

Abstract
Untargeted profiling of small-molecule metabolites from microbial culture supernatants (metabolic footprinting) has great potential as a phenotyping tool. We used time-resolved metabolic footprinting to compare one Escherichia coli and three Pseudomonas aeruginosa strains growing on complex media and show that considering metabolite changes over the whole course of growth provides much more information than analyses based on data from a single time point. Most strikingly, there was pronounced selectivity in metabolite uptake, even when the bacteria were growing apparently exponentially, with certain groups of metabolites not taken up until others had been entirely depleted from the medium. In addition, metabolite excretion showed some complex patterns. Fitting nonlinear equations (four-parameter sigmoids) to individual metabolite data allowed us to model these changes for metabolite uptake and visualize them by back-projecting the curve-fit parameters onto the original growth curves. These “uptake window” plots clearly demonstrated strain differences, with the uptake of some compounds being reversed in order between different strains. Comparison of an undefined rich medium with a defined complex medium designed to mimic cystic fibrosis sputum showed many differences, both qualitative and quantitative, with a greater proportion of excreted to utilized metabolites in the defined medium. Extending the strain comparison to a more closely related set of isolates showed that it was possible to discriminate two species of the Burkholderia cepacia complex based on uptake dynamics alone. We believe time-resolved metabolic footprinting could be a valuable tool for many questions in bacteriology, including isolate comparisons, phenotyping deletion mutants, and as a functional complement to taxonomic classifications.
The increasing speed of gene discovery has exceeded our ability to understand gene function, and one of the bottlenecks is the need for new, high-throughput tools to evaluate cellular phenotypes (22). Even in bacterial genomes, less than 70% of genes have an assigned putative function, and fewer still have been characterized biochemically. Metabolic profiling approaches have shown great promise for providing these tools for functional genomics and hypothesis generation (1, 6, 10, 18, 28, 43, 49) because they offer complementary information to transcriptomics and proteomics, in particular giving an integrated picture of information downstream of the genome (51). Various aspects of cellular physiology, such as the levels of transcripts, proteins, or protein activity, are altered in response to environmental cues or metabolite concentrations themselves. In return, these changes are amplified in the metabolome to give an accumulated—and highly sensitive—description of the physiological state of the organism or cellular compartment (26, 45, 49). This extends to natural populations that have multiple uncharacterized genetic changes, such as an accumulation of mutations, as well as sometimes extensive genetic differences such as pathogenicity islands (21), which may interact to give complex phenotypes. Molecular phylogenetic methods based on gene sequences have proved successful in classifying bacteria into taxonomic groupings, but these may not always correspond to easily identifiable phenotypes or ecotypes (29, 33, 48). Hence, additional methods for strain assessment that could be related to function would still be valuable.
Metabolomics gives an integrated measurement of cellular phenotype and is highly suited to quantitative analysis and description. In a microbial context, metabolomics offers the additional advantage that there is only a single cell type and little compartmentation (at least in comparison to the equivalent problem in a multicellular organism). However, sampling intracellular metabolites without either changing their relative concentrations or introducing contamination from supernatant metabolites is not straightforward, and research methods are still under active development by different groups (7, 12, 15, 59, 62). In contrast, exometabolome or supernatant profiling (“metabolic footprinting”) is simple, and extracellular metabolites can exhibit very large changes in pool size (1, 27, 40, 45). These multiple advantages mean that exometabolome analysis has already been used for a number of diverse applications, such as phenotyping of both single-gene deletion mutants and isolates from natural populations, although thus far mostly for fungi rather than bacteria (1, 2, 9, 25, 40, 48).
Because metabolism integrates information from gene expression and a wealth of environmental cues, each organism will exhibit a distinct response, i.e., metabolic pattern that takes into account all of these factors. It is therefore unsurprising that these patterns change with growth phase (1, 30). Despite this fact, it is currently common practice to sample only at one or two time points, mostly the end of growth, in stationary phase (see, for example, reference 48) and/or in mid-exponential phase (41, 52). In contrast, there is ample evidence that cellular biochemistry changes during growth (1, 3, 8, 39). Vertebrate studies have shown that explicitly considering “through time” responses (metabolic trajectories) adds considerably to the description and understanding of biological events (16, 23, 58). We therefore argue that new approaches that are capable of integrating metabolic phenotypes over a range of conditions could be extremely beneficial for microbiology.
In the present study we have developed such an approach and evaluated it by monitoring metabolic changes over the course of time in growing batch cultures. Time-resolved metabolic footprinting (TReF) was used to compare the well-studied organisms Escherichia coli and Pseudomonas aeruginosa. We demonstrate that TReF is considerably more data-rich and informative than sampling at single time points and show the usefulness of the approach in hypothesis generation and as a phenotyping tool. We also show that TReF distinguishes isolates from the closely related Burkholderia cepacia complex (BCC) at the species level for B. cepacia and B. cenocepacia, which is not the case for single time point analysis. The approach is very general and would therefore benefit the broader application of metabolomics to bacterial systems.
MATERIALS AND METHODS
Bacterial strains.
We used the following strains in the present study: E. coli MG1655; the P. aeruginosa wild-type strains PA01 and PA14 (50); P. aeruginosa PA0381 leu-38 str-2, a leucine auxotroph derived from PA01 (17); B. cenocepacia LMG 16654; B. cenocepacia LMG 16659; B. cenocepacia LMG 18830; B. cenocepacia LMG 16656 (J2315); B. cenocepacia LMG 18863; B. cepacia LMG; B. cepacia LMG 6963; B. cepacia LMG 6988; and B. cepacia LMG 18821. Starter cultures for four biological replicates were set up by inoculating single colonies into 5 ml of LB medium (10 g of tryptone, 5 g of yeast extract, and 5 g of NaCl/liter), followed by growth overnight at 37°C, with shaking at 150 rpm. The growth of PA01 was compared under the same conditions in synthetic cystic fibrosis medium (SCFM), a complex defined medium designed to model nutrient status in sputum (46). These cultures were used to inoculate 20 ml of LB or SCFM in 250-ml conical flasks and then grown for 24 h at 37°C with shaking at 150 rpm.
Sampling.
Portions (1 ml) were taken from the culture at 0, 2, 4, 5, 6, 7, 8, 9, 10, 11, 12, and 24 h for E. coli, P. aeruginosa PA01, and all Burkholderia strains. The P. aeruginosa PA14 and P. aeruginosa PA0381 cultures were sampled at 0, 2, 3, 4, 5, 6, 8, 10, 12, 14, 16, and 24 h. (It should be noted that the total volumes sampled from each culture would potentially change the cell physiology compared to an unsampled flask; however, we were not aiming to model an unsampled culture.) For each sample, 0.1 ml was mixed with 0.9 ml of culture medium for determination of the cell density (i.e., the optical density at 600 nm [OD600]). The remainder of the sample was centrifuged (16,000 × g, room temperature), and 0.75 ml of the supernatant was mixed with 0.2 ml of nuclear magnetic resonance (NMR) buffer (25 mM sodium azide, 0.25 M phosphate buffer [pH 7], 5 mM sodium 3-trimethylsilyl-2,2,3,3-2H4-propionate [TSP] in 2H2O). The 2H2O provided a field frequency lock for the spectrometer, and the TSP served as an internal chemical shift reference.
1H NMR measurement.
Spectra were acquired on a Bruker Avance DRX600 NMR spectrometer (Bruker BioSpin, Rheinstetten, Germany), with a magnetic field strength of 14.1 T and a resulting 1H resonance frequency of 600 MHz, equipped with a 5-mm inverse flow probe. Samples were introduced by using a Gilson flow-injection autosampler. Spectra were acquired following the approach described by Beckonert et al. (4). Briefly, a one-dimensional NOESY pulse sequence was used for water suppression; data were acquired into 32 K data points over a spectral width of 12 kHz, with 8 dummy scans and 64 scans per sample, and an additional longitudinal relaxation recovery delay of 3.5 s per scan, giving a total recycle time of 5 s.
Spectral processing and data analysis.
Spectra were processed in iNMR 2.5 (Nucleomatica, Molfetta, Italy). Free induction decays were multiplied by an exponential apodization function equivalent to 0.5 Hz line broadening, followed by Fourier transformation. Spectra were manually phased and automated first order baseline correction was applied. Spectral data between −0.5 and 10 ppm were then imported into Matlab 2007b (MathWorks, Cambridge, United Kingdom) and normalized to the integral of the TSP signal. Metabolites were assigned using in-house data, the Chenomx NMR Suite 3.1 (Chenomx, Inc., Edmonton, Alberta, Canada) and the Biological Magnetic Resonance Databank metabolomics database (14). Signature peaks, i.e., well-resolved resonances that could be easily assigned to one compound, were identified from the spectra. Difference spectra were calculated in order to eliminate the influence of (nonbiological) variation in media composition. For this, the spectrum at time point 0 h was subtracted from the spectra of other time points of the same strain-replicate pair (i.e., all spectra sampled from the same flask). In addition to full-resolution spectra, all analyses were carried out on binned integrals representing the dominant resonances detected in fresh, noninoculated medium. A total of 153 integrals were fitted for LB, and 130 integrals were fitted for SCFM. For the heatmap plots, the overall range of the resonance intensity changes was set to 1, and the changes were expressed relative to the starting values.
Modeling and pattern recognition analysis.
We tested two different approaches to monitor the time-dependent changes in metabolite concentration. (i) Linear regression analysis was carried out with both the OD600 and time as an x variable. A cutoff value for goodness of fit (R2 = 0.6) was determined by visual inspection of the fits. (ii) Nonlinear regression of the data against time using a sigmoid curve model (equation 1) was carried out using “nlinfit” (Matlab statistics toolbox). This resulted in fitting each variable with four parameters, the amplitude of the curve, the “half-life” (t50), and the width of the decrease. Cutoffs for t50 (1 to 24 h), width (0 to 12 h), and relative error (<0.6) were imposed.
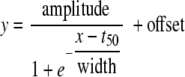 |
(1) |
The width is defined as the time that elapses for the exponent of e to go from 1 to −1. Growth rate differences (E. coli grows faster than the Pseudomonas strains) manifest themselves in higher t50 values for slower-growing than for faster-growing strains, and these quantitative growth rate effects complicate the elucidation of qualitative differences that are particularly interesting for strain comparison purposes. Therefore, the sigmoid parameters were corrected for growth curve bias before pattern recognition: the OD values were also fitted to the same nonlinear model (equation 1). The amplitude was divided by the amplitude of the OD, and the t50 was expressed relative to the t50 of the growth curve by subtracting the t50 of each individual growth curve and dividing the resulting values by the width of the growth curve (equation 2).
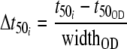 |
(2) |
The fitting parameters were then mean-centered and used as inputs for hierarchical principal component analysis (H-PCA) (61). As a first step for H-PCA, PCA was carried out on the corrected amplitude, the corrected “half-life,” and the width. To account for the missing values introduced by using cutoff values, a nonlinear iterative partial least squares-PCA algorithm was used. The three resulting scores blocks were normalized by division by their highest values to give each “score block” equal importance and used as input variables for a second-level PCA.
RESULTS
TReF provides additional biological information compared to single time point analysis.
Initially, we monitored changes in Luria broth culture supernatant during the growth of the widely studied gram-negative bacteria E. coli (wild-type MG1655) and P. aeruginosa (wild-type PA01 and PA14 and the leucine auxotroph PA0381, which was derived from PA01 [17]). In addition, growth of PA01 in a defined medium (SCFM) (46) was compared. The 1H NMR spectra showed a complex mixture of small molecules, the majority of which could be readily assigned by comparison of their multiplicity and chemical shift to published or online values (Table 1). There were also a smaller number of resonances, which we have not yet assigned (0.91d, 1.07d, 1.19m, 1.27m, 1.36d, 2.69m, 3.81s, 5.85d, 5.88d, 6.03d, 6.08d, 6.15d, 6.30d, and 6.86m).
TABLE 1.
Assigned NMR-visible resonances in LBa
| Compoundb | Assigned resonance frequency(ies) (ppm) |
|---|---|
| Acetate* | 1.92 |
| Acetaldehyde* | 2.24 |
| Adenosine* | 6.08, 8.26, 8.34 |
| Alanine* | 1.48, 3.79 |
| Arginine* | 1.69, 1.73, 1.75, 1.91, 3.25, 3.78 |
| Asparagine† | 1.72, 2.86, 2.96, 4.00 |
| Aspartate* | 2.68, 2.82, 3.91 |
| Formate* | 8.46 |
| Glucose* | 3.39, 5.24 |
| Glutamate* | 2.07, 2.35, 3.74 |
| Glycine* | 3.57 |
| Glycine-betaine* | 3.27, 3.90 |
| Histidine (not fitted)* | 3.11, 3.14, 3.31, 7.07, 7.88 |
| 6-Hydroxynicotinate† | 6.62, 8.07 |
| Indole§ | 6.61, 7.18, 7.27, 7.42, 7.56, 7.72 |
| Isoleucine* | 0.94, 1.01, 1.25, 1.26, 3.68, 3.74 |
| Lactate‡ | 1.33, 4.12 |
| Leucine* | 0.96, 0.97, 1.72, 3.74 |
| Lysine* | 1.46, 1.48, 1.73, 1.89, 1.91, 3.03 |
| Methionine* | 2.12, 2.14, 2.65 |
| Methionine-S-oxide† | 2.74, 2.76, 2.93 |
| Nicotinic acid* | 8.61, 8.94 |
| Pyrimidine nucleotide* | 5.91 |
| Ornithine‡ | 3.81 |
| Phenylalanine* | 3.11, 3.28, 4.01, 7.33, 7.39, 7.43 |
| Pyroglutamate† | 2.06, 2.39, 2.42, 2.51, 7.98 |
| Serine* | 3.79, 3.85, 3.96 |
| Succinate† | 2.41 |
| Threonine* | 1.33, 3.59, 4.26 |
| Trehalose* | 3.46, 3.65, 3.83, 3.86, 3.88, 5.20 |
| Tryptophan† | 3.31, 7.29, 7.55, 7.74 |
| Tyrosine* | 3.073.22, 3.31, 3.94, 6.90, 7.20 |
| Uracil* | 5.82, 7.55 |
| Valine* | 0.99, 1.05, 2.28, 3.62 |
| Unassigned metabolite (potential quinolone) | 7.68, 8.10 |
Note that the metabolites listed here may have other resonances: the table includes only the most characteristic and well-resolved resonances. Resonances in boldface were used for nonlinear fitting of compounds.
*, Observed in both LB and SCFM; †, observed in LB only; ‡, observed in SCFM only; §, observed for E. coli only (not tested in SCFM).
Over the course of growth there were major changes in the metabolite composition of the growth media. This is illustrated in Fig. 1A, which shows the chemical shift region from 2 to 4 ppm of one LB-grown culture of P. aeruginosa PA01 over time. At the compound level, TReF revealed differences in the rates of uptake of individual compounds, as shown for three amino acids in P. aeruginosa PA01 LB cultures. Alanine was taken up first from the medium, followed by threonine and then leucine (Fig. 1B). This clear time separation shows different modes of compound utilization during growth, and this differential compound utilization was observed for multiple compounds and in all investigated isolates. Further, the order in which compounds were utilized varied, but was reproducible at the isolate level. These differences would have been missed by single time point profiling at 12 or 24 h and clearly indicate that comparative metabolomics would benefit from the application of TReF-based approaches, since differences can be highly growth phase dependent.
FIG. 1.

(A) Section of 600 MHz 1H NMR spectra (4 to 2 ppm) for a single P. aeruginosa PA01 culture over a growth curve. Time-specific metabolic changes are clearly seen. (B) Single compound utilization data for three selected metabolites for P. aeruginosa PA01. Error bars indicate the standard errors of the means (n = 4).
Figure 2 provides a summary of the changes that were observed in the investigated cultures over time. Figure 2A to E is a heatmap representation of averaged difference spectra depicting both uptake and secretion at compound level, clearly showing patterns of metabolite secretion and uptake that differed greatly between the different strains and media. Four different modes were identified: (i) constant depletion, where the majority of metabolites in the medium decreased constantly over time (e.g., Fig. 1 and 3B); (ii) transient excretion, followed by depletion, where some compounds (e.g., acetate [Fig. 2F]) were excreted during one growth phase and taken up during another; (iv) transient depletion, followed by excretion, in which all Pseudomonas strains first took up formate, only to excrete it at later time points (Fig. 2H); and (iv) constant excretion, where some compounds increased in a sigmoid fashion, e.g., an as-yet-unassigned doublet resonance at δ 1.10 ppm (probable methyl group signal from an organic acid [Fig. 2G]). As shown in Fig. 2, the growth medium itself has a large influence on the metabolite utilization and depletion patterns, with major differences between P. aeruginosa PA01 grown in LB and in SCFM. The uptake behavior at the compound level is summarized in Fig. 2 and Table 2. Based on these first observations, the differences in compound utilization and excretion were further investigated and are discussed below.
FIG. 2.

Metabolite changes in different media and different strains across the course of growth. (A to E) Heatmaps. Each row represents a metabolite or a peak from an as-yet unassigned metabolite. Blue indicates a decrease in concentration, and red indicates an increase in concentration. Note that panels B to E can be directly compared visually but that the metabolites in panel A do not line up directly with B to E. The row for metabolite “6HN/indole” represents 6-hydroxynicotinate for P. aeruginosa strains and indole for E. coli. (A) P. aeruginosa PA01, SCFM; (B) P. aeruginosa PA01, LB; (C) P. aeruginosa PA14, LB; (D) P. aeruginosa PA0381, LB; (E) E. coli, LB. Selected metabolites with different modes of utilization and/or production are then shown in detail in the bottom half of the figure (error bars represent ± the standard errors of the mean). (F) Acetate, E. coli, showing a transient increase in metabolite concentration; (G) unassigned metabolite, E. coli, showing a peak at δ 1.10 ppm and a steady increase in metabolite concentration; (H) formate, P. aeruginosa PA01, LB, showing a transient decrease in metabolite concentration followed by subsequent production. (I and J) Overall comparison of the different modes shown as pie charts (indicating the percentage of assigned metabolites that changed in some way during growth). (I) P. aeruginosa PA01, SCFM; (J) P. aeruginosa PA01, LB.
FIG. 3.

(A) Schematic showing the parameters for nonlinear curve fitting. (B [inset]) Curve fit for a representative compound (pyroglutamate) for P. aeruginosa PA01. Solid line indicates the sigmoid fit; the dotted line indicates much poorer linear fit.
TABLE 2.
Comparison of fitted metabolite t50 values for P. aeruginosa PA01 grown in LB and SCFM
| Amino acid | Metabolite t50 values (h)a in:
|
Difference (h)b | |
|---|---|---|---|
| LB | SCFM | ||
| Tyrosine | 14.8 | 6.5 | -8.3 |
| Valine | 15.8 | 8.5 | -7.3 |
| Phenylalanine | 8.8 | 5.0 | -3.8 |
| Lysine | 11.3 | 8.0 | -3.3 |
| Leucine | 9.6 | 7.0 | -2.5 |
| Isoleucine | 8.1 | 7.0 | -1.2 |
| Aspartate | 3.1 | 2.2 | -0.9 |
| Arginine | 4.0 | 3.5 | -0.5 |
| Glycine | 5.3 | 5.0 | -0.3 |
| Glutamate | 2.9 | 2.8 | -0.2 |
| Alanine | 3.5 | 3.4 | -0.1 |
| Serine | 2.7 | 4.3 | 1.6 |
| Threonine | 5.8 | 7.7 | 2.0 |
| Asparagine | 2.3 | NO | |
| Methionine | NU | 11.8 | |
| Ornithine | NO | 4.0 | |
| Tryptophan | 4.7 | NO | |
NU, not utilized; NO, not observed.
“Difference” refers to the difference between the t50 value in SCFM compared to that in LB medium, i.e., the lower the value, the earlier the metabolite was taken up in SCFM compared to LB.
Nonlinear regression modeling can be used to describe metabolite utilization over time.
Most of the NMR-detectable resonances decreased over the course of growth (Fig. 2I and J). In order to further describe the changes in exometabolome composition over time, the concentration changes of individual metabolites were modeled by regression. Linear regression against time was a poor descriptor of metabolite consumption. Most of the NMR intensities did not describe a straight line when plotted against time, and thus each modeled variable usually contained an unacceptable amount of fitting error (as an example, the dotted line in Fig. 3B shows the linear fit of the pyroglutamate resonance at 2.40 ppm in one P. aeruginosa PA01 culture). Only about one-third of the fitted resonances had an R2 value of greater than 0.6. For many compounds, the change in the resonance intensities roughly mirrored a growth curve and thus more closely resembled a straight line when plotted against OD (data not shown). The fits were indeed slightly improved when cell density (OD600) rather than time was used as the x variable: about half of the fitted resonances had R2 values greater than 0.6. However, the average correlation across all resonances was still poor for both time and the OD (R = 0.48 for time, 0.56 for OD). Instead, fits were significantly improved by using an appropriate nonlinear model. A bacterial growth curve typically describes a sigmoid shape over time. Although the intensities of most NMR resonances did not exactly mirror this growth curve, they did decrease in a sigmoid fashion. Consequently, fitting sigmoid curves to the evolution of the resonances over time markedly decreased errors for “real” peaks as opposed to noise (the solid line in Fig. 3B). Even after imposing stringent cutoff values for fit (see Materials and Methods), the data set still contained about two-thirds of the resonances. Nonlinear fitting is well suited to study media depletion but was less useful for secreted metabolites.
For the data successfully fitted by nonlinear modeling, the time course of each metabolite resonance was described by four parameters (Fig. 3A). Three parameters summarize the uptake characteristics for each metabolite: the relative decrease of the resonance with time (amplitude), the time of uptake (t50), and the duration of uptake (width). The fourth parameter, the offset, i.e., the intensity at the start of the experiment, does not represent meaningful information in this case, since we used difference spectra for the analysis. Hence, the offset was zero (for the original data) or close to zero (fitted data).
Uptake window plots visualize compound utilization.
The parameters of the sigmoid equation can be used to obtain physiological information for individual compounds. Both the t50 and the width are in units of time, with the t50 defining the time point at which the amplitude has reached its half-way point, i.e., when half of the compound has been utilized. The width is defined as the time that elapses for the exponent of e to go from 1 to −1 (see Materials and Methods) and roughly translates to the duration in which the compound is taken up at the maximum rate; the width thus defines a time span or “uptake window” for any fitted compound or resonance lying around the compound t50 value (Fig. 3). These “uptake windows” can be projected onto the OD600 growth curves of the individual strains to visualize differential compound uptake. Figures 4A to D show the projections of the uptake windows of seven compounds (alanine, leucine, threonine, asparagine, valine, succinate, and the disaccharide trehalose) onto the growth curves of the E. coli and the three P. aeruginosa strains for LB. Each circle represents the t50 value of one compound for one biological replicate, with the bars on either side representing the width of the same compound.
FIG. 4.

Uptake window plots for seven example compounds for all four bacterial strains. The compound t50 is back-projected upon the actual culture growth curve, i.e., all biological replicates are shown. The “error bars” represent the calculated width (see Fig. 2 for an illustration of t50 and width). Note that both the abscissa (time) and ordinate (OD600) have been scaled such that growth curve maxima are set at 100%, to facilitate comparison across different strains. (A) E. coli; (B) P. aeruginosa PA01; (C) P. aeruginosa PA14; (D) P. aeruginosa PA0381. The remaining two panels compare uptake windows for P. aeruginosa PA01 for two different media: (E) LB and (F) SCFM. Note that glucose is plotted (not trehalose as for panel A), since glucose is higher concentration in SCFM.
These uptake window plots illustrate how TReF is able to elucidate similarities and differences in compound utilization of strains, as summarized with a single plot. Not only did the uptake windows differ dramatically for the individual metabolites, but there was very clear separation between them, i.e., the different amino acids fell into different “utilization groups,” which were separated along the growth curve. For example, P. aeruginosa PA01 (grown in LB) did not take up threonine until after the simultaneous depletion of alanine and asparagine. Leucine was then taken up after threonine had been removed. This order was also observed for the two other P. aeruginosa strains but was different in E. coli, with trehalose taken up before alanine and leucine not taken up at all.
In addition, the plots provide evidence for significant differences between the three P. aeruginosa strains. PA14 does not take up succinate in a sigmoidal fashion; however, the compound was quickly removed from the medium in all strains. Interestingly, PA0381, originally derived from PA01, was shown to have lost its ability to utilize trehalose. This loss of function could be a side effect of the leucine auxotrophy causing a metabolic network rearrangement. However, a more parsimonious explanation is that the nonspecific mutagenesis used to obtain the leucine auxotroph phenotype (53) also affected one of the genes necessary for trehalose breakdown (the transporter or the trehalase).
Transient changes in the exometabolome and metabolite excretion.
Apart from metabolite uptake, a large proportion of the detected resonances transiently increased or decreased during growth in both LB and SCFM (Fig. 2). As a positive confirmation, we detected acetate production by E. coli, a known example of overflow metabolism. Acetate is a fermentation product that accumulates at high growth rates, probably due to a rate bottleneck in aerobic metabolism (38); it was by far the clearest example of overflow metabolism in the present study. When grown in LB, all Pseudomonas strains transiently excreted the amino acids valine and tyrosine. In PA01 cultures, a singlet resonance at 2.24 ppm (putatively assigned as acetaldehyde) showed excretion dynamics similar to those of valine. Interestingly, this was not observed for the other Pseudomonas strains. In contrast, formate (Fig. 2H) was taken up from the medium during the first couple of hours of growth but was excreted in the stationary phase. In addition to these transient changes, a number of resonances increased proportionally to cell number over the course of growth, including 6-hydroxynicotinate (all Pseudomonas strains), indole (E. coli), and uracil (all strains).
Compound utilization and excretion are dramatically influenced by the constituents of the growth media.
It could be argued that the complexity of the responses we observed was partly due to our using a complex and undefined growth medium. To that end, we compared the exometabolome of P. aeruginosa PA01 grown in LB to that grown in SCFM, a defined medium designed and shown to mimic conditions and utilization dynamics in cystic fibrosis sputum (46). Even though the cell density (i.e., the OD600) in different media did not differ greatly (data not shown), the choice of growth medium had a dramatic effect on the dynamics of the exometabolome, affecting both compound uptake and excretion (Fig. 2 and 4E and F). Concerning compound utilization, a comparison of the uptake windows for selected amino acids in the two media (Table 2 and Fig. 4E and F) revealed several trends. Some amino acids, such as lysine, phenylalanine, and leucine, were taken up later from LB than from SCFM, which might hint at some sort of catabolite repression-like regulation in LB (see the Discussion). In contrast, the uptake dynamics of alanine, glutamate, aspartate, and arginine were relatively unaffected since they were taken up at an early stage in both cultures. In terms of compound secretion, many more resonances increased when PA01 was grown in SCFM compared to when PA01 was grown in LB. The transient increases in tyrosine and valine were also not observed in SCFM, but other resonances (1.07d, 2.51s, and 2.53s) were observed to increase transiently. Finally, the pattern of formate change (transient decrease, followed by an increase) was even more pronounced in SCFM.
Potential application of TReF as a functional genomics tool.
Pattern recognition algorithms like PCA are widely used for multivariate data to visualize and summarize metabolic differences by dimension reduction. It was possible to separate E. coli and all P. aeruginosa strains using PCA on stationary-phase samples, and the approach very clearly showed the metabolites responsible for the strain differences (Fig. 5). However, the plots also show how single time point profiling would miss the “big picture,” i.e., the metabolite concentration changes that occur at other time points. If, for example, the cultures were sampled at 12 h, valine would appear to be excreted only by PA01 (Fig. 5E). In fact, PA14 and PA0381 also excrete valine at earlier time points. Had the exometabolome been sampled at 24 h only, valine would appear to be utilized by all three P. aeruginosa strains to roughly the same extent. In addition, the strains' leucine (Fig. 5F) utilization would look roughly equivalent after 12 h, whereas, in fact, leucine uptake was slower and had a slightly greater amplitude in PA01 cultures. Of the discriminatory metabolites at 12 h, only trehalose (Fig. 5D) showed the same qualitative difference between the strains at all time points. One advantage of the nonlinear metabolite fitting is that the fit parameters summarize key biological endpoints (e.g., compound uptake rates) in a compact way. Thus, by using the fit parameters as input for the multivariate analyses, it is possible to compare data in a principled way from different strains, which might have slightly different growth rates, lag phases, etc. Naturally, each parameter could be analyzed separately, but it is also possible to combine these in a single hierarchical model (Fig. 5C).
FIG. 5.

(A) PCA for 12-h data, score plot of axes 1 and 2. Symbols: •, E. coli; ○, P. aeruginosa PA01; ▵, P. aeruginosa PA14; □, P. aeruginosa PA0381. Principal components 1 and 2 explained 87 and 8% of the variance in the data, respectively. (B) Loadings plot for the analysis shown in panel A. Variables corresponding to assigned metabolite bins for leucine, valine, and trehalose are labeled directly on the plot. (C) H-PCA of fitted time course data. Figure symbols are the same as for panel A. Principal components 1 and 2 explained 50 and 33% of the variance in the data, respectively. (D) Trehalose utilization during growth for four strains. The solid black line represents E. coli, the solid gray line represents P. aeruginosa PA01, the dashed line (long dashes) represents P. aeruginosa PA14, and the dashed line (short dashes) represents P. aeruginosa PA0381. (E) Valine utilization during growth for four strains. Lines are as defined for panel D. (F) Leucine utilization during growth for four strains. Lines are as defined for panel D.
As a test case for the resolution the TReF/H-PCA approach could offer, we analyzed culture supernatants of two species (nine strains in total) of the closely related BCC. Single time point profiling like that shown for stationary phase (t = 24 h) samples only provided some possible species separation, but with considerable overlap between the species groups (Fig. 6A, similar results for other time points [data not shown]). An added complication for this data set was that the strains showed large variations in growth rate, which were picked up by standard multivariate methods. However, the nonlinear H-PCA approach showed a separation of B. cepacia and B. cenocepacia along PC1 (Fig. 6B). Thus, while “standard” footprinting based on single time points may be adequate for showing large metabolic differences, it failed to fully represent the subtle metabolic differences between the BCC strains, which required the nonlinear fit data. (We also tested H-PCA alone, i.e., a hierarchical model based on PCA for individual time points without any curve fitting, but this offered no advantages in comparison to analyzing single time points, and failed to separate the BCC species; data not shown.)
FIG. 6.

Comparison of single-time-point and nonlinear fitted metabolite data for four B. cepacia (open symbols) and five B. cenocepacia (filled symbols) isolates as principal component score plots, axis 1 versus axis 2. Different symbol shapes represent different individual isolates. (A) Single-time-point analysis does not discriminate all isolates into species. Principal components 1 and 2 explained 55 and 39% of the variance in the data, respectively. (B) Fitted metabolite data (TReF) show that species are discriminated along principal component 1 across different isolates. Principal components 1 and 2 explained 35 and 25% of the variance in the data, respectively.
DISCUSSION
We have developed a TReF approach for bacteria that should be widely applicable. Considering changes in the culture medium over the whole course of growth provides information that would be lost in a single time point analysis.
Bacteria show “multiauxic” uptake behavior on complex media.
TReF revealed differential compound uptake for all investigated strains, and for both a rich and a defined medium (LB and SCFM). The existence of a complex regulatory network leading to highly adaptable uptake dynamics is not surprising. In rich (or defined multicompound) media, expression and translation of the transporter systems and catabolic pathways need to be controlled. The genomes of the Pseudomonas species group contain over 300 known or putative nutrient uptake systems (56). Expressing all inducible transporters and catabolic pathways at once will not be energetically favorable, and so a form of multiauxic growth and sequential compound uptake, like that observed here, is the likely outcome—although the extent of the differentiation between compound utilization classes during apparently exponential growth was surprising. A number of previous studies, albeit mostly not using rich media, have hinted at the complexity of the regulation at hand (see, for example, references 3, 19, and 30).
Catabolite repression is a generic mechanism for regulation of substrate usage, and, for example, succinate represses arginine catabolism in Pseudomonas aeruginosa (42). ArgR controls the aerobic catabolism of arginine in P. aeruginosa (47) and also controls the levels of OprD, a porin for basic amino acids (44) and a serine transporter (32). It is therefore logical that our data show that succinate depletion precedes the utilization of not only arginine but also a number of other amino acids (Fig. 4). As a second example, lysine was depleted at an earlier growth phase in SCFM than in LB. Lysine can be imported by the specific permease LysP (54) but also by the putative basic amino acid ABC transporter PA5152-PA5155 (24). Transposon mutants within this operon were severely impaired in growth on ornithine as a single carbon source (24), so this transporter clearly contributes to P. aeruginosa's ability to use ornithine. Hence, it is highly probable that the high concentrations of ornithine in SCFM would induce expression of PA5152-PA5155, thereby potentially simultaneously increasing the potential rate of lysine uptake. These examples show how an untargeted approach can generate eminently testable hypotheses.
Influence of medium composition on uptake and excretion.
In addition to utilization, we studied compound excretion. Various compounds, such as acetate, valine, and tyrosine, were excreted transiently, whereas others, such as 6-hydroxy nicotinic acid or indole, constantly increased over the course of growth for P. aeruginosa and E. coli, respectively. Formate had a particularly surprising utilization profile, with depletion followed by subsequent excretion; the precise reason is not clear at this moment. Compound excretion is a well-known phenomenon for biotechnologically interesting compounds in bacteria such as Corynebacterium glutamicum (11, 40). A number of fundamental principles that lead to compound excretion have been formulated (11). The obvious explanation for a compound entering the culture medium is excretion of a product that bacteria “want” to excrete. This is the case for signaling molecules such as quorum-sensing (QS) signals. Our data show excretion of indole in E. coli, which was suggested to have extracellular signaling properties (60). It should be noted that P. aeruginosa in particular is known for producing a suite of QS metabolites, which might be expected to be visible in the medium; the reason that we do not identify more QS-related changes is probably just that NMR has relatively high detection limits. However, the TReF principle would be identical if a more sensitive analytical platform was used, such as many techniques based on mass spectrometry. In addition, compounds might also be excreted because of overflow metabolism, limited catabolism, and deregulated anabolism (11). This “relief-valve” function has previously been suggested for the aromatic amino acid exporter (ArAE, formerly AaeAB) in E. coli (57). This transporter has recently been functionally annotated in P. aeruginosa (24), so tyrosine could be excreted by P. aeruginosa when grown in LB to relieve intracellular stress. Valine might be excreted due to similar reasons by an as-yet-unindentified transporter. Interestingly, tyrosine and valine were only excreted into LB and not in SCFM. Finally, the increase of 6-hydroxynicotinic acid in P. aeruginosa cultures is probably due to limited catabolism of NAD or niacin and has been used as a diagnostic marker of P. aeruginosa infection (20).
Species discrimination in BCC.
An important and general question is to what degree phenotypic metabolomic data is informative about the genotype, i.e., strain relatedness, as opposed to, say, the ecotype, which could be a convergent result of adaptation. Previous studies have used both endo- and exometabolome profiling to address this in yeast and bacteria (33, 34, 48); it is clearly a complex question, since metabolomic data have shown both apparent clustering by ecotype, with additional genetic within-cluster separation, and also high between-strain metabolic variability that mostly correlated with genotype divisions.
The P. aeruginosa and E. coli profiles were dramatically different, with the order of uptake of specific metabolites reversed (Fig. 4). However, this is perhaps not surprising given these are very different organisms. We decided to carry out a more realistic test: whether differences could still be observed for a set of much more closely related bacteria. We chose two species of the BCC (B. cepacia and B. cenocepacia, represented here by four and five independent isolates, respectively) as a model comparison. The BCC is a collection of genotypically distinct but phenotypically similar species within the genus Burkholderia (13, 37). Some BCC members are opportunistic pathogens that can cause serious infections in patients with chronic granulomatous disease or cystic fibrosis (35, 37), while some are found in the rhizosphere of important crops such as maize and can protect these plants from fungal infection (5). Species-level identification of BCC members is difficult, and species are still frequently misidentified, especially using commercial identification systems (31). Single-gene phylogenies showed that B. cepacia and B. cenocepacia are especially similar genetically even within the BCC (36, 55), meaning these two species formed a stringent test for our approach. The nonlinear fitting TReF approach was nevertheless able to discriminate the isolates into species groups. It cannot be concluded at this point that this could therefore be used as a general tool for BCC taxonomy (more isolates would need to be tested to derive robust conclusions about metabolic differences in these species) but serves as a proof-of-principle that our approach of modeling the full-time course of metabolic changes can provide additional and biologically meaningful data over single-time-point analyses.
Conclusion.
We have shown potential microbiological applications of time-dependent exometabolome profiling. Modeling of the amino acid utilization of E. coli and P. aeruginosa demonstrated an unexpected complexity of regulation. In addition, the same approach was shown to have clear advantages over single-time-point profiling. TReF allowed comparison of the physiology of bacteria in different nutritional environments, and our data clearly demonstrate that marked differences could be found. We believe time-dependent metabolic profiling could be a valuable addition to the fields of bacterial physiology, functional genomics, and as a tool for strain comparison, both as a complement to traditional taxonomies and also for investigating properties such as strain-specific virulence. It is still likely that single-time-point metabolic footprinting will be preferred for many studies, simply because it requires analysis of fewer replicates. We see TReF having a complementary role, for in-depth phenotype analysis of a smaller number of strains—which might well, for instance, have been initially selected through single-time-point profiling.
Acknowledgments
V.B. was funded by a divisional studentship.
Footnotes
Published ahead of print on 13 February 2009.
REFERENCES
- 1.Allen, J., H. M. Davey, D. Broadhurst, J. K. Heald, J. J. Rowland, S. G. Oliver, and D. B. Kell. 2003. High-throughput classification of yeast mutants for functional genomics using metabolic footprinting. Nat. Biotechnol. 21:692-696. [DOI] [PubMed] [Google Scholar]
- 2.Allen, J., H. M. Davey, D. Broadhurst, J. J. Rowland, S. G. Oliver, and D. B. Kell. 2004. Discrimination of modes of action of antifungal substances by use of metabolic footprinting. Appl. Environ. Microbiol. 70:6157-6165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baev, M. V., D. Baev, A. J. Radek, and J. W. Campbell. 2006. Growth of Escherichia coli MG1655 on LB medium: determining metabolic strategy with transcriptional microarrays. Appl. Microbiol. Biotechnol. 71:323-328. [DOI] [PubMed] [Google Scholar]
- 4.Beckonert, O., H. C. Keun, T. M. Ebbels, J. Bundy, E. Holmes, J. C. Lindon, and J. K. Nicholson. 2007. Metabolic profiling, metabolomic and metabonomic procedures for NMR spectroscopy of urine, plasma, serum and tissue extracts. Nat. Protoc. 2:2692-2703. [DOI] [PubMed] [Google Scholar]
- 5.Bevivino, A., V. Peggion, L. Chiarini, S. Tabacchioni, C. Cantale, and C. Dalmastri. 2005. Effect of Fusarium verticillioides on maize-root-associated Burkholderia cenocepacia populations. Res. Microbiol. 156:974-983. [DOI] [PubMed] [Google Scholar]
- 6.Bino, R. J., R. D. Hall, O. Fiehn, J. Kopka, K. Saito, J. Draper, B. J. Nikolau, P. Mendes, U. Roessner-Tunali, M. H. Beale, R. N. Trethewey, B. M. Lange, E. S. Wurtele, and L. W. Sumner. 2004. Potential of metabolomics as a functional genomics tool. Trends Plant Sci. 9:418-425. [DOI] [PubMed] [Google Scholar]
- 7.Bolten, C. J., P. Kiefer, F. Letisse, J. C. Portais, and C. Wittmann. 2007. Sampling for metabolome analysis of microorganisms. Anal. Chem. 79:3843-3849. [DOI] [PubMed] [Google Scholar]
- 8.Buckstein, M. H., J. He, and H. Rubin. 2008. Characterization of nucleotide pools as a function of physiological state in Escherichia coli. J. Bacteriol. 190:718-726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bundy, J. G., B. Papp, R. Harmston, R. A. Browne, E. M. Clayson, N. Burton, R. J. Reece, S. G. Oliver, and K. M. Brindle. 2007. Evaluation of predicted network modules in yeast metabolism using NMR-based metabolite profiling. Genome Res. 17:510-519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bundy, J. G., T. L. Willey, R. S. Castell, D. J. Ellar, and K. M. Brindle. 2005. Discrimination of pathogenic clinical isolates and laboratory strains of Bacillus cereus by NMR-based metabolomic profiling. FEMS Microbiol. Lett. 242:127-136. [DOI] [PubMed] [Google Scholar]
- 11.Burkovski, A., and R. Kramer. 2002. Bacterial amino acid transport proteins: occurrence, functions, and significance for biotechnological applications. Appl. Microbiol. Biotechnol. 58:265-274. [DOI] [PubMed] [Google Scholar]
- 12.Canelas, A. B., C. Ras, A. ten Pierick, J. C. van Dam, J. J. Heijnen, and W. M. van Gulik. 2008. Leakage-free rapid quenching technique for yeast metabolomics. Metabolomics 4:226-239. [Google Scholar]
- 13.Chiarini, L., A. Bevivino, C. Dalmastri, S. Tabacchioni, and P. Visca. 2006. Burkholderia cepacia complex species: health hazards and biotechnological potential. Trends Microbiol. 14:277-286. [DOI] [PubMed] [Google Scholar]
- 14.Cui, Q., I. A. Lewis, A. D. Hegeman, M. E. Anderson, J. Li, C. F. Schulte, W. M. Westler, H. R. Eghbalnia, M. R. Sussman, and J. L. Markley. 2008. Metabolite identification via the Madison Metabolomics Consortium Database. Nat. Biotechnol. 26:162-164. [DOI] [PubMed] [Google Scholar]
- 15.de Koning, W., and K. van Dam. 1992. A method for the determination of changes of glycolytic metabolites in yeast on a subsecond time scale using extraction at neutral pH. Anal. Biochem. 204:118-123. [DOI] [PubMed] [Google Scholar]
- 16.Ebbels, T. M., H. C. Keun, O. P. Beckonert, M. E. Bollard, J. C. Lindon, E. Holmes, and J. K. Nicholson. 2007. Prediction and classification of drug toxicity using probabilistic modeling of temporal metabolic data: the consortium on metabonomic toxicology screening approach. J. Proteome Res. 6:4407-4422. [DOI] [PubMed] [Google Scholar]
- 17.Fyfe, J. A., and J. R. Govan. 1980. Alginate synthesis in mucoid Pseudomonas aeruginosa: a chromosomal locus involved in control. J. Gen. Microbiol. 119:443-450. [DOI] [PubMed] [Google Scholar]
- 18.Goodacre, R., S. Vaidyanathan, W. B. Dunn, G. G. Harrigan, and D. B. Kell. 2004. Metabolomics by numbers: acquiring and understanding global metabolite data. Trends Biotechnol. 22:245-252. [DOI] [PubMed] [Google Scholar]
- 19.Gschaedler, A., N. Thi Le, and J. Boudrant. 1994. Glucose and acetate influences on the behavior of the recombinant strain Escherichia coli HB 101 (GAPDH). J. Ind. Microbiol. 13:225-232. [DOI] [PubMed] [Google Scholar]
- 20.Gupta, A., M. Dwivedi, G. A. Nagana Gowda, A. Ayyagari, A. A. Mahdi, M. Bhandari, and C. L. Khetrapal. 2005. 1H NMR spectroscopy in the diagnosis of Pseudomonas aeruginosa-induced urinary tract infection. NMR Biomed. 18:293-299. [DOI] [PubMed] [Google Scholar]
- 21.Hacker, J., and J. B. Kaper. 2000. Pathogenicity islands and the evolution of microbes. Annu. Rev. Microbiol. 54:641-679. [DOI] [PubMed] [Google Scholar]
- 22.Hocquette, J. F. 2005. Where are we in genomics? J. Physiol. Pharmacol. 56(Suppl. 3):37-70. [PubMed] [Google Scholar]
- 23.Holmes, E., F. W. Bonner, B. C. Sweatman, J. C. Lindon, C. R. Beddell, E. Rahr, and J. K. Nicholson. 1992. Nuclear magnetic resonance spectroscopy and pattern recognition analysis of the biochemical processes associated with the progression of and recovery from nephrotoxic lesions in the rat induced by mercury(II) chloride and 2-bromoethanamine. Mol. Pharmacol. 42:922-930. [PubMed] [Google Scholar]
- 24.Johnson, D. A., S. G. Tetu, K. Phillippy, J. Chen, Q. Ren, and I. T. Paulsen. 2008. High-throughput phenotypic characterization of Pseudomonas aeruginosa membrane transport genes. PLoS Genet. 4:e1000211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kaderbhai, N. N., D. I. Broadhurst, D. I. Ellis, R. Goodacre, and D. B. Kell. 2003. Functional genomics via metabolic footprinting: monitoring metabolite secretion by Escherichia coli tryptophan metabolism mutants using FT-IR and direct injection electrospray mass spectrometry. Comp. Funct. Genom. 4:376-391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kell, D. B. 2006. Systems biology, metabolic modeling, and metabolomics in drug discovery and development. Drug Discov. Today 11:1085-1092. [DOI] [PubMed] [Google Scholar]
- 27.Kell, D. B., M. Brown, H. M. Davey, W. B. Dunn, I. Spasic, and S. G. Oliver. 2005. Metabolic footprinting and systems biology: the medium is the message. Nat. Rev. Microbiol. 3:557-565. [DOI] [PubMed] [Google Scholar]
- 28.Kell, D. B., and S. G. Oliver. 2004. Here is the evidence, now what is the hypothesis? The complementary roles of inductive and hypothesis-driven science in the post-genomic era. Bioessays 26:99-105. [DOI] [PubMed] [Google Scholar]
- 29.Konstantinidis, K. T., A. Ramette, and J. M. Tiedje. 2006. The bacterial species definition in the genomic era. Philos. Trans. R. Soc. Lond. B Biol. Sci. 361:1929-1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee, R., A. S. Ptolemy, L. Niewczas, and P. Britz-McKibbin. 2007. Integrative metabolomics for characterizing unknown low-abundance metabolites by capillary electrophoresis-mass spectrometry with computer simulations. Anal. Chem. 79:403-415. [DOI] [PubMed] [Google Scholar]
- 31.LiPuma, J. J. 2007. Update on Burkholderia nomenclature and resistance. Clin. Microbiol. Newsl. 29:65-69. [Google Scholar]
- 32.Lu, C. D., Z. Yang, and W. Li. 2004. Transcriptome analysis of the ArgR regulon in Pseudomonas aeruginosa. J. Bacteriol. 186:3855-3861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mackenzie, D. A., M. Defernez, W. B. Dunn, M. Brown, L. J. Fuller, S. R. de Herrera, A. Gunther, S. A. James, J. Eagles, M. Philo, R. Goodacre, and I. N. Roberts. 2008. Relatedness of medically important strains of Saccharomyces cerevisiae as revealed by phylogenetics and metabolomics. Yeast 25:501-512. [DOI] [PubMed] [Google Scholar]
- 34.Maharjan, R. P., and T. Ferenci. 2005. Metabolomic diversity in the species Escherichia coli and its relationship to genetic population structure. Metabolomics 1:235-242. [Google Scholar]
- 35.Mahenthiralingam, E., A. Baldwin, and C. G. Dowson. 2008. Burkholderia cepacia complex bacteria: opportunistic pathogens with important natural biology. J. Appl. Microbiol. 104:1539-1551. [DOI] [PubMed] [Google Scholar]
- 36.Mahenthiralingam, E., J. Bischof, S. K. Byrne, C. Radomski, J. E. Davies, Y. Av-Gay, and P. Vandamme. 2000. DNA-Based diagnostic approaches for identification of Burkholderia cepacia complex, Burkholderia vietnamiensis, Burkholderia multivorans, Burkholderia stabilis, and Burkholderia cepacia genomovars I and III. J. Clin. Microbiol. 38:3165-3173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mahenthiralingam, E., T. A. Urban, and J. B. Goldberg. 2005. The multifarious, multireplicon Burkholderia cepacia complex. Nat. Rev. Microbiol. 3:144-156. [DOI] [PubMed] [Google Scholar]
- 38.Majewski, R. A., and M. M. Domach. 1990. Simple constrained-optimization view of acetate overflow in Escherichia coli. Biotechnol. Bioeng. 35:732-738. [DOI] [PubMed] [Google Scholar]
- 39.Majors, P. D., J. S. McLean, and J. C. Scholten. 2008. NMR bioreactor development for live in-situ microbial functional analysis. J. Magn. Reson. 192:159-166. [DOI] [PubMed] [Google Scholar]
- 40.Mapelli, V., L. Olsson, and J. Nielsen. 2008. Metabolic footprinting in microbiology: methods and applications in functional genomics and biotechnology. Trends Biotechnol. 26:490-497. [DOI] [PubMed] [Google Scholar]
- 41.Mas, S., S. G. Villas-Boas, M. E. Hansen, M. Akesson, and J. Nielsen. 2007. A comparison of direct infusion MS and GC-MS for metabolic footprinting of yeast mutants. Biotechnol. Bioeng. 96:1014-1022. [DOI] [PubMed] [Google Scholar]
- 42.Mercenier, A., J. P. Simon, D. Haas, and V. Stalon. 1980. Catabolism of l-arginine by Pseudomonas aeruginosa. J. Gen. Microbiol. 116:381-389. [DOI] [PubMed] [Google Scholar]
- 43.Nicholson, J. K., J. Connelly, J. C. Lindon, and E. Holmes. 2002. Metabonomics: a platform for studying drug toxicity and gene function. Nat. Rev. Drug Discov. 1:153-161. [DOI] [PubMed] [Google Scholar]
- 44.Ochs, M. M., C. D. Lu, R. E. Hancock, and A. T. Abdelal. 1999. Amino acid-mediated induction of the basic amino acid-specific outer membrane porin OprD from Pseudomonas aeruginosa. J. Bacteriol. 181:5426-5432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Oliver, S. G., M. K. Winson, D. B. Kell, and F. Baganz. 1998. Systematic functional analysis of the yeast genome. Trends Biotechnol. 16:373-378. [DOI] [PubMed] [Google Scholar]
- 46.Palmer, K. L., L. M. Aye, and M. Whiteley. 2007. Nutritional cues control Pseudomonas aeruginosa multicellular behavior in cystic fibrosis sputum. J. Bacteriol. 189:8079-8087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Park, S. M., C. D. Lu, and A. T. Abdelal. 1997. Cloning and characterization of argR, a gene that participates in regulation of arginine biosynthesis and catabolism in Pseudomonas aeruginosa PAO1. J. Bacteriol. 179:5300-5308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pope, G. A., D. A. MacKenzie, M. Defernez, M. A. Aroso, L. J. Fuller, F. A. Mellon, W. B. Dunn, M. Brown, R. Goodacre, D. B. Kell, M. E. Marvin, E. J. Louis, and I. N. Roberts. 2007. Metabolic footprinting as a tool for discriminating between brewing yeasts. Yeast 24:667-679. [DOI] [PubMed] [Google Scholar]
- 49.Raamsdonk, L. M., B. Teusink, D. Broadhurst, N. S. Zhang, A. Hayes, M. C. Walsh, J. A. Berden, K. M. Brindle, D. B. Kell, J. J. Rowland, H. V. Westerhoff, K. van Dam, and S. G. Oliver. 2001. A functional genomics strategy that uses metabolome data to reveal the phenotype of silent mutations. Nat. Biotechnol. 19:45-50. [DOI] [PubMed] [Google Scholar]
- 50.Rahme, L. G., M. W. Tan, L. Le, S. M. Wong, R. G. Tompkins, S. B. Calderwood, and F. M. Ausubel. 1997. Use of model plant hosts to identify Pseudomonas aeruginosa virulence factors. Proc. Natl. Acad. Sci. USA 94:13245-13250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ryan, D., and K. Robards. 2006. Metabolomics: the greatest omics of them all? Anal. Chem. 78:7954-7958. [DOI] [PubMed] [Google Scholar]
- 52.Sariyar-Akbulut, B., A. Salman-Dilgimen, S. Ceylan, S. Perk, A. A. Denizci, and D. Kazan. 2008. Preliminary phenotypic characterization of newly isolated halophilic microorganisms by footprinting: a rapid metabolome analysis. Arch. Microbiol. 189:19-26. [DOI] [PubMed] [Google Scholar]
- 53.Stanisich, V., and B. W. Holloway. 1969. Conjugation in Pseudomonas aeruginosa. Genetics 61:327-339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Steffes, C., J. Ellis, J. Wu, and B. P. Rosen. 1992. The lysP gene encodes the lysine-specific permease. J. Bacteriol. 174:3242-3249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tabacchioni, S., L. Ferri, G. Manno, M. Mentasti, P. Cocchi, S. Campana, N. Ravenni, G. Taccetti, C. Dalmastri, L. Chiarini, A. Bevivino, and R. Fani. 2008. Use of the gyrB gene to discriminate among species of the Burkholderia cepacia complex. FEMS Microbiol. Lett. 281:175-182. [DOI] [PubMed] [Google Scholar]
- 56.Tamber, S., and R. E. Hancock. 2003. On the mechanism of solute uptake in Pseudomonas. Front. Biosci. 8:s472-s483. [DOI] [PubMed] [Google Scholar]
- 57.Van Dyk, T. K., L. J. Templeton, K. A. Cantera, P. L. Sharpe, and F. S. Sariaslani. 2004. Characterization of the Escherichia coli AaeAB efflux pump: a metabolic relief valve? J. Bacteriol. 186:7196-7204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Viant, M. R., J. G. Bundy, C. A. Pincetich, J. S. de Ropp, and R. S. Tjeerdema. 2005. NMR-derived developmental metabolic trajectories: an approach for visualizing the toxic actions of trichloroethylene during embryogenesis. Metabolomics 1:149-158. [Google Scholar]
- 59.Villas-Boas, S. G., and P. Bruheim. 2007. Cold glycerol-saline: the promising quenching solution for accurate intracellular metabolite analysis of microbial cells. Anal. Biochem. 370:87-97. [DOI] [PubMed] [Google Scholar]
- 60.Wang, D., X. Ding, and P. N. Rather. 2001. Indole can act as an extracellular signal in Escherichia coli. J. Bacteriol. 183:4210-4216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Westerhuis, J. A., T. Kourti, and J. F. MacGregor. 1998. Analysis of multiblock and hierarchical PCA and PLS models. J. Chemometrics 12:301-321. [Google Scholar]
- 62.Winder, C. L., W. B. Dunn, S. Schuler, D. Broadhurst, R. Jarvis, G. M. Stephens, and R. Goodacre. 2008. Global metabolic profiling of Escherichia coli cultures: an evaluation of methods for quenching and extraction of intracellular metabolites. Anal. Chem. 80:2939-2948. [DOI] [PubMed] [Google Scholar]