined by Western blotting) to 450 μl diluent containing sterile PBS plus 1 mg BSA ml−1.
Scrapie inoculation and diagnosis
Three-week-old female Syrian golden hamsters were injected with 50 μl of each inoculum using 28G disposable hypodermic needles inserted into the parietal lobe. After inoculation, hamsters were examined daily for neurological dysfunction, and standard diagnostic criteria were used to identify animals exhibiting signs of scrapie (Fraser & Dickinson, 1967). Animals at the end stage of disease were killed, and their brains were removed for histological and biochemical analyses.
PrPSc detection
Unless otherwise specified, protease-digested (+PK) samples were incubated with 50 μg PK ml−1 for 1 h at 37 °C. An equal volume of 2× SDS sample buffer was then added, and samples were boiled for 10 min at 95 °C. SDS-PAGE was performed on 1.5 mm 12 % polyacrylamide gels with an acrylamide/bisacrylamide ratio of 29 : 1 (Bio-Rad). After electrophoresis, the proteins were transferred to a methanol-charged, buffer-equilibrated PVDF membrane (Millipore) by using a Transblot SD SemiDry Transfer Cell (Bio-Rad) set at 2 mA cm−2 for 30 min.
To visualize PrP signals, Western blot membranes were first treated with 3 M GdnSCN (Roche) at room temperature for 30 min. Membranes were then rinsed with TBST (10 mM Tris pH 7.2, 150 mM NaCl, 0.1 % Tween 20) and blocked for 1 h in skimmed milk buffered with TBST. The blocked membrane was incubated overnight at 4 °C with 3F4 monoclonal antibody (Signet Laboratories) diluted 1 : 5000 in TBST. After this incubation, the membrane was washed three times for 10 min in TBST and incubated for 1 h at 4 °C with horseradish peroxidase-labelled anti-mouse IgG secondary antibody conjugate (GE Healthcare) diluted 1 : 5000 in TBST. The membrane was washed four times for 10 min with TBST. The blot was developed by using West Femto chemiluminescence substrate (Pierce), sealed in plastic covers, and captured digitally by using a Fuji (Fujifilm) LAS-3000 chemiluminescence documentation system. Digital images were captured using Image Reader version 2.0 (Fujifilm). Relative molecular masses were based on migration of pre-stained standards from either Fermentas or Bio-Rad.
Enzymic deglycosylation
To deglycosylate PrPSc molecules, 50 μl of each sample was incubated with 40 μl PK in PBS, 1 % (final concentration of 10 μg ml−1) for 30 min at 37 °C. A stock solution of 0.3 M PMSF dissolved in ethanol was added to each sample to achieve a final concentration of 1 mM. Samples were then diluted to a final volume of 750 μl containing PBS and 10 % Sarkosyl (Sigma), and centrifuged at 100 000 g for 1 h at 4 °C. Supernatants were removed and pellets were resuspended in 100 μl 1× glycoprotein denaturing buffer (New England Biolabs) and boiled at 95 °C for 10 min. After boiling, 10× G7 reaction buffer and 10 % NP-40 (both from New England Biolabs) were added to a final concentration of 1× and 1 %, respectively. Next, 3 μl PNGase F enzyme or storage buffer (20 mM Tris/HCl pH 7.5, 50 mM NaCl, 5 mM EDTA) was added to tubes and samples were incubated at 37 °C for 2 h. Reactions were stopped by boiling samples in SDS sample buffer.
Protease resistance assay
Three scrapie-infected brains from each group were homogenized as described above and pooled for use in protease resistance and guanidine denaturation assays. Semi-quantitative Western blots were run to estimate the amount of PrPSc present in each brain pool, and samples were adjusted to normalize PrPSc concentration by using 10 % Prnp0/0 brain homogenate as a diluent.
To determine protease resistance, normalized homogenates were digested for 1 h at 37 °C with increasing amounts of PK. The following concentrations were used: 0.316, 1, 3.16, 10, 31.6, 100, 316 and 1000 μg ml−1. Digestion was terminated by boiling samples in SDS sample buffer. Following SDS-PAGE and Western blotting, PrP signals for samples in each group were normalized against the signal obtained at 0.316 μg PK ml−1.
Guanidine denaturation assay
Guanidine denaturation assays were performed as described previously (Deleault et al., 2007). All centrifugation was done at 4 °C. Briefly, samples containing 40 μl normalized brain homogenate were brought up to 100 μl with 10 % Triton X-100 and varying concentrations of GdnHCl to achieve a final concentration of 1 % Triton X-100 and [GdnHCl] from 1–4 M (note that GdnHCl treatments started at 1 M due to inconsistencies of PrPSc recovery at lower concentrations). Samples were incubated for 90 min at 37 °C at 800 r.p.m. in an Eppendorf Thermomixer (Fisher), and 800 μl dilution buffer [10 mM Tris pH 8.0, 150 mM NaCl, 0.5 % NP-40, 0.5 % DOC (Sigma)] was added. The final GdnHCl concentration of each sample was adjusted to 0.4 M by adding 50 μl water or solutions containing varying concentrations of GdnHCl. Samples were then digested with 20 μg PK ml−1 as described above, and PMSF (dissolved in 100 % ethanol) was added to a final concentration of 1 mM to inactivate PK. Samples were then centrifuged at 100 000 g for 30 min. Supernatants were removed and pellets were resuspended in 1 ml dilution buffer. Samples were then vortexed, and centrifuged at 100 000 g for 20 min. Supernatants were removed, and the pellets were resuspended in 100 μl dilution buffer. An equal volume of 2×SDS sample buffer was added, and samples were boiled for 10 min. Following SDS-PAGE and Western blotting, PrP signals for samples in each group were normalized against the signal obtained at 1 M GdnHCl.
PrPSc aggregation assay
Samples were prepared by incubating 20 μl 10 % (w/v) scrapie-infected brain homogenates with 0.65 ml PBS, 1 % N-lauryl sarcosine with shaking at 800 r.p.m. at 37 °C for 20 min. Duplicate aliquots each containing 0.3 ml were mixed with 0.7 ml PBS, 1 % Triton X-100 and centrifuged at 100 000 g at 4 °C for 1 h. Supernatant fractions were discarded, the pellet was washed with 1 ml PBS, 1 % Triton X-100, resuspended in 0.7 ml Triton X-100, and then either shaken at 800 r.p.m. at 37 °C for 1 h in an Eppendorf Thermomixer (control samples) or subjected to three times 30 s bursts of sonication separated by two times 30 min incubations in a Misonix 3000-MPD programmable sonicator (sonicated samples). Following each respective treatment, samples were vortexed vigorously for 20 s and layered over a 0.3 ml cushion of 30 % sucrose, and centrifuged at 10 000 g for 30 min. Supernatant fractions were collected and precipitated with sodium phosphotungstic acid as described previously (Safar et al., 1998). Pellet fractions were resuspended by boiling in 60 μl SDS sample buffer for 10 min. For each sample, we calculated the per cent of PrPSc in the supernatant=Western blot signal of PrPSc in supernatant/(PrPSc in supernatant + PrPSc in pellet).
Quantification and statistical analyses of biochemical assays
Digitally captured images of Western blots were subjected to densitometric analysis using the program Image Gauge version 4.22 (Fuji). For each condition, replicates were performed (n=3) and values were averaged. Mean values generated for the PK and GdnHCl curves were best fitted using the two parameter exponential decay equation and three parameter sigmoidal equation, respectively, by using the program SigmaPlot version 10.0 (Systat Software). Error bars indicate standard error of the mean (SEM).
Neuropathology and statistical analyses
Brains were removed at the time of sacrifice by using new, sterile-packaged dissection instruments and disposable surfaces to avoid cross-contamination. They were fixed by immersion in 10 % buffered formalin for 1–2 days, cut into approximately 3 mm thick coronal sections, and placed in a tissue-processing cassette. Cassettes were treated with 88 % formic acid for 1–1.5 h to disinfect, and then switched back into 10 % formalin for 1–3 days. The tissue was processed for paraffin embedding, and representative slides were stained with haematoxylin and eosin. Slides from adjacent sections were also prepared for antigen retrieval [CitraPlus (Biogenex), pH 6.23 and pressure steaming to 126C/16 p.s.i. (110.4 kPa)] followed by immunostaining with 3F4 anti-PrP monoclonal antibody (DakoCytomation or Signet, final working concentrations 1 μg ml−1) using a Biogenix 6000 automated stainer. Immunodeposition was visualized using HRP/DAB MultiLink detection kit (Biogenex) following the manufacturer’s recommended protocol.
A single neuropathologist (B. Harris), who was blind to the identities of both experimental groups and the overall design of the study, examined all of the slides, which were labelled with arbitrary six digit numbers. Each brain was scored for a degree of vacuolation in five brain regions: frontal cortex, parietal cortex, hippocampus CA1/2, cerebellum and medulla. Scoring was done using ordinal variables as described previously (Deleault et al., 2007): 0, no lesions; 0.5, minimum vacuolation (2–3 vacuoles in half a × 40 objective field); 1.0, little vacuolation (3–5 vacuoles in half a field); 2.0, moderate vacuolation (several vacuoles evenly scattered); 3.0, extensive vacuolation (many vacuoles distributed in half a field); 4.0, severe vacuolation (numerous vacuoles often coalescing). 3F4 immunostaining was scored as follows in the same brain regions: 0, no deposition; 1.0, minimal deposition; 2.0, moderate deposition; 3.0, extensive deposition.
Data were analysed using Stata 9.0 (Stata Statistical Software: Release 9.0. College Station, TX). For each of the experimental groups, we compared vacuolation and immunohistochemistry characteristics in five brain regions using the non-parametric Mann–Whitney test. For each strain, we compared control versus sonicated and control versus PrP27–30. We also compared incubation times for inoculation in each of the three strains tested, using the Mann–Whitney test to compare control versus sonicated and control versus PrP27–30. We defined P<0.05 as statistically significant.
RESULTS
We chose to use the hamster scrapie isolates Sc237, 139H and DY as starting materials for this study because sPrPSc molecules have been most thoroughly characterized in hamster strains (Safar et al., 1998), and because these strains have been previously characterized pathologically and biochemically (DeArmond et al., 1993; Peretz et al., 2001). To prepare experimental inocula, we separately subjected brain homogenates from all three strains to either digestion with PK or indirect sonication. To generate PrP27–30 samples, we chose to treat homogenates with 10 μg PK ml−1 at 37 °C for 1 h. For the purposes of this study, we define rPrPSc molecules as PrPSc molecules that resist proteolysis with 10 μg PK ml−1 at 37 °C for 1 h, and sPrPSc molecules as those that are degraded by this treatment. This arbitrary definition is operational; at a molecular level, there may exist a spectrum of PrPSc isoforms that exhibit finer differences in their levels of protease resistance. N-terminal truncation of the PrP27–30 molecules was confirmed by Western blot (Fig. 1), and based on densitometry measurements of PK-resistant bands, samples were subsequently adjusted so that the PrP concentrations in all inocula were similar. The disaggregation of sonicated PrPSc molecules was confirmed by a sucrose-cushion centrifugation assay (Supplementary Fig. S1 available in JGV Online).
Fig. 1.

Western blot analysis of brain homogenate samples inoculated into Syrian hamsters. Three different scrapie strains were used to generate samples: DY, 139H and Sc237. Control, PrP27–30 and sonicated samples were generated as described in Methods.
Intracerebral inoculation of control and experimental samples into wild-type Syrian hamsters caused scrapie in all groups tested. For all three prion strains, the incubation periods produced by PrP27–30 and sonicated inocula did not differ statistically from those produced by control inocula (Table 1). As expected, hamsters inoculated with DY samples had incubation periods approximately twice as long as animals inoculated with 139H or Sc237 samples (Table 1).
Table 1. Scrapie transmission data.
n/no = No. of hamsters diagnosed with scrapie/no. of hamsters inoculated. Ref, Reference sample.
| Strain | Inoculum | n/no | IP★ | P value† |
|---|---|---|---|---|
| DY | Control | 6/7 | 259±59 | Ref |
| PrP27–30 | 8/8 | 246±34 | 0.897 | |
| Sonicated | 5/8 | 219±16 | 0.518 | |
| 139H | Control | 7/7 | 106±12 | Ref |
| PrP27–30 | 7/8 | 150±73‡ | 0.158 | |
| Sonicated | 7/7 | 110±14 | 0.700 | |
| Sc237 | Control | 8/8 | 106±26 | Ref |
| PrP27–30 | 7/8 | 110±21 | 0.559 | |
| Sonicated | 8/8 | 116±44 | 0.787 |
Mean incubation period (IP) of scrapie sick animals ± SD.
Mann–Whitney test comparing incubation period for each inoculum against controls within each strain.
The large SD in this group was attributable to two animals with incubation times of 214 and 288 days; excluding these animals, the incubation period of the remaining hamsters in this group was 110 ± 5 days.
The clinical signs displayed by hamsters inoculated with PrP27–30 or sonicated inocula were indistinguishable from the signs displayed by animals inoculated with the control inoculum for each strain. Specifically, all pre-terminal DY-inoculated animals displayed marked lethargy and diminished responsiveness to various stimuli, while all pre-terminal hamsters inoculated with Sc237 or 139H samples displayed ataxic gait, circling movements, trembling and hyper-responsiveness.
The strain-dependent regional distribution of neuropathological changes in scrapie-infected hamsters was measured by double blind scoring for vacuolation (Fig. 2a) and PrP immunohistochemistry (Fig. 2b). The results of these analyses show that the PrP27–30 and sonicated inocula produced patterns of vacuolation and PrP deposition that were very similar to the patterns produced by control inocula for all three strains (Fig. 2). We next performed a series of comparisons using the non-parametric Mann–Whitney test to determine whether any statistically significant differences could be identified between the different groups in individual brain regions. Among the 60 separate comparisons made, we found one statistically significant difference, between the cerebellar vacuolation of DY control compared with DY sonicated (P=0.04). The other nine comparisons between DY control and DY sonicated showed no statistically significant differences. Based on a significance level of P=0.05, we would expect to see three statistically significant differences by chance alone, and we conclude that the difference in cerebellar vacuolation in DY control versus DY sonicated was most probably due to chance variation.
Fig. 2.

Neuropathology of scrapie-infected hamsters. (a) Vacuolation profile scores of animals inoculated with samples derived from different prion strains, as indicated. (b) PrP immunohistochemistry profiles of animals inoculated with samples derived from different prion strains, as indicated. (a and b) Symbols: black squares, animals inoculated with control scrapie brain homogenate; open circles, animals inoculated with PrP27–30 samples; grey triangles, animals inoculated with sonicated samples. Brain regions: FC, frontal cortex; PC, parietal cortex; H, hippocampus; C, cerebellum; M, medulla. The mean values (n=5–8 animals per group) are shown ± SEM.
Next, we characterized the biochemical properties of PrPSc molecules present in the brains of scrapie-infected animals from each group. Western blotting revealed no differences in the glycoform distribution or electrophoretic mobility of PrPSc molecules produced by control and experimental inocula for all three strains (Fig. 3a). The relative electrophoretic mobility of PrPSc molecules was also examined with increased resolution by enzymic deglycosylation of samples with PNGase F prior to Western blot. The results confirmed that all DY-derived samples (control, PrP27–30 and sonicated) contained PrPSc molecules with a slightly increased mobility compared with PrPSc molecules from all Sc237- and 139H-derived samples (Fig. 3b). To measure the structural stability of PrPSc molecules in scrapie-infected animals from each group, we performed PrPSc protease resistance (Fig. 4a) and guanidine denaturation assays (Fig. 4b). For all three strains, no statistically significant deviations in the protease resistance or guanidine stability of PrPSc molecules were found between the control, PrP27–30 and the sonicated groups. All DY-derived samples displayed greater susceptibility to protease digestion (Fig. 4a) and guanidine denaturation (Fig. 4b) than all Sc237- and 139H-derived samples. The (Hopkins)1/2 values for the control samples were approximately 30, 300 and 200 μg ml−1 for DY, 139H and Sc237, respectively, confirming the relative susceptibility of the DY strain to proteolysis previously reported by other workers (Bessen & Marsh, 1994). The [GdnHCl]1/2 values for the control samples were approximately 1.9, 2.1 and 2.2 M for DY, 139H and Sc237, respectively. This rank order of stability to GdnHCl denaturation (DY<139H<Sc237) mirrored the GdnHCl stability rank order of the same strains previously determined by ELISA (Peretz et al., 2001).
Fig. 3.
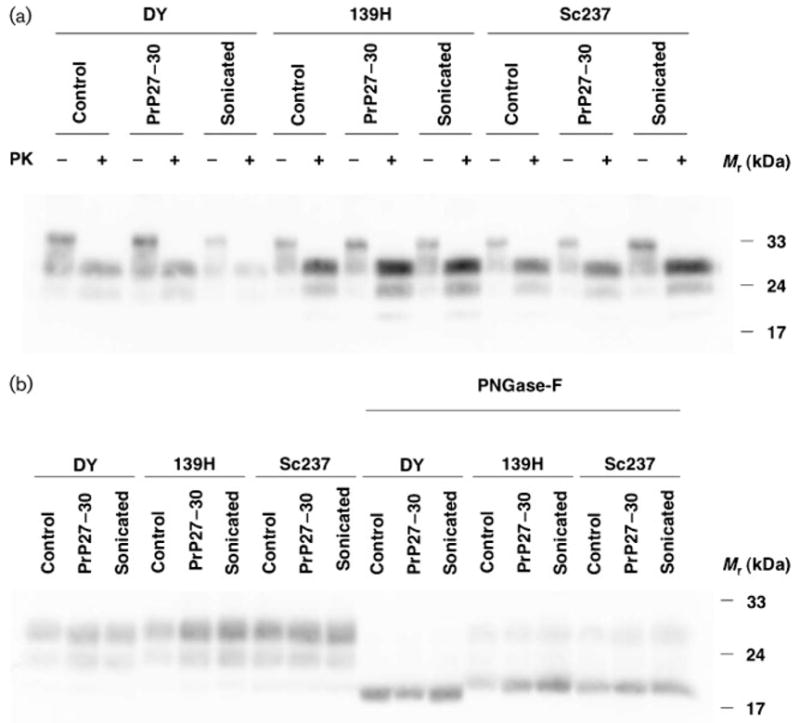
Biochemical analyses of scrapie-infected hamster brains. (a) Western blot analysis of brain homogenate samples prepared from animals inoculated with samples derived from different prion strains, as indicated. Twenty-five microlitres was loaded for each sample not treated with PK (−), and 50 μl was loaded for each sample subjected to digestion with 50 μg PK ml−1 (+). (b) Western blot analysis of brain homogenate samples prepared from animals inoculated with samples derived from different prion strains, as indicated. All samples were incubated with 10 μg PK ml−1 for 30 min at 37 °C. Samples on the right side of the blot were treated with the enzyme PNGase F, as indicated.
Fig. 4.

Biochemical analysis of PrPSc stability. (a) Protease resistance assay showing PrPSc levels in samples of brain homogenates taken from animals inoculated with samples derived from different prion strains, as indicated, and incubated with various concentrations of PK as described in Methods. (b) Guanidine denaturation assay showing PrPSc levels in samples of brain homogenates taken from animals inoculated with samples derived from different prion strains, as indicated, and incubated with various concentrations of guanidine hydrochloride, as described in Methods. (a and b) Symbols: black squares, animals inoculated with control scrapie brain homogenate; open circles, animals inoculated with PrP27–30 samples; grey triangles, animals inoculated with sonicated samples. The mean values of replicates (n=3) are shown for each point ± SEM.
DISCUSSION
Many advances in our understanding of prion disease pathogenesis have been gained over the past few decades, especially with regard to the central role of PrP conformational change. Furthermore, studies of fungal prions in model systems have confirmed the basic principle that self-propagating protein structure can encode heritable and infectious phenotypes (reviewed by Wickner et al., 2007). However, the intriguing question as to how specific PrPSc conformers structurally encode strain-specific mammalian prion disease phenotypes currently remains unanswered. The experiments described in this manuscript allow us to make several specific conclusions about the physical basis of scrapie strain variation.
sPrPSc molecules are not required to maintain strain properties
Using a CDI, Safar et al. (1998) found that native prions are composed of a mixture of sPrPSc and rPrPSc molecules. Furthermore, CDI analysis revealed that different strains of native hamster scrapie prions contain different ratios of sPrPSc : rPrPSc molecules (Safar et al., 1998). However, the relative contributions of sPrPSc and rPrPSc molecules to prion infectivity and strain specificity have remained unknown.
Our results show that proteolytic removal of sPrPSc molecules does not alter the clinical, pathological or biochemical characteristics of three different strains of hamster prions. In addition, physical disruption of PrPSc aggregates by sonication also did not affect prion strain properties. Taken together, these results indicate that rPrPSc molecules are sufficient to encode all the known strain-specific characteristics of mammalian prions. Thus, variations in sPrPSc : rPrPSc ratio between different prion strains appear to be a by-product rather than a determinant of strain properties.
It is worth noting that our experiments necessarily defined rPrPSc and sPrPSc molecules operationally as infectious prions that are either resistant or sensitive to treatment with 10 μg PK ml−1 for 1 h, respectively. It is certainly possible that PrPSc molecules with intermediate levels of protease resistance exist, and additional studies would be needed to study whether such forms might be necessary to maintain prion strain properties. We also advise that our studies do not provide any confirmation or information about the existence, infectious properties or strain-encoding capabilities of sPrPSc molecules. Recent developments in isolating autocatalytic sPrPSc molecules should facilitate characterization of this conformer (Pastrana et al., 2006). Studies in prion-infected Tg(MoPrP P101L) mice have shown that high titres of prion infectivity can be generated in the absence of either sPrPSc or rPrPSc conformers (Barron et al., 2001, 2007). Interestingly, it was observed that strain-dependent incubation times and neuronal targeting were significantly altered in Tg(MoPrP P101L) mice lacking PrPSc molecules compared with wild-type mice containing PrPSc molecules (Barron et al., 2001), suggesting that PrPSc conformers may be required to maintain strain-specific information. Alternatively, the loss of strain information upon passage in Tg(MoPrP P101L) mice may be attributable to mutation in the primary sequence of PrP, and additional studies will be required to distinguish between these two possibilities.
The N-terminal domain of PrPSc molecules is not required to maintain strain properties
Digestion of rPrPSc molecules with PK forms truncated PrP27–30 molecules in which approximately 70 N-terminal residues are removed (Prusiner et al., 1984). In full-length PrPC, this unstructured, polybasic region contains multiple octapeptide domains, and binds both copper ions and polyanionic molecules (Burns et al., 2003; Jackson et al., 2001; Jones et al., 2005; Viles et al., 1999; Warner et al., 2002). In addition, the extreme N-terminal residues 23–27 (KKRPK) are required for dominant-negative inhibition of PrPSc formation by mutant PrPC molecules in neuroblastoma cells (Zulianello et al., 2000), and for interaction with LDLR1 during endocytosis of PrPC (Taylor & Hooper, 2007). Samples containing non-denatured PrP27–30 molecules are known to be infectious (Prusiner et al., 1984), but it was previously unknown whether these truncated molecules can maintain prion strain properties. Our results show, for the first time, that the protease-resistant core of rPrPSc (i.e. PrP27–30) molecules is sufficient to encipher strain properties, including native patterns of neurotropism. Thus, we can conclude that the accessible approximately 70 N-terminal residues of PrP do not participate in coding hamster scrapie strain properties.
Disaggregation of PrPSc molecules by sonication does not alter prion strain properties
Finally, our results also showed that PrPSc disaggregation by sonication did not alter the infectious strain properties of the three hamster prion strains tested, despite disrupting PrPSc aggregates. Thus, we can conclude that strain variation is likely not to be encoded by differences in the quaternary state of PrPSc aggregates. These results are consistent with previous work showing that the strain properties of hamster 263K prions are not altered by protein misfolding cyclic amplification, a prion amplification technique that employs intermittent sonication (Castilla et al., 2005).
Conclusion
Using animal bioassays, we have formally shown that neither protease digestion nor sonication of three different hamster scrapie isolates alters the infectious strain properties of the treated prions. These results indicate that PrPSc aggregation, sPrPSc molecules and the N terminus of native PrPSc molecules are all unnecessary for the maintenance of prion strain properties in vivo. We conclude that prion strain variation is most likely to be encoded by PrP27–30 molecules, either alone or in conjunction with accessory non-proteinaceous molecules.
Acknowledgments
We thank Todd Bissonnette and David McGuire for helping to provide expert veterinary care. The National Institutes of Health provided financial support for this study.
Footnotes
A supplementary figure is available with the online version of this paper.
References
- Barron RM, Thomson V, Jamieson E, Melton DW, Ironside J, Will R, Manson JC. Changing a single amino acid in the N-terminus of murine PrP alters TSE incubation time across three species barriers. EMBO J. 2001;20:5070–5078. doi: 10.1093/emboj/20.18.5070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barron RM, Campbell SL, King D, Bellon A, Chapman KE, Williamson RA, Manson JC. High titers of transmissible spongiform encephalopathy infectivity associated with extremely low levels of PrPScin vivo. J Biol Chem. 2007;282:35878–35886. doi: 10.1074/jbc.M704329200. [DOI] [PubMed] [Google Scholar]
- Bessen RA, Marsh RF. Biochemical and physical properties of the prion protein from two strains of the transmissible mink encephalopathy agent. J Virol. 1992;66:2096–2101. doi: 10.1128/jvi.66.4.2096-2101.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bessen RA, Marsh RF. Distinct PrP properties suggest the molecular basis of strain variation in transmissible mink encephalopathy. J Virol. 1994;68:7859–7868. doi: 10.1128/jvi.68.12.7859-7868.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown P, Gajdusek DC. Survival of scrapie virus after 3 years’ interment. Lancet. 1991;337:269–270. doi: 10.1016/0140-6736(91)90873-n. [DOI] [PubMed] [Google Scholar]
- Bruce ME. Scrapie strain variation and mutation. Br Med Bull. 1993;49:822–838. doi: 10.1093/oxfordjournals.bmb.a072649. [DOI] [PubMed] [Google Scholar]
- Burns CS, Aronoff-Spencer E, Legname G, Prusiner SB, Antholine WE, Gerfen GJ, Peisach J, Millhauser GL. Copper coordination in the full-length, recombinant prion protein. Biochemistry. 2003;42:6794–6803. doi: 10.1021/bi027138+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson GA. Prion strains. Curr Top Microbiol Immunol. 1996;207:35–47. doi: 10.1007/978-3-642-60983-1_4. [DOI] [PubMed] [Google Scholar]
- Castilla J, Saa P, Hetz C, Soto C. In vitro generation of infectious scrapie prions. Cell. 2005;121:195–206. doi: 10.1016/j.cell.2005.02.011. [DOI] [PubMed] [Google Scholar]
- Collinge J, Sidle KC, Meads J, Ironside J, Hill AF. Molecular analysis of prion strain variation and the aetiology of ‘new variant’ CJD. Nature. 1996;383:685–690. doi: 10.1038/383685a0. [DOI] [PubMed] [Google Scholar]
- DeArmond SJ, Yang SL, Lee A, Bowler R, Taraboulos A, Groth D, Prusiner SB. Three scrapie prion isolates exhibit different accumulation patterns of the prion protein scrapie isoform. Proc Natl Acad Sci U S A. 1993;90:6449–6453. doi: 10.1073/pnas.90.14.6449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deleault NR, Harris BT, Rees JR, Supattapone S. Formation of native prions from minimal components in vitro. Proc Natl Acad Sci U S A. 2007;104:9741–9746. doi: 10.1073/pnas.0702662104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickinson AG, Fraser H. Modification of the pathogenesis of scrapie in mice by treatment of the agent. Nature. 1969;222:892–893. doi: 10.1038/222892a0. [DOI] [PubMed] [Google Scholar]
- Endo T, Groth D, Prusiner SB, Kobata A. Diversity of oligosaccharide structures linked to asparagines of the scrapie prion protein. Biochemistry. 1989;28:8380–8388. doi: 10.1021/bi00447a017. [DOI] [PubMed] [Google Scholar]
- Fraser H, Dickinson AG. Distribution of experimentally induced scrapie lesions in the brain. Nature. 1967;216:1310–1311. doi: 10.1038/2161310a0. [DOI] [PubMed] [Google Scholar]
- Gabizon R, McKinley MP, Groth DF, Kenaga L, Prusiner SB. Properties of scrapie prion protein liposomes. J Biol Chem. 1988;263:4950–4955. [PubMed] [Google Scholar]
- Glatzel M, Stoeck K, Seeger H, Luhrs T, Aguzzi A. Human prion diseases: molecular and clinical aspects. Arch Neurol. 2005;62:545–552. doi: 10.1001/archneur.62.4.545. [DOI] [PubMed] [Google Scholar]
- Holada K, Simak J, Vostal JG. Transmission of BSE by blood transfusion. Lancet. 2000;356:1772. doi: 10.1016/S0140-6736(05)71968-1. [DOI] [PubMed] [Google Scholar]
- Hopkins TR. Physical and chemical cell disruption for the recovery of intracellular proteins. Bioprocess Technol. 1991;12:57–83. [PubMed] [Google Scholar]
- Jackson GS, Murray I, Hosszu LL, Gibbs N, Waltho JP, Clarke AR, Collinge J. Location and properties of metal-binding sites on the human prion protein. Proc Natl Acad Sci U S A. 2001;98:8531–8535. doi: 10.1073/pnas.151038498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones CE, Klewpatinond M, Abdelraheim SR, Brown DR, Viles JH. Probing copper2+ binding to the prion protein using diamagnetic nickel2+ and 1H NMR: the unstructured N terminus facilitates the coordination of six copper2+ ions at physiological concentrations. J Mol Biol. 2005;346:1393–1407. doi: 10.1016/j.jmb.2004.12.043. [DOI] [PubMed] [Google Scholar]
- Ligios C, Sigurdson CJ, Santucciu C, Carcassola G, Manco G, Basagni M, Maestrale C, Cancedda MG, Madau L, Aguzzi A. PrPSc in mammary glands of sheep affected by scrapie and mastitis. Nat Med. 2005;11:1137–1138. doi: 10.1038/nm1105-1137. [DOI] [PubMed] [Google Scholar]
- Locht C, Chesebro B, Race R, Keith JM. Molecular cloning and complete sequence of prion protein cDNA from mouse brain infected with the scrapie agent. Proc Natl Acad Sci U S A. 1986;83:6372–6376. doi: 10.1073/pnas.83.17.6372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathiason CK, Powers JG, Dahmes SJ, Osborn DA, Miller KV, Warren RJ, Mason GL, Hays SA, Hayes-Klug J, et al. Infectious prions in the saliva and blood of deer with chronic wasting disease. Science. 2006;314:133–136. doi: 10.1126/science.1132661. [DOI] [PubMed] [Google Scholar]
- Pastrana MA, Sajnani G, Onisko B, Castilla J, Morales R, Soto C, Requena JR. Isolation and characterization of a proteinase K-sensitive PrPSc fraction. Biochemistry. 2006;45:15710–15717. doi: 10.1021/bi0615442. [DOI] [PubMed] [Google Scholar]
- Peretz D, Scott MR, Groth D, Williamson RA, Burton DR, Cohen FE, Prusiner SB. Strain-specified relative conformational stability of the scrapie prion protein. Protein Sci. 2001;10:854–863. doi: 10.1110/ps.39201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peretz D, Supattapone S, Giles K, Vergara J, Freyman Y, Lessard P, Safar JG, Glidden DV, McCulloch C, et al. Inactivation of prions by acidic sodium dodecyl sulfate. J Virol. 2006;80:322–331. doi: 10.1128/JVI.80.1.322-331.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science. 1982;216:136–144. doi: 10.1126/science.6801762. [DOI] [PubMed] [Google Scholar]
- Prusiner SB, McKinley MP, Bowman KA, Bolton DC, Bendheim PE, Groth DF, Glenner GG. Scrapie prions aggregate to form amyloid-like birefringent rods. Cell. 1983;35:349–358. doi: 10.1016/0092-8674(83)90168-x. [DOI] [PubMed] [Google Scholar]
- Prusiner SB, Groth DF, Bolton DC, Kent SB, Hood LE. Purification and structural studies of a major scrapie prion protein. Cell. 1984;38:127–134. doi: 10.1016/0092-8674(84)90533-6. [DOI] [PubMed] [Google Scholar]
- Safar J, Wille H, Itri V, Groth D, Serban H, Torchia M, Cohen FE, Prusiner SB. Eight prion strains have PrPSc molecules with different conformations. Nat Med. 1998;4:1157–1165. doi: 10.1038/2654. [DOI] [PubMed] [Google Scholar]
- Seeger H, Heikenwalder M, Zeller N, Kranich J, Schwarz P, Gaspert A, Seifert B, Miele G, Aguzzi A. Coincident scrapie infection and nephritis lead to urinary prion excretion. Science. 2005;310:324–326. doi: 10.1126/science.1118829. [DOI] [PubMed] [Google Scholar]
- Somerville RA, Carp RI. Altered scrapie infectivity estimates by titration and incubation period in the presence of detergents. J Gen Virol. 1983;64:2045–2050. doi: 10.1099/0022-1317-64-9-2045. [DOI] [PubMed] [Google Scholar]
- Stahl N, Borchelt DR, Hsiao K, Prusiner SB. Scrapie prion protein contains a phosphatidylinositol glycolipid. Cell. 1987;51:229–240. doi: 10.1016/0092-8674(87)90150-4. [DOI] [PubMed] [Google Scholar]
- Supattapone S, Muramoto T, Legname G, Mehlhorn I, Cohen FE, DeArmond SJ, Prusiner SB, Scott MR. Identification of two prion protein regions that modify scrapie incubation time. J Virol. 2001;75:1408–1413. doi: 10.1128/JVI.75.3.1408-1413.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor DM, Fernie K. Exposure to autoclaving or sodium hydroxide extends the dose-response curve of the 263K strain of scrapie agent in hamsters. J Gen Virol. 1996;77:811–813. doi: 10.1099/0022-1317-77-4-811. [DOI] [PubMed] [Google Scholar]
- Taylor DR, Hooper NM. The low-density lipoprotein receptor-related protein 1 (LRP1) mediates the endocytosis of the cellular prion protein. Biochem J. 2007;402:17–23. doi: 10.1042/BJ20061736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telling GC, Parchi P, DeArmond SJ, Cortelli P, Montagna P, Gabizon R, Mastrianni J, Lugaresi E, Gambetti P, Prusiner SB. Evidence for the conformation of the pathologic isoform of the prion protein enciphering and propagating prion diversity. Science. 1996;274:2079–2082. doi: 10.1126/science.274.5295.2079. [DOI] [PubMed] [Google Scholar]
- Turk E, Teplow DB, Hood LE, Prusiner SB. Purification and properties of the cellular and scrapie hamster prion proteins. Eur J Biochem. 1988;176:21–30. doi: 10.1111/j.1432-1033.1988.tb14246.x. [DOI] [PubMed] [Google Scholar]
- Viles JH, Cohen FE, Prusiner SB, Goodin DB, Wright PE, Dyson HJ. Copper binding to the prion protein: structural implications of four identical cooperative binding sites. Proc Natl Acad Sci U S A. 1999;96:2042–2047. doi: 10.1073/pnas.96.5.2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warner RG, Hundt C, Weiss S, Turnbull JE. Identification of the heparan sulfate binding sites in the cellular prion protein. J Biol Chem. 2002;277:18421–18430. doi: 10.1074/jbc.M110406200. [DOI] [PubMed] [Google Scholar]
- Wickner RB, Edskes HK, Shewmaker F, Nakayashiki T. Prions of fungi: inherited structures and biological roles. Nat Rev Microbiol. 2007;5:611–618. doi: 10.1038/nrmicro1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zulianello L, Kaneko K, Scott M, Erpel S, Han D, Cohen FE, Prusiner SB. Dominant-negative inhibition of prion formation diminished by deletion mutagenesis of the prion protein. J Virol. 2000;74:4351–4360. doi: 10.1128/jvi.74.9.4351-4360.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]