

Abstract
Sulfonyl chlorides substituted with functional groups having high proton affinity can serve as derivatization reagents to enhance the sensitivity for steroidal estrogens in liquid chromatography electrospray ionization tandem mass spectrometry (LC-ESI-MS/MS). The most commonly used reagent for derivatization of estrogens for LC-ESI-MS/MS is dansyl chloride. In this study, we compared dansyl chloride, 1,2-dimethylimidazole-4-sulfonyl (DMIS) chloride, pyridine-3-sulfonyl (PS) chloride, and 4-(1H-pyrazol-1-yl)benzenesulfonyl (PBS) chloride for derivatization of 17β-estradiol (E2) prior to LC-ESI-MS/MS. The product ion spectra of the dansyl and DMIS derivatives were dominated by ions representing derivatization reagent moieties. In contrast, the product ion spectrum of the PS derivative of E2 and, to a lesser extent, the PBS derivative, showed analyte-specific fragment ions. Derivatization with PS chloride was therefore chosen for further investigation. The product ion spectrum of the PS derivative of E2 showed intense ions at m/z 272, assigned to the radical E2 cation, and at m/z 350, attributed to the loss of SO2 from the [M+H]+ ion. Third-stage mass spectrometry of the PS derivative of E2 with isolation and collisional activation of the m/z 272 ion resulted in steroid C and D ring cleavages analogous to those observed in electron ionization mass spectrometry. The product ion spectra of the PS derivatives of estrone, 17α-ethinylestradiol, equilin, and equilenin showed similar estrogen-specific ions. Using derivatization with PS chloride, we developed an LC-ESI-MS/MS method with multiple reaction monitoring of primary and confirmatory precursor-to-product ion transitions for the determination of E2 in serum.
Keywords: liquid chromatography electrospray ionization tandem mass spectrometry, chemical derivatization of estrogens, pyridine sulfonyl derivatives, serum estradiol
Introduction
The accurate and precise measurement of 17β-estradiol (E2)1 in human serum is important in evaluating ovarian function, fertility, menopausal status, and cancer risk [1,2]. The major analytical challenge is to measure the very low levels of E2 that are found in physiological samples. The E2 levels in serum of cycling women only reach about 300 pg/mL during the peak of menstrual cycle, and are considerably lower in serum of men, children and postmenopausal women [1]. Various immunoassays are used to detect E2 in human urine and serum, but the accuracy, precision, and reproducibility of them are often of concern [3–5]. The values measured by immunoassays are in some cases significantly higher than the values measured in parallel assays by mass spectrometry (MS), and the differences have been attributed to antibody cross-reactivity and various interferences [6,7].
Numerous methods employing gas chromatography-MS and tandem mass spectrometry (MS/MS) have been developed for steroid analysis [7–10], although in recent years increasing numbers of analysts are opting for the use of liquid chromatography (LC)-MS and LC-MS/MS techniques to determine these compounds. A major obstacle in steroid analysis by LC-MS and LC-MS/MS had been that many steroids are weakly ionizable in the electrospray ionization (ESI) and atmospheric pressure chemical ionization techniques of LC-MS and LC-MS/MS, leading to low inherent sensitivity. Over the years chemical derivatization techniques have been developed to enhance the ionization of various analytes in LC-MS [11–14]. The deficiency of poor ionization of steroids in LC-MS has now been largely overcome; a number of derivatization procedures that involve either the introduction of a group carrying a formal charge [15–18] or the attachment of a group with high proton affinity [14,19–22] are now used to enhance ionization efficiency for positive ion detection in steroid analysis by LC-MS and LC-MS/MS.
For the analysis of steroidal estrogens by LC-ESI-MS/MS, pre-column derivatization by reaction with dansyl chloride at the C-3 hydroxyl groups of these compounds is by far the most popular technique, and is now widely used in both biomedical [14,23–29] and environmental [30–32] applications. There are, however, aspects of the analysis of estrogens as dansyl derivatives that are not desirable. With dansyl derivatization and LC-ESI-MS/MS of steroidal estrogens, the vast majority of the ion current after collision-induced dissociation (CID) of the [M+H]+ ion is carried by a single ion derived from the protonated 5-(dimethylamino)-naphthyl moiety [14,23–26], and very little of it by estrogen-derived ions, thereby making MS/MS and third stage MS (MS3) analyses structurally uninformative. The identification of unknown estrogens and their metabolites is therefore not significantly aided by the formation and analysis of dansyl derivatives. For quantitative LC-ESI-MS/MS determinations of the dansyl derivative of a typical steroidal estrogen, the analyst is usually confined to recording the intensity of a single MS/MS transition that does not involve the measurement of an estrogen-specific product ion. Since no confirmatory MS/MS transitions are readily available, interferences from matrix components may be problematic in the quantitative analysis of estrogens as dansyl derivatives [30].
In a recent study, we investigated the use of 1,2-dimethylimidazole-4-sulfonyl (DMIS) chloride as a derivatization reagent for the analysis of several phenolic compounds, including steroidal estrogens. We found that the product ion spectra of most of the DMIS derivatives showed predominantly ions representing the DMIS moiety; however, for the DMIS derivatives of some of the compounds, such as 3-phenanthrol, the product ion spectra contained peaks that we assigned to radical ions derived from the parent phenolic compounds [20]. These results prompted us to investigate additional sulfonyl chlorides as potential derivatization reagents, with the goal of identifying ones that produce derivatives that give rise to analyte-specific fragment ions in MS/MS. In this study, we compared the properties of several sulfonyl chlorides substituted with functional groups having high proton affinity as derivatization reagents for steroidal estrogens: dansyl chloride, DMIS chloride, 4-(1H-pyrazol-1-yl)benzenesulfonyl (PBS) chloride, and pyridine-3-sulfonyl (PS) chloride. While each of these reagents effectively enhanced the ionization efficiency for E2 analysis by LC-ESI-MS/MS, we found that the product ion spectra of the PS derivatives of steroidal estrogens showed abundant estrogen-specific ions. Derivatization with PS chloride was therefore chosen for further investigation with additional steroidal estrogens, and for development of a quantitative analytical method for E2.
Materials and methods
Materials
E2, 17α-ethinylestradiol (EE2), and dansyl chloride were purchased from Sigma-Aldrich (St. Louis, MO). Estrone (E1), equilin (EQ) and equilenin (EQN) were purchased from Steraloids (Wilton, NH). [2,4,16,16,17-2H5]17β-Estradiol (E2-d5) was synthesized from E1 as described [33], with the exception that the reduction at C-17 was performed with sodium borodeuteride. DMIS chloride was purchased from Oakwood Products (West Columbia, SC). PS chloride was obtained from Matrix Scientific (Columbia, SC). PBS chloride was purchased from Maybridge of Thermo Fisher Scientific (Morris Plains, NJ). HPLC-grade acetonitrile and methylene chloride were purchased from J.T. Baker (Phillipsburg, NJ). Analytical-grade acetone and methanol were purchased from Mallinckrodt Baker (Paris, KY). Charcoal-stripped fetal bovine serum (CS-FBS) was purchased from Hyclone (Logan, UT). The water used was purified using a MilliQ system (Millipore, Billerica, MA).
Derivatization
Aliquots of estrogen standards in methanol or serum extracts were evaporated to dryness in 3-mL reacti-vials (Pierce, Rockford, IL) under N2. Eighty microliters of sodium bicarbonate buffer (0.1 M, pH 10) were added to the vials, followed by 80 µL of one of the derivatization reagents, which were 1.0 mg/mL solutions in acetone of either dansyl chloride, DMIS chloride, PBS chloride, or PS chloride. The vials were vortexed, placed in a heater block preset at 60 °C, and allowed to react for 15 min for preparation of dansyl, DMIS, and PS derivatives, or for 30 min for preparation of PBS derivatives. The reaction mixtures were then cooled on ice for 10 min and were then transferred to 250-µL inserts in autosampler vials for LC-ESI-MS/MS analysis. The reaction of PS chloride with E2 is depicted in Scheme 1.
Scheme 1.
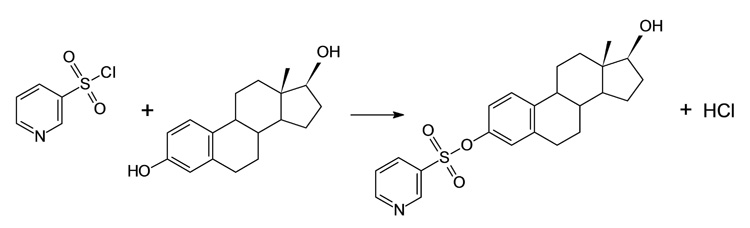
Derivatization of E2 by reaction with PS chloride.
Analysis by LC-ESI-MS, -MS/MS, and -MS3
The two analytical systems used in this study consisted of (1) an Applied Biosystems/MDS Sciex API 2000 triple quadrupole mass spectrometer operating with Analyst software, version 1.4.1, and interfaced with an Agilent (Santa Clara, CA) 1100 Series liquid chromatograph, and (2) an Applied Biosystems/MDS Sciex API 4000 Q TRAP mass spectrometer operating with Analyst software, version 1.4.2, and interfaced with an Agilent 1200 Series liquid chromatograph. Both systems were equipped with turbo ion spray sources. LC-ESI-MS with full scanning on quadrupole 1 (Q1) and LC-ESI-MS/MS with product ion scanning on Q3 were performed in the positive ion mode on system 1. LC-ESI-MS was performed over the range of m/z 200 to 500 with 1.5-s scans. In LC-ESI-MS/MS product ion scanning, [M+H]+ ions identified in previous LC-ESI-MS analyses were isolated by Q1, and were subjected to CID in Q2; Q3 was operated in the scanning mode with 1.5-s scans from m/z 70 to 50 m/z above the value of the [M+H]+ ion under analysis. Both Q1 and Q3 were operated with unit mass resolution. Nitrogen was used as the curtain gas (at setting 30), gas 1 (at setting 50), gas 2 (at setting 60), and the collision gas (at setting 9). The declustering potential, entrance potential, collision energy, and collision exit potential were 90, 7, 40, and 10 V, respectively. On both systems, the electrospray voltage was set at 5000 V and the turbo gas temperature was set at 500 °C.
LC-ESI-MS3 and LC-ESI-MS/MS with multiple reaction monitoring (MRM) were performed in the positive ion mode on system 2. In MS3 scan functions performed on the API 4000 Q TRAP, product ions produced in Q2 were isolated with Q3 functioning as a linear ion trap, and these ions were subjected to further CID and mass analysis. The final MS3 spectrum was obtained by a 1.5-s scan of Q3 over the range of m/z 50 to 300. For the analysis of E2 as its PS derivative by LC-ESI-MS/MS with MRM, the ion source and instrumental parameters were optimized for E2-PS, monitoring the m/z 414 → 272 MS/MS transition, and E2-d5-PS, monitoring the m/z 419 → 277 MS/MS transition, using direct infusion. The dwell times were 100 ms, and both Q1 and Q3 were operated with unit mass resolution. Nitrogen was used as the curtain gas (setting 30), gas 1 (setting 50), gas 2 (setting 60), and the collision gas (setting high). The declustering potential, entrance potential, collision energy, and collision exit potential were 91, 7, 35 and 15 V, respectively. In experimental protocols, the MS/MS transition of m/z 414 → 350 was also monitored, to confirm the presence of PS-E2. The electrospray voltage was set at 5000 V, and the turbo gas temperature was set at 500 °C.
High-performance liquid chromatography (HPLC) was performed using a Luna 3µ Phenyl-Hexyl column (2.0 × 150 mm, 3-µm particle size; Phenomenex, Torrance, CA). The injection volume was routinely 20 µL. Unless otherwise indicated, the mobile phase program consisted of an initial hold at 60% solvent A (0.1% formic acid in H2O)/40% solvent B (0.1% formic acid in 95/5 acetonitrile/H2O) for 1 min, followed by a linear gradient to 20% A/80% B over 18 min, a linear gradient returning to 60% A over 1 min, and a re-equilibration hold at 60% A/40% B for 8 min prior to the next sample injection. The flow rate was 0.2 mL/min, and the column was at ambient temperature.
Preparation of E2 standard solutions and calibration curves
A methanolic stock solution of E2 at 0.2 mg/mL was used to prepare working standards of 1, 10, 100, and 10,000 ng/mL by serial dilution into methanol. All standards were stored in amber vials at −80 °C. CS-FBS was used as the matrix to prepare the calibration standards. These standards were prepared by addition of the appropriate amount of working standards to 2-mL aliquots of CS-FBS, to give the final concentrations of 10, 25, 50, 100, 250, 500, and 1000 ng/L.
Analysis of E2 in serum as its PS derivative
To each sample, standard, and blank, a constant amount of internal standard, E2-d5 (1.8 ng), was added. These samples were extracted with 12 mL of methylene chloride for 30 min at room temperature with continuous rotation on a Barnstead/Thermolyne Labquake (Dubuque, IA) rotating shaker. The lower organic phases were then recovered and transferred to new glass tubes, the solvent was evaporated to about 2 mL, and the extracts were transferred into 3-mL reacti-vials. The organic solvent then was evaporated to dryness under N2, and the samples were derivatized with PS chloride and analyzed by LC-ESI-MS/MS with MRM as described above.
Lower Limit of Quantitation (LLOQ) and internal standard % recovery
The U.S. Food and Drug Administration LLOQ recommendations2 of (1) the analyte peak signal-to-noise ratio ≥ 5, (2) the relative standard deviation of <20% for replicate analyses, and (3) the accuracy of the analysis within 20% of the theoretical value were evaluated for the determination of the LLOQ for the analysis E2 in serum as its PS derivative. Recovery of the E2-d5 internal standard was determined as the ratio of the peak areas recorded for the E2-d5 internal standard, monitoring the MS/MS transition of m/z 419 → 277, in samples in which it was spiked into 2-mL CS-FBS aliquots prior to extraction with methylene chloride to those recorded when the internal standards were spiked into the methylene chloride phases that were recovered after extraction of 2-mL CS-FBS aliquots.
Method validation
For inter-day validation of the method for E2 analysis in serum, aliquots (2 mL) of CS-FBS were spiked with appropriate amounts of working standards of E2, in quadruplicate, to achieve concentrations of 50 ng/L (low level), 250 ng/L (medium level), and 800 ng/L (high level). To each of these samples, 1.8 ng of E2-d5 was added as the internal standard. The samples were extracted and derivatized as described above prior to analysis by LC-ESI-MS/MS on the API 4000 Q TRAP using MRM. Three validation batches were prepared, to assess the accuracy and precision this method, and each batch was analyzed on a different day. Each batch included a blank, a set of calibration standards with duplicates at each E2 concentration, and four replicates of samples spiked at the low, medium, and high levels.
Results and discussion
LC-ESI-MS/MS analysis of four aryl-sulfonyl derivatives of E2
The reconstructed ion chromatograms from analysis of the dansyl, PBS, PB, and DMIS derivatives of E2 by LC-ESI-MS/MS, each using the same HPLC gradient program but recorded with product ion scan functions specific for each particular derivative, are shown in Fig. 1. The retention times increased for chromatography of the DMIS, PS, PBS, and dansyl derivatives, respectively, reflecting the relative hydrophobicities of the reagent moieties. The Q1 full-scan mass spectrum of each of the four derivatives of E2 showed an intense [M+H]+ ion (data not shown). Upon CID, the product ion spectra recorded for the four derivatives of E2 indicated fundamentally different types of fragmentation processes (Fig. 2). The product ion spectrum of the [M+H]+ ion of the DMIS derivative of E2 at m/z 431 (Fig. 2A) is dominated by the ions of m/z 159, m/z 144, and m/z 96, which are assigned respectively to DMIS, methylimidzaole sulfonyl, and dimethylimidazole ions, as discussed previously [20]. In contrast to the product ion spectrum of the DMIS derivative, in which the ion current was almost entirely carried by ions originating from the DMIS moiety, the product ion spectrum of the PS derivative of E2 (Fig. 2B) shows a series of intense ions that appear to have originated from the E2 moiety. Most notable of these is the prominent ion at m/z 272, which we have assigned to a radical E2 cation produced by the loss of the PS group as a neutral radical from the [M+H]+ ion, or [M+H−C5H4NSO2]+. (Scheme 2).
Fig. 1.
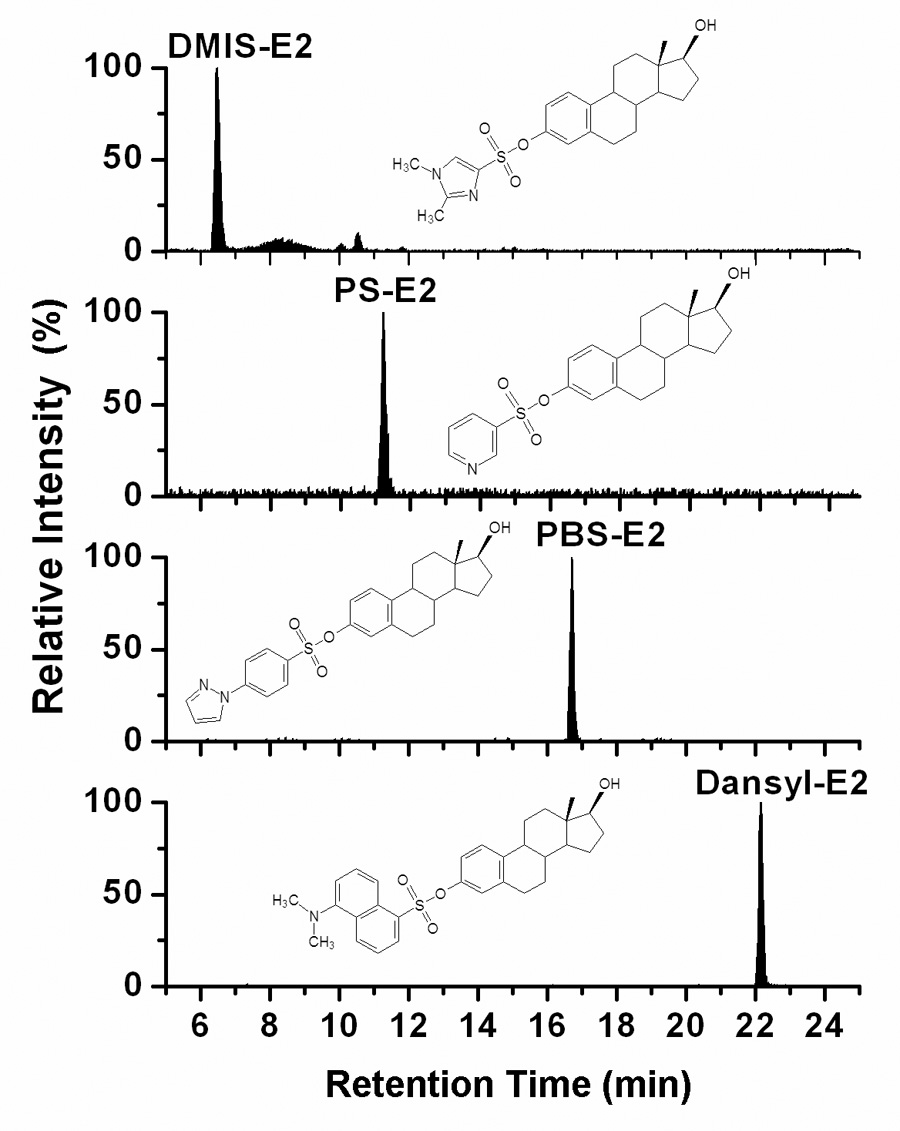
Analysis of the DMIS, PS, PBS, and dansyl derivatives of E2 by LC-ESI-MS/MS with product ion scanning. The HPLC mobile phase program for each analysis consisted of an initial hold at 50% solvent A (0.1% formic acid in H2O)/50% solvent B (0.1% formic acid in 95/5 acetonitrile/H2O) for 1 min, followed by a linear gradient to 10% A/90% B over 22 min, a linear gradient returning to 50% A over 1 min. The product ion scan functions were m/z 431 → scan m/z 50 to 550, m/z 414 → scan m/z 50 to 550, m/z 479 → scan m/z 50 to 550, and m/z 506 → scan m/z 50 to 550, for the detection of the DMIS, PS, and PBS and dansyl derivatives, respectively.
Fig. 2.
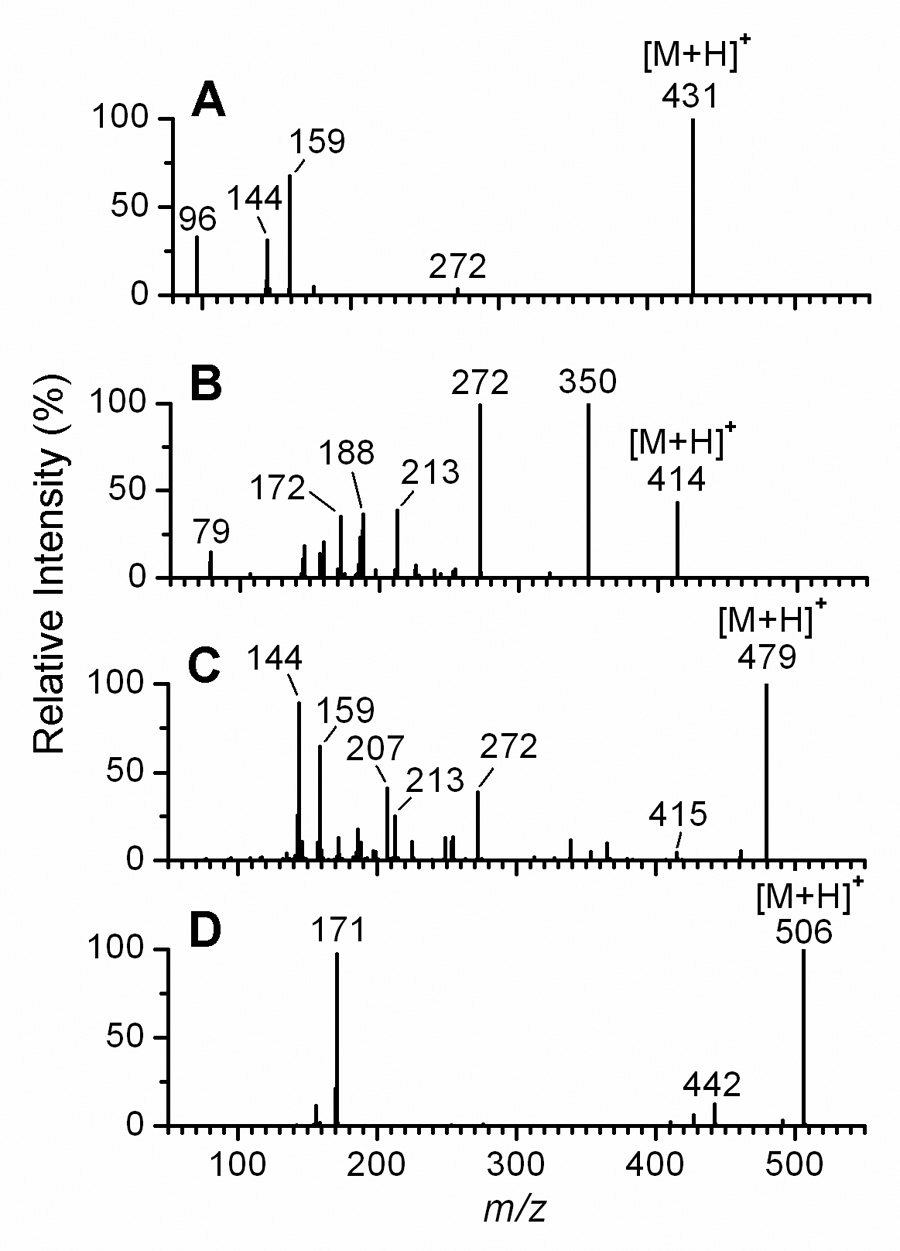
LC-ESI-MS/MS product ion spectra of the [M+H]+ ions of derivatized E2. Shown are product ion spectra of (A) DMIS-E2; (B) PS-E2; (C) PBS-E2; and (D) dansyl-E2 at 40 eV collision energy.
Scheme 2.
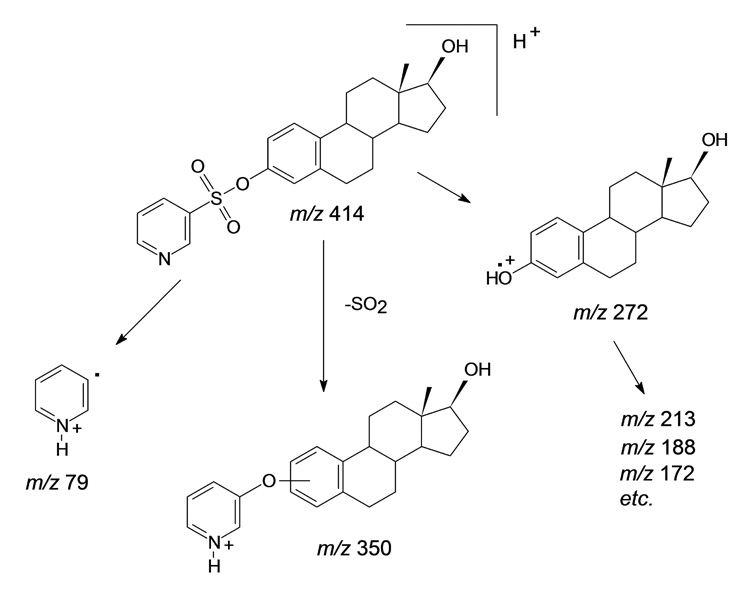
Proposed pathways for the fragmentation of the [M+H]+ ion of the PS derivative of E2 under CID.
Another prominent ion in the product ion spectrum of the [M+H]+ ion of the PS derivative of E2 is observed at m/z 350. We propose that this ion is formed by the loss of SO2 from the [M+H]+ ion, with rearrangement (Scheme 2); it can be designated as [M+H−SO2]+. Peaks representing the loss of 64 Da, attributable to the loss of SO2, are apparent in the product ion spectra of the dansyl derivatives of some phenolic compounds [34,35] and for some sulfonamides in the negative ion mode [36]. The relatively minor peak at m/z 79 is assigned to the radical pyridinium ion formed by homolytic cleavage of the bond between the protonated pyridine ring and the sulfonyl group. Other ions in the product ion spectrum, notably those at m/z 254, 213, 188, and 186, appear to arise from the E2 portion of the molecule as a result of ring cleavages of the steroid nucleus.
Like the product ion spectrum of the E2 PS derivative, the product ion spectrum of the [M+H]+ ion of the E2 PBS derivative at m/z 479 shows a peak at m/z 272, assigned to the radical E2 cation (Fig. 2C). However, the three more abundant ions at m/z 207, 159, and 144 appear to arise from the PBS moiety. We propose that the ion at m/z 207, assigned to [C9H7N2SO2]+, is a highly resonance-stabilized ion formed by heterolytic cleavage of the S-O bond between the PBS moiety and the estrogen A ring. The ion at m/z 159 is assigned to [C9H7NO]+ and appears to arise from cleavage of the S-O bond after rearrangement of the sulfonyl group, analogous to the fragmentation observed in the product ion spectra of benzenesulfonamides [37]. We propose that the ion at m/z 144 represents a radical pyrazolylbenzyl ion, [C9H8N2]+., arising from homolytic cleavage of the C-S bond between the protonated pyrazolylbenzene moiety and the sulfonyl group. The product ion spectrum of the dansyl derivative of E2 showed the dominant ion of m/z 171 (Fig. 2D), as has been previously observed and assigned to an ion representing the 5-(dimethylamino)naphthyl moiety, formed by homolytic cleavage of the C-S bond between the naphthalene and sulfonyl groups [14,23].
Based on our observation that, upon CID, the PS derivative of E2 produced the highest relative abundance of E2-specific fragment ions of the four derivatives – a property that could engender more selective and specific MS/MS methods – we chose PS chloride for further investigation as a derivatization reagent for the analysis of E2 and other steroidal estrogens.
LC-ES-MS3 of the PS derivative of E2
The ion at m/z 272 in the product ion spectrum of the PS derivative of E2 (Fig. 2B) appears to be structurally identical to the molecular ion, M+., that is produced in the mass spectrometric analysis of underivatived E2 with electron ionization (EI). While taking into account the caveat that the processes in EI and CID that lead to elevated internal energy of the ions are distinct, we nonetheless hypothesized that CID of the ion at m/z 272 that is produced in LC-ESI-MS/MS of the PS derivative of E2 may result in a spectrum that is analogous to the EI spectrum of underivatized E2. To investigate this possibility, we performed MS3 experiments in which the [M+H]+ ion of the PS derivative at m/z 414 was isolated and collisionally activated to produce the m/z 272 ion, which was in turn isolated and collisionally activated, with the resulting fragment ions then analyzed by scanning the mass spectrum over the range of m/z 50 to 300. This final mass spectrum from the MS3 analysis (Fig. 3) shows a high degree of similarity to the EI mass spectrum of E2 found in the National Institute of Standards and Technology database3. While they differ considerably in their relative intensities, all of the annotated peaks in Fig. 3 are also observed in the EI mass spectrum of E2, suggesting common fragmentation pathways in the two MS techniques.
Fig. 3.
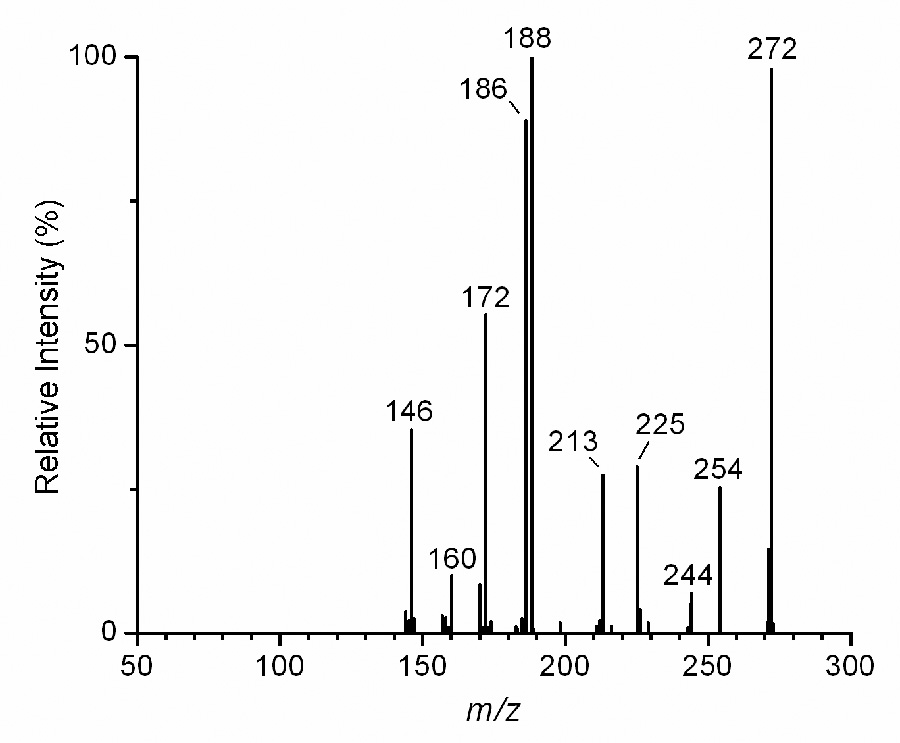
LC-ESI-MS3 analysis of the PS derivative of E2. The MS3 sequence, m/z 414 → 272 → scan m/z 50–300, was performed, producing the final mass spectrum shown.
Based on extensive studies of the EI mass spectra of underivzatized estrogens and estrogen analogs [38–41], several fragmentation pathways were proposed by these previous authors, leading to the structural assignments for prominent ions depicted in Scheme 3. The peak at m/z 254, one that is prominent in the MS3 spectrum of the PS derivative of E2 but is of very low intensity in the EI spectrum of E2, is most likely formed by the loss of H2O from the C-17 hydroxyl group, a process that may be more favorable in CID rather than in EI-induced fragmentation. The two prominent peaks at m/z 186 and 188 (Fig. 3) appear to represent cleavages of the steroid C and D rings. In analogy to the EI spectrum of E2, the formation of the ion at m/z 172 may be produced by the cleavage of the C and D rings with hydrogen migration, and the peak at m/z 146 may be produced by cleavage of the C ring, as has been proposed [38,39].
Scheme 3.
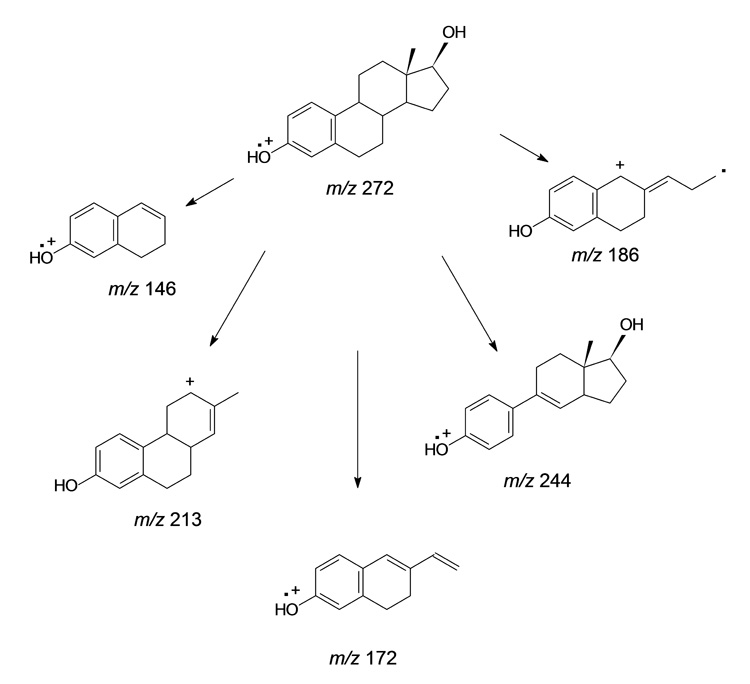
Analogous fragmentation of the M+. of E2 under EI conditions and the radical E2 cation in the MS3 spectrum of the PS deriviatve of E2. Structural assignments for the ions of m/z 146, 213, 172, 186, and 244 are those proposed in previous studies [38–41] based on the MS of steroidal estrogens under EI conditions. When the PS derivative of E2 was analyzed by LC-ESI-MS3 with the scan function m/z 414 → 272 → scan 50–300, ions of m/z 146, 213, 172, 186, and 244 were also produced (Fig. 3), suggesting that common fragmentation mechanisms are occurring in the two techniques.
LC-ESI-MS/MS of the PS derivatives of E1, EE2, EQ, and EQN
We next investigated the broader utility of PS derivatization by applying the technique to additional steroidal estrogens. The PS derivatives of E1, a major endogenous estrogen, EE2, a synthetic estrogen widely used in birth control formulations [42], and the equine-derived, B-ring unsaturated estrogens, EQ and EQN, which are present in pharmaceutical preparations used for estrogen replacement therapy [43], were prepared and analyzed by LC-ESI-MS/MS. The product ion spectrum of the PS derivative of E1 showed a fragmentation pattern analogous to that of the PS derivative of E2; the peak at m/z 270, assigned to the radical E1 cation, and the peak at m/z 348, assigned to the [M+H−SO2]+ ion, are the two most abundant in the spectrum (Fig. 4A). The radical pyridinium ion at m/z 79 is of relatively low abundance. The ions at m/z 146, 172, 185 and 213 all appear to arise from E1, and all are also prominent in the EI spectrum of E1 [41]. Their formation has been discussed in detail in earlier reports [38–41]. The product ion spectrum of the PS derivative of EE2 shows a significant peak at m/z 374, assigned to [M+H−SO2]+, while the radical EE2 cation at m/z 296 and the radical pyridinium ion at m/z 79 are of relatively low intensity (Fig. 4B). The base peak in the spectrum of the PS derivative of EE2 is at m/z 213, and is thought to arise from a D ring cleavage (Scheme 3) [38] as discussed above.
Fig. 4.
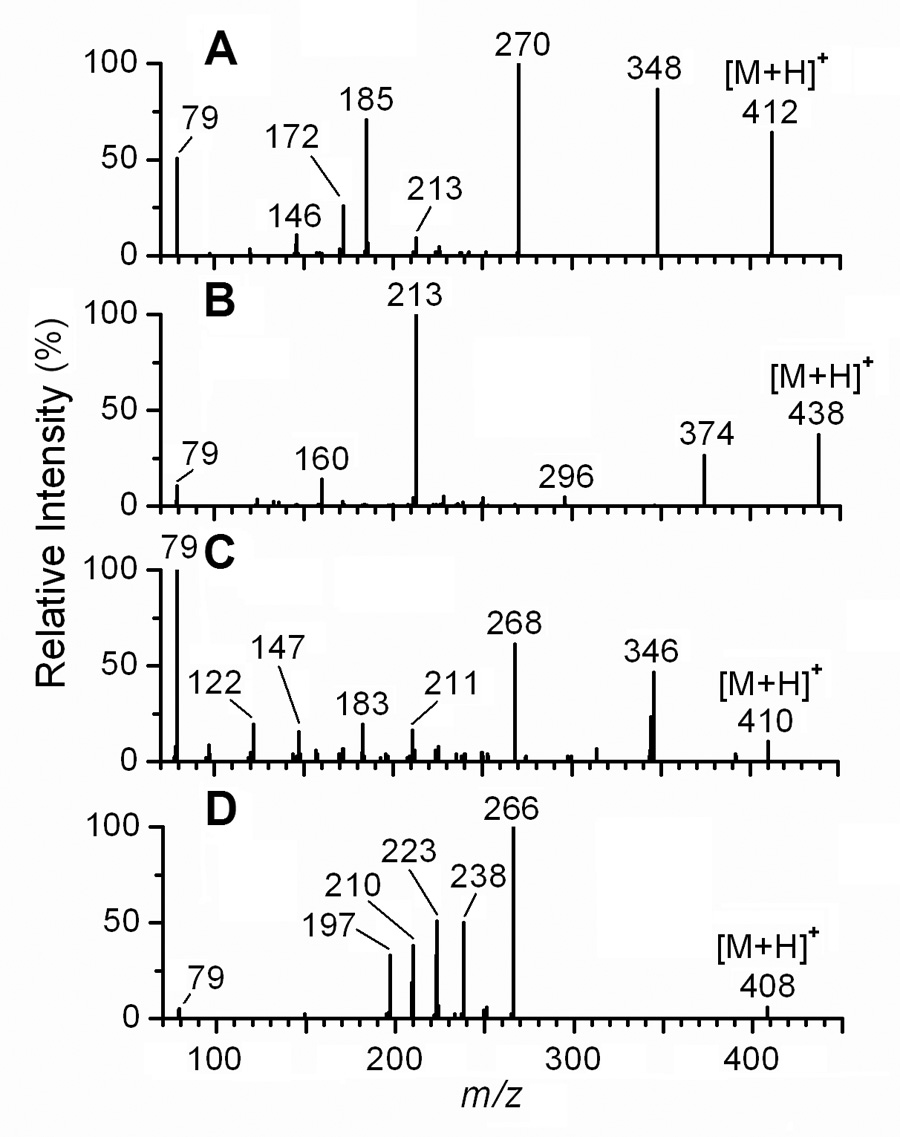
LC-ESI-MS/MS product ion spectra of the [M+H]+ ions of the PS derivatives of E1, EE2, EQ, and EQN. The [M+H]+ ions of the PS derivatives of (A) E1, (B) EE2, (C) EQ, and (D) EQN at m/z 412, 438, 408, and 410, respectively, were collisionally activated at 40 eV, and the resulting product ion spectra are shown.
Intense peaks representing the radical EQ cation at m/z 268 and the [M+H−SO2]+ ion at m/z 346 are observed in the product ion spectrum of the PS derivative of EQ; however, the base peak is at m/z 79, representing the radical pyridinium ion (Fig. 4C). Other characteristic fragment ions, including those at m/z 211, 183, 147, and 122, which appear to arise from the EQ moiety, are relatively minor. The ions at m/z 211 and 183 have also been observed and discussed in previous studies of EI mass spectrum of EQ [38]. The fragment ions at m/z 147 and 122 are likely formed by cleavage of C and B rings, respectively, followed by hydrogen rearrangement.
Unlike the spectra of the PS derivatives of the other estrogens investigated in this study, the product ion spectrum of the PS derivative of EQN did not show a peak representing the [M+H−SO2]+ ion, which would have been observed at m/z 344 (Fig. 4D). The fully aromatic A and B ring system may contribute to this phenomenon by stabilizing the radical EQN ion at m/z 266, leading to the preferential formation of this ion, as opposed to the loss of SO2 from the [M+H]+ ion. The ions at m/z 238, 223, 210, and 197 appear to arise from various C and D steroid ring cleavages of the radical EQN ion, and analogous ions have been observed in the EI mass spectrum of EQN-3-methyl ether [38]. We propose that the fragment ion at m/z 238 is formed by loss of CO at C-17. The fragment ion at m/z 223 may then be formed by the loss of a methyl radical from the m/z 238 ion, and the ion at m/z 210 by loss of carbons 15 and 16 as C2H4, also from the m/z 238 ion.
Analysis of E2 in serum as its PS derivative
For the quantitative analysis of E2 in serum, seven-point calibration curves in duplicate with increasing E2 concentrations over the range of 10–1000 pg/mL showed excellent linearity (r2 > 0.997), when responses for both the m/z 414 → 272 and m/z 414 → 350 MS/MS transitions relative to the responses to the E2-d5 internal standard transition of m/z 419 → 277 were plotted against increasing E2 concentration (Fig. 5). The ratio of the slope of the calibration curve for the m/z 414 → 272 transition to the slope of the m/z 414 → 350 curve is 2.11, indicating that this ratio of the peak areas of the two MS/MS transitions at the retention time of the PS derivative of E2 is confirmatory for the presence of E2, and this ratio should be observed in determinations of E2 in this analysis. The mean recovery of the E2-d5 internal standard determined for the procedure used to extract E2 from serum was 94.4% (n = 8).
Fig. 5.
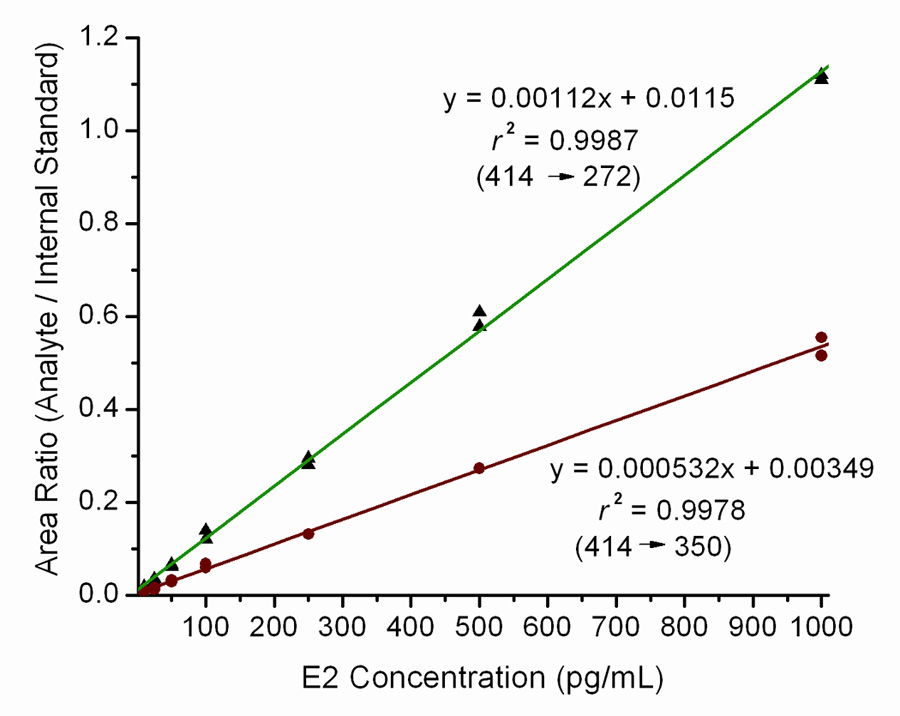
Calibration curves for the analysis of E2 in serum as its PS derivative. Duplicate standards of 10, 20, 50, 100, 250, 500 and 1000 pg/mL E2, each containing 1800 pg/mL E2-d5, in 2-mL aliquots of CS-FBS as the matrix were extracted, derivatized with PS chloride, and analyzed by LS-ESI-MS/MS with MRM of m/z 414 → 272 and m/z 414 → 350 for PS derivative of E2 and m/z 419 → 277 for the PS derivative of E2-d5, with a collision energy of 35 eV. The ratios of the responses for m/z 414 → 272 to those for m/z 419 → 277 and for m/z 414 → 350 to those for m/z 419 → 277 are plotted as functions of increasing E2 concentration.
The results of the inter-day validation assays at the spike levels of 50, 250, and 800 pg/mL E2 in CS-FBS are presented in Table 1. The accuracy of the method was evaluated by determination of the mean relative errors, and the precision by evaluation of the relative standard deviations. The accuracies for each of the three spike levels on each of the three days were within 9.0%, and the precision ranged from 1.9 to 8.6 %. For these determinations, the ratios of the peaks areas for the m/z 414 → 272 MS/MS transition to peak areas for the m/z 414 → 350 transition ranged from 1.96 to 2.09, with a mean value of 2.02 ± 0.04 (n = 12). Under our LC–ESI-MS/MS conditions for the analysis of the PS derivative of E2, the reconstructed ion chromatograms showed no significant interferences in the analysis of the CS-FBS blank for either the m/z 414 → 272 or the m/z 414 → 350 transition (Fig. 6). The signal-to-noise ratios for the analyte peaks in the 10 pg/mL E2 sample exceeded 5 for both MS/MS transitions. Analysis of a set of CS-FBS samples spiked with E2 at 10 pg/mL (n = 8) resulted in a relative standard deviation of 19.8%, a mean relative error of −8.1%, and a mean ratio of the peak areas for the m/z 414 → 272 to m/z 414 → 350 MS/MS transitions of 2.19 ± 0.20. These results indicate that a LLOQ of 10 pg/mL was achieved by this method, representing an instrumental sensitivity of about 2 pg (7 fmol) of E2 on column. The ultimate sensitivity of this technique will depend on the instrumentation used. There are a number of commercially available mass spectrometers employing ESI that significantly exceed the sensitivity of the systems used in this study.
Table 1.
Inter-day assay values, accuracy, and precision for the analysis of CS-FBS samples spiked with E2.
| Addition of |
||||
|---|---|---|---|---|
| 50 pg/mL E2 | 250 pg/mL E2 | 800 pg/mL E2 | ||
| Day 1 | Mean, pg/mL | 45.5 | 254.7 | 818.2 |
| (n = 4) | Relative error, % | −9.0 | 1.9 | 2.3 |
| Relative SD, % | 5.4 | 4.6 | 3.5 | |
| Day 2 | Mean, pg/mL | 46.8 | 256.2 | 781.5 |
| (n = 4) | Relative error, % | −6.4 | 2.5 | −2.3 |
| Relative SD, % | 7.9 | 8.6 | 4.4 | |
| Day 3 | Mean, pg/mL | 51.8 | 256.2 | 849.7 |
| (n = 4) | Relative error, % | 3.5 | 2.5 | 6.2 |
| Relative SD, % | 6.6 | 7.6 | 1.3 | |
| Inter-day | Mean, pg/mL | 48.0 | 255.8 | 816.5 |
| (n = 12) | Relative error, % | −4.0 | 2.3 | 2.1 |
| Relative SD, % | 8.4 | 6.5 | 4.6 | |
Fig. 6.
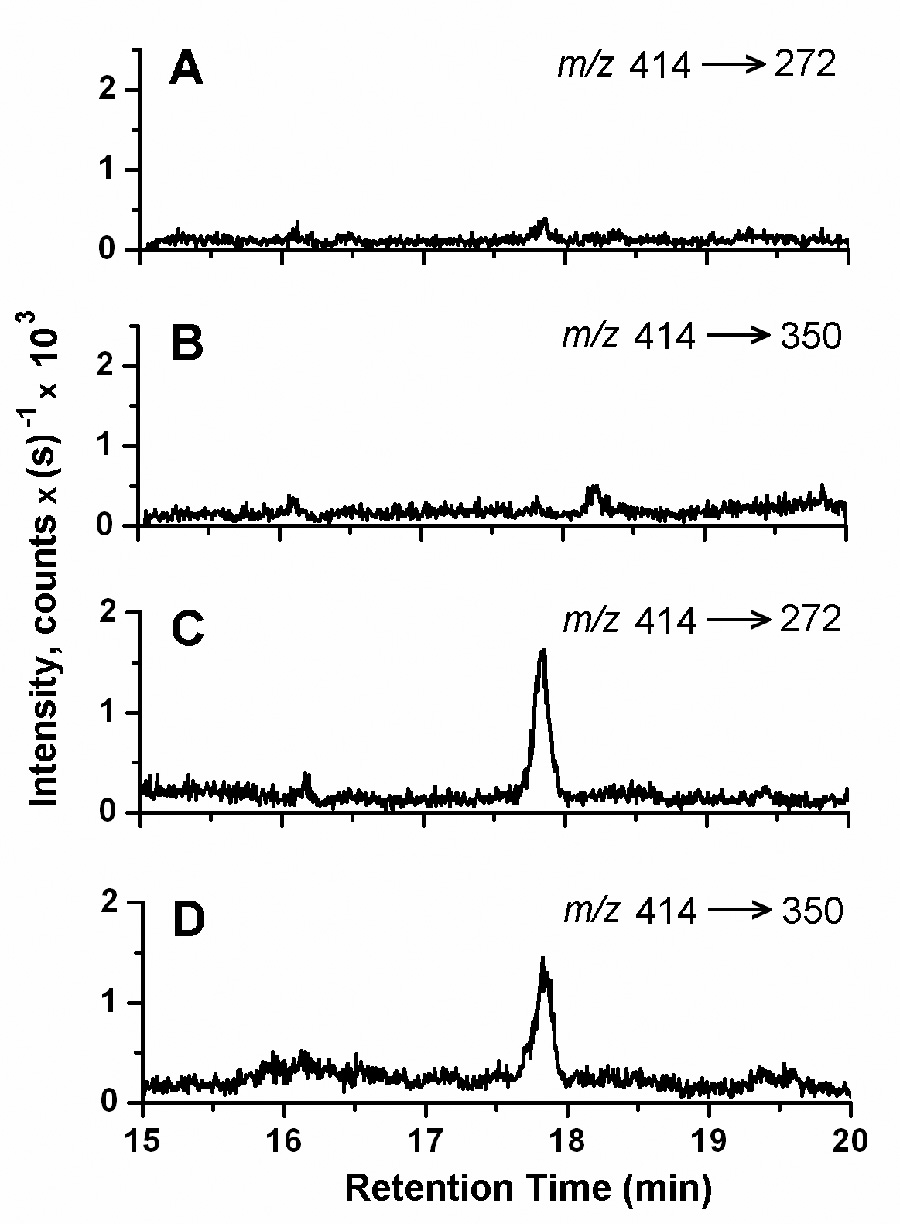
MRM analysis of the CS-FBS blank and for E2 in serum at the LLOQ. The reconstructed ion chromatograms for (A) m/z 414 → 272 and (B) m/z 414 → 350 from the analysis of a CS-FBS blank and for (C) m/z 414 → 272 and (D) m/z 414 → 350 of a CS-FBS sample spiked at 10 pg/mL are shown. The signal-to-noise ratios for the peaks representing the PS derivative of E2 at 17.8 min in (C) and (D) are 8.2 and 5.7, respectively.
Conclusions
In our comparison of dansyl, DMIS, PBS, and PS chlorides as derivatization reagents for the enhancement of the sensitivity for the analysis of estrogens in LC-ESI-MS/MS, we found that each effectively enhanced the ionization of steroidal estrogens under ESI conditions. These reagents also offer differing hydrophobicities of the resultant estrogen derivatives, allowing the analyst to opt for shorter retention times in reversed-phase HPLC by selecting one of the less hydrophobic derivatives. We chose derivatization with PS chloride for further development not only because of the chromatographic properties of the derivatives, but mainly because of the analyte-specific and highly informative MS/MS product ion spectra obtained from them, which are beneficial in both qualitative and quantitative analyses. We found that, by the use of derivatization with PS chloride and LC-ESI-MS3, “EI-like” mass spectra of estrogens can be recorded under ESI conditions. These mass spectra will undoubtedly be useful in structural identification and confirmation of estrogens and their metabolites, and perhaps in the analysis of other phenolic compounds. This will particularly be the case when the product ions of the PS derivatives are analyzed using MS instrumentation that is capable of high mass accuracy determinations. For quantitative determinations by MRM, the analysis of E2 as its PS derivative allows recording of primary and confirmatory MS/MS transitions that involve detection of analyte-specific fragment ions, which may lessen the probability of matrix interferences over recording the intensity of a single reagent-derived ion. The monitoring of two MS/MS transitions for E2 analysis offers an additional degree of analytical specificity, since the ratio of the responses of the two MS/MS transitions should be constant, and deviation from the expected ratio would be indicative of interference.
Acknowledgements
This research was supported by NIH grant CA81243. We thank Drs. Kenneth M. Aldous and Xinxin Ding of the Wadsworth Center for making instrumentation in their laboratories available for these studies.
Footnotes
Abbreviations used: CID, collision-induced dissociation; CS-FBS, charcoal-stripped fetal bovine serum; DMIS, 1,2-dimethylimidazole-4-sulfonyl; E1, estrone; E2, 17β-estradiol; E2-d5, [2,4,16,16,17-2H5]17β-estradiol; EE2, 17α-ethinylestradiol; EI, electron ionization; EQ, equilin; EQN, equilenin; ESI, electrospray ionization; HPLC, high-performance liquid chromatography; LC, liquid chromatography; LLOQ, lower limit of quantitation; MS, mass spectrometry; MS/MS tandem mass spectrometry; MS3, third-stage mass spectrometry; MRM, multiple reaction monitoring; PBS, 4-(1H-pyrazol-1-yl)benzenesulfonyl; PS, pyridine-3-sulfonyl; Q1, Q2, and Q3, quadrupoles 1, 2, and 3.
Available at: http://www.fda.gov/cder/guidance/4252fnl.htm
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Haymond S, Gronowski AM. Reproductive related disorders. In: Burtis CA, Ashwood ER, Bruns DE, editors. Tietz Textbook of Clinical Chemistry and Molecular Diagnostics. 4th ed. St. Louis: Elsevier Saunders; 2006. pp. 2097–2152. [Google Scholar]
- 2.Tamimi RM, Byrne C, Colditz GA, Hankinson SE. Endogenous hormone levels mammographic density, and subsequent risk of breast cancer in postmenopausal women. J. Natl. Cancer Inst. 2007;99:1178–1187. doi: 10.1093/jnci/djm062. [DOI] [PubMed] [Google Scholar]
- 3.Giese RW. Measurement of endogenous estrogens: analytical challenges and recent advances. J. Chromatogr. A. 2003;1000:401–412. doi: 10.1016/s0021-9673(03)00306-6. [DOI] [PubMed] [Google Scholar]
- 4.Hsing AW, Stanczyk FZ, Belanger A, Schroeder P, Chang L, Falk RT, Fears TR. Reproducibility of serum sex steroid assays in men by RIA and mass spectrometry Cancer Epidemiol. Biomarkers Prev. 2007;16:1004–1008. doi: 10.1158/1055-9965.EPI-06-0792. [DOI] [PubMed] [Google Scholar]
- 5.Cao Z, Swift TA, West CA, Rosano TG, Rej R. Immunoassay of estradiol: unanticipated suppression by unconjugated estriol. Clin. Chem. 2004;50:160–165. doi: 10.1373/clinchem.2003.023325. [DOI] [PubMed] [Google Scholar]
- 6.Lee JS, Ettinger B, Stanczyk FZ, Vittinghoff E, Hanes V, Cauley JA, Chandler W, Settlage J, Beattie MS, Folkerd E, Dowsett M, Grady D, Cummings SR. Comparison of methods to measure low serum estradiol levels in postmenopausal women. J. Clin. Endocrinol. Metab. 2006;91:3791–3797. doi: 10.1210/jc.2005-2378. [DOI] [PubMed] [Google Scholar]
- 7.Santen RJ, Demers L, Ohorodnik S, Settlage J, Langecker P, Blanchett D, Goss PE, Wang S. Superiority of gas chromatography/tandem mass spectrometry assay(GC/MS/MS) for estradiol for monitoring of aromatase inhibitor therapy. Steroids. 2007;72:666–671. doi: 10.1016/j.steroids.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 8.Biancotto G, Angeletti R, Traldi P, Silvestri M, Saccon M, Guidugli F. Determination of 17β-estradiol in bovine plasma: development of a highly sensitive technique by ion trap gas chromatography-tandem mass spectrometry using negative chemical ionization. J. Mass Spectrom. 2000;37:1266–1271. doi: 10.1002/jms.397. [DOI] [PubMed] [Google Scholar]
- 9.Biddle S, Teale P, Robinson A, Bowman J, Houghton E. Gas chromatography-mass spectrometry/mass spectrometry analysis to determine natural and post-administration levels of oestrogens in bovine serum and urine. Anal. Chim. Acta. 2007;586:115–121. doi: 10.1016/j.aca.2006.10.044. [DOI] [PubMed] [Google Scholar]
- 10.Wu H, Ramsay C, Ozaeta P, Liu L, Aboleneen H. Serum estradiol quantified by isotope dilution-gas chromatography/mass spectrometry. Clin. Chem. 2002;48:364–366. Erratum in: Clin.Chem. 48 (2002) 686. [PubMed] [Google Scholar]
- 11.Voyksner RD, Bush ED, Brent D. Derivatization to improve thermospray HPLC/MS sensitivity for the determination of prostaglandins and thromboxane B2. Biomed Environ. Mass Spectrom. 1987;14:523–531. doi: 10.1002/bms.1200140908. [DOI] [PubMed] [Google Scholar]
- 12.Quirke JME, Adams CL, Van Berkel GJ. Chemical derivatization for electrospray ionization mass spectrometry. 1. Alkyl halides, alcohols, phenols, thiols, and amines. Anal. Chem. 1994;66:1302–1315. [Google Scholar]
- 13.Singh G, Gutierrez A, Xu K, Blair IA. Liquid chromatography/electron capture atmospheric pressure chemical ionization/mass spectrometry: analysis of pentafluorobenzyl derivatives of biomolecules and drugs in the attomole range. Anal. Chem. 2000;72:3007–3013. doi: 10.1021/ac000374a. [DOI] [PubMed] [Google Scholar]
- 14.Anari MR, Bakhtiar R, Zhu B, Huskey S, Franklin RB, Evans DC. Derivatization of ethinylestradiol with dansyl chloride to enhance electrospray ionization: application in trace analysis of ethinylestradiol in rhesus monkey plasma. Anal. Chem. 2002;74:4136–4144. doi: 10.1021/ac025712h. [DOI] [PubMed] [Google Scholar]
- 15.Higashi T, Shimada K. Derivatization of neutral steroids to enhance their detection characteristics in liquid chromatography-mass spectrometry. Anal. Bioanal. Chem. 2004;378:875–882. doi: 10.1007/s00216-003-2252-z. [DOI] [PubMed] [Google Scholar]
- 16.Higashi T, Nagahama A, Otomi N, Shimada K. Studies on neurosteroids XIX. Development and validation of liquid chromatography-tandem mass spectrometric method for determination of 5α-reduced pregnane-type neurosteroids in rat brain and serum. J. Chromatogr. B. 2007;848:188–199. doi: 10.1016/j.jchromb.2006.10.036. [DOI] [PubMed] [Google Scholar]
- 17.Higashi T, Shibayama Y, Shimada K. Determination of salivary dehydroepiandrosterone using liquid chromatography--tandem mass spectrometry combined with charged derivatization. J. Chromatogr. B. 2007;846:195–201. doi: 10.1016/j.jchromb.2006.08.050. [DOI] [PubMed] [Google Scholar]
- 18.Borges CR, Miller N, Shelby M, Hansen M, White C, Slawson MH, Monti K, Crouch DJ. Analysis of a challenging subset of World Anti-Doping Agency-banned steroids and antiestrogens by LC-MS-MS. J. Anal. Toxicol. 2007;31:125–131. doi: 10.1093/jat/31.3.125. [DOI] [PubMed] [Google Scholar]
- 19.Xu X, Keefer LK, Waterhouse DJ, Saavedra JE, Veenstra TD, Ziegler RG. Measuring seven endogenous ketolic estrogens simultaneously in human urine by high-performance liquid chromatography-mass spectrometry. Anal. Chem. 2004;76:5829–5836. doi: 10.1021/ac049405i. [DOI] [PubMed] [Google Scholar]
- 20.Xu L, Spink DC. 1,2-Dimethylimidazole-4-sulfonyl chloride, a novel derivatization reagent for the analysis of phenolic compounds by liquid chromatography electrospray tandem mass spectrometry: Application to 1-hydroxypyrene in human urine. J.Chromatogr. B. 2007;855:159–165. doi: 10.1016/j.jchromb.2007.04.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yamashita K, Kobayashi S, Tsukamoto S, Numazawa M. Synthesis of pyridine-carboxylate derivatives of hydroxysteroids for liquid chromatography-electrospray ionization-mass spectrometry. Steroids. 2007;72:50–59. doi: 10.1016/j.steroids.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 22.Yamashita K, Okuyama M, Watanabe Y, Honma S, Kobayashi S, Numazawa M. Highly sensitive determination of estrone and 17β-estradiol in human serum by liquid chromatography-electrospray ionization tandem mass spectrometry. Steroids. 2007;72:819–827. doi: 10.1016/j.steroids.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 23.Nelson RE, Grebe SK, OKane DJ, Singh RJ. Liquid chromatography-tandem mass spectrometry assay for simultaneous measurement of estradiol and estrone in human plasma. Clin. Chem. 2004;50:373–384. doi: 10.1373/clinchem.2003.025478. [DOI] [PubMed] [Google Scholar]
- 24.Shou WZ, Jiang X, Naidong W. Development and validation of a high-sensitivity liquid chromatography/tandem mass spectrometry (LC/MS/MS) method with chemical derivatization for the determination of ethinyl estradiol in human plasma. Biomed. Chromatogr. 2004;18:414–421. doi: 10.1002/bmc.329. [DOI] [PubMed] [Google Scholar]
- 25.Li W, Li YH, Li AC, Zhou S, Naidong W. Simultaneous determination of norethindrone and ethinyl estradiol in human plasma by high performance liquid chromatography with tandem mass spectrometry – experiences on developing a highly selective method using derivatization reagent for enhancing sensitivity. J. Chromatogr. B. 2005;825:223–232. doi: 10.1016/j.jchromb.2005.01.012. [DOI] [PubMed] [Google Scholar]
- 26.Xu X, Veenstra TD, Fox SD, Roman JM, Issaq HJ, Falk R, Saavedra JE, Keefer LK, Ziegler RG. Measuring fifteen endogenous estrogens simultaneously in human urine by high-performance liquid chromatography-mass spectrometry. Anal. Chem. 2005;77:6646–6654. doi: 10.1021/ac050697c. [DOI] [PubMed] [Google Scholar]
- 27.Tai SS-C, Welch MJ. Development and evaluation of a reference measurement procedure for the determination of estradiol-17β in human serum using isotope-dilution liquid chromatography-tandem mass spectrometry. Anal. Chem. 2005;77:6359–6363. doi: 10.1021/ac050837i. [DOI] [PubMed] [Google Scholar]
- 28.Licea-Perez H, Wang S, Bowen CL, Yang E. A semi-automated 96-well plate method for the simultaneous determination of oral contraceptives concentrations in human plasma using ultra performance liquid chromatography coupled with tandem mass spectrometry. J. Chromatogr. B. 2007;852:69–76. doi: 10.1016/j.jchromb.2006.12.052. [DOI] [PubMed] [Google Scholar]
- 29.Xu X, Roman JM, Issaq HJ, Keefer LK, Veenstra TD, Ziegler RG. Quantitative measurement of endogenous estrogens and estrogen metabolites in human serum by liquid chromatography-tandem mass spectrometry. Anal. Chem. doi: 10.1021/ac070494j. in press. [DOI] [PubMed] [Google Scholar]
- 30.Lin YH, Chen CY, Wang GS. Analysis of steroid estrogens in water using liquid chromatography/tandem mass spectrometry with chemical derivatizations. Rapid Commun. Mass Spectrom. 2007;21:1973–1983. doi: 10.1002/rcm.3050. [DOI] [PubMed] [Google Scholar]
- 31.Salvador A, Moretton C, Piram A, Faure R. On-line solid-phase extraction with on support derivatization for high-sensitivity liquid chromatography tandem mass spectrometry of estrogens in influent/effluent of wastewater treatment plants. J. Chromatogr. A. 2007;1145:102–109. doi: 10.1016/j.chroma.2007.01.055. [DOI] [PubMed] [Google Scholar]
- 32.Zhang F, Bartels MJ, Brodeur JC, McClymont EL, Woodburn KB. Quantitation of 17α-ethinylestradiol in aquatic samples using liquid-liquid phase extraction, dansyl derivatization, and liquid chromatography/positive electrospray tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2004;18:2739–2742. doi: 10.1002/rcm.1690. [DOI] [PubMed] [Google Scholar]
- 33.Dehennin L, Reiffsteck A, Scholler R. Simple methods for the synthesis of twenty different, highly enriched deuterium labeled steroids, suitable as internal standards for isotope dilution mass spectrometry. Biomed. Mass Spectrom. 1980;7:493–499. [Google Scholar]
- 34.Beaudry F, Guenette SA, Vachon P. Determination of eugenol in rat plasma by liquid chromatography-quadrupole ion trap mass spectrometry using a simple off-line dansyl chloride derivatization reaction to enhance signal intensity. Biomed. Chromatogr. 2006;20:1216–1222. doi: 10.1002/bmc.687. [DOI] [PubMed] [Google Scholar]
- 35.Li Y, Li AC, Shi H, Zhou S, Shou WZ, Jiang X, Naidong W, Lauterbach JH. The use of chemical derivatization to enhance liquid chromatography/tandem mass spectrometric determination of 1-hydroxypyrene, a biomarker for polycyclic aromatic hydrocarbons in human urine, Rapid Commun. Mass Spectrom. 2005;19:3331–3338. doi: 10.1002/rcm.2196. [DOI] [PubMed] [Google Scholar]
- 36.Wang Z, Hop C, Kim MS, Huskeyn SW, Baillie TA, Guan Z. The unanticipated loss of SO2 from sulfonamides in collision-induced dissociation. Rapid Commun. Mass Spectrom. 2003;17:81–86. doi: 10.1002/rcm.877. [DOI] [PubMed] [Google Scholar]
- 37.Wang HY, Zhang X, Guo YL, Dong XC, Tang QH, Long L. Sulfonamide bond cleavage in benzenesulfonamides and rearrangement of the resulting p-aminophenyl-sulfonyl cations: application to a 2-pyrimidinyloxybenzylamino-benzenesulfonamide herbicide. Rapid Commun. Mass Spectrom. 2005;19:1696–1702. [Google Scholar]
- 38.Djerassi C, Wilson JM, Budzikiewicz H, Chamberlin JW. Mass spectrometry in structural and stereochemical problems. XIV. Steroids with one or two aromatic rings. J Am. Chem. Soc. 1962;84:4544–4552. [Google Scholar]
- 39.Budzikiewicz H, Djerassi C, Williams DH. Structure elucidation of natural products by mass spectrometry volume II: steroids, terpenoids, sugars, and miscellaneous classes. San Francisco: Holden-Day Inc.; 1964. pp. 50–63. [Google Scholar]
- 40.Okerholm RA, Clark SJ, Wotiz HH. Effect of derivative formation on the mass spectra of the estrogens. Anal. Biochem. 1971;44:1–11. doi: 10.1016/0003-2697(71)90340-x. [DOI] [PubMed] [Google Scholar]
- 41.Smith DH, Buchanan BG, Engelmore RS, Duffield AM, Yeo A, Feigenbaum EA, Lederberg J, Djerassi C. Applications of artificial intelligence for chemical inference VIII. An approach to the computer interpretation of the high resolution mass spectra of complex molecules. Structure elucidation of estrogenic steroids. J. Amer. Chem. Soc. 1972;94:5962–5971. doi: 10.1021/ja00772a005. [DOI] [PubMed] [Google Scholar]
- 42.Sulak P, Lippman J, Siu C, Massaro J, Godwin A. Clinical comparison of triphasic norgestimate/35 micrograms ethinyl estradiol and monophasic norethindrone acetate/20 micrograms ethinyl estradiol. Cycle control, lipid effects, and user satisfaction. Contraception. 1999;59:161–166. doi: 10.1016/s0010-7824(99)00026-8. [DOI] [PubMed] [Google Scholar]
- 43.Bhavnani BR. The saga of the ring B unsaturated equine estrogens. Endocrine Rev. 1988;9:396–416. doi: 10.1210/edrv-9-4-396. [DOI] [PubMed] [Google Scholar]