

Abstract
Multiple sclerosis (MS) is a common inflammatory disease of the central nervous system with a poorly defined and complex immunopathogenesis. Although initiated by reactive T cells, persistent inflammation is evident throughout the disease course. A contribution from complement has long been suspected, based on the results of pathological and functional studies which have demonstrated complement activation products in MS brain and biological fluids. However, the extent and nature of complement activation and its contribution to disease phenotype and long-term outcome remain unclear. Furthermore, functional polymorphisms in components and regulators of the complement system which cause dysregulation, and are known to contribute to other autoimmune inflammatory disorders, have not been investigated to date in MS in any detail. In this paper we review evidence from pathological, animal model and human functional and genetic studies, implicating activation of complement in MS. We also evaluate the potential of complement components and regulators and their polymorphic variants as biomarkers of disease, and suggest appropriate directions for future research.
Keywords: biomarker, complement system, encephalomyelitis, inflammation, multiple sclerosis
Introduction
Multiple sclerosis (MS) is the most common cause of neurological disability in young adults in northern European Caucasian populations, with an approximate lifetime risk of one in 400 [1]. Its exact aetiology remains unclear, but it is likely that diverse immunological factors interact with those from genes and the environment in individual patients to contribute to its characteristically variable pathology, phenotypic presentation, disease course and outcome [1,2]. The precise impact of these components of disease aetiology on clinical features is not well understood and largely unpredictable at disease onset [3], which creates considerable problems in disease management. Objective evidence from simple, easily repeatable, sensitive biomarkers are largely unavailable but would be invaluable in providing significant help to clinicians with diagnosis and assessment of prognosis, as well as the monitoring of treatment response in this complex disorder.
The fundamental concept underlying the perceived pathogenesis of MS is of wide arrays of inflammatory mediators interacting to cause both demyelination and axonal damage which, in turn, results in a variety of neurological deficits [4] (Fig. 1). However, this simplified overview may not explain entirely the pathogenic mechanism of progressive disease, attributed originally to multiple acute inflammatory events causing chronic axonal degeneration, and considered more recently to be an independent degenerative process possibly secondary to low levels of chronic background inflammation [5].
Fig. 1.
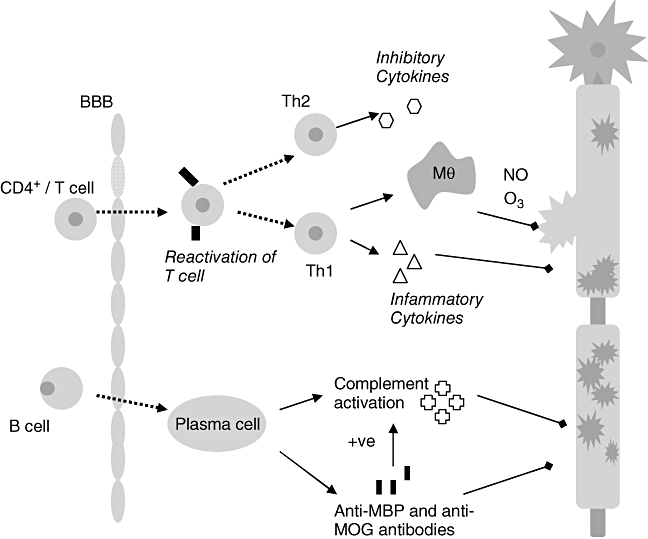
The pathogenic cascade in multiple sclerosis (MS): the initiation of inflammation central to the pathogenesis of MS depends on the peripheral activation of T cells via foreign antigens. Activated T cells express integrins which bind to up-regulated cell adhesion molecules on the blood–brain barrier (BBB) endothelial surface allowing diapedesis of T cells into the central nervous system (CNS). Within the CNS T cells re-encounter specific antigen and are reactivated setting up an inflammatory cascade which results in conduction block and demyelination. Myelin and axon damage is dependent on various effector mechanisms; the extent of involvement of each is thought to vary between individuals. Activated T cells bind directly to myelin epitopes leading to activation of macrophages (Mθ) which can phagocytose myelin directly or, along with T cells and glial cells, can release cytotoxic products and mediators such as proteases [122], inflammatory cytokines, e.g. tumour necrosis factor-α, interferon-γ[123], soluble reactive oxygen (03) and nitrogen (NO) intermediates [124,125], free radicals and glutamic acid. Further attack to myelin comes from antibodies and complement components – mainly the terminal complement complex – C5b–9 [126] triggered principally by infiltrating B cells and myelin antibodies.
Understanding of the role of complement (C) in the central nervous system (CNS) and in systemic inflammatory disorders is now evolving at a considerable pace. Although first suggested in the early 1970s to play a role in the pathogenesis of MS [6], the extent of C activation in subgroups of MS patients, the mechanisms of activation and the role of C in inflammation, axonal damage and neuroprotection remain unclear. In particular, the involvement of inflammatory mediators, including C, to the progressive phase of disease caused by axonal injury remains a point for debate and, without good animal models of axonal injury, remains difficult to investigate.
In part because of limited access to pathological human tissue, the animal model of MS, experimental autoimmune encephalomyelitis (EAE), has proved useful in developing insights into the role of C in MS. Indeed, in a recent review it has been suggested that animal models of MS may provide the best and broadest understanding of underlying disease processes and should be the primary focus of research [7]. However, there are many well-rehearsed problems in extrapolating models of animal disease to the human context, and pathological, functional and genetic studies in man will continue to provide a valuable contribution that should not be overlooked.
Although C activation is not specific to MS, patterns of activation, assessed in combination with other inflammatory and immune markers, may be of value as biomarkers in MS subgroups. Here we review the evidence from pathological work, animal model studies and human functional and genetic studies, implicating C in the pathogenesis of MS, and discuss the use of C activation products, components, regulators and their polymorphic variants as biomarkers of disease.
Complement activation and regulation
Complement plays a central role in the innate immune system, providing an important defence against infection and immune complex disease. The system consists of approximately 30 circulating and membrane expressed proteins which collaborate to provide protection from infection [8,9]. The physiological actions of C are mediated through production of opsonins (molecules that enhance the ability of macrophages and neutrophils with C receptors to phagocytose material – C3b, iC3b, C4b, etc.), anaphylatoxins (peptides that induce local and systemic inflammatory responses, increasing the permeability of blood vessels and attracting neutrophils through their chemotactic properties – C3a, C4a and C5a) and through direct killing of organisms by the terminal complement complex (TCC/C5b–9) which disrupts and forms pores in the phospholipid bilayer of a target cell. Although the C components in plasma are synthesized mainly by hepatocytes in the liver, within the CNS it is clear that glial cells and neurones can produce the majority of C proteins and expression is increased in response to inflammation [10–12].
The C system is activated via three initiating pathways (classical, alternative and lectin), all of which converge on a common effector pathway with formation of the TCC (Fig. 2). Inappropriate activation of C leading to depletion of the C components is normally prevented by C inhibitors present either in the fluid-phase or membrane-bound [13,14] (Fig. 2). Regulation in the alternative pathway is particularly important because the pathway is characterized by a constant ‘tick-over’ activation of C3, which can be amplified in pathology. Regulation in plasma is provided by factor H (fH) and deficiency of fH allows unregulated amplification with breakdown of C3 leading to C3 deficiency.
Fig. 2.
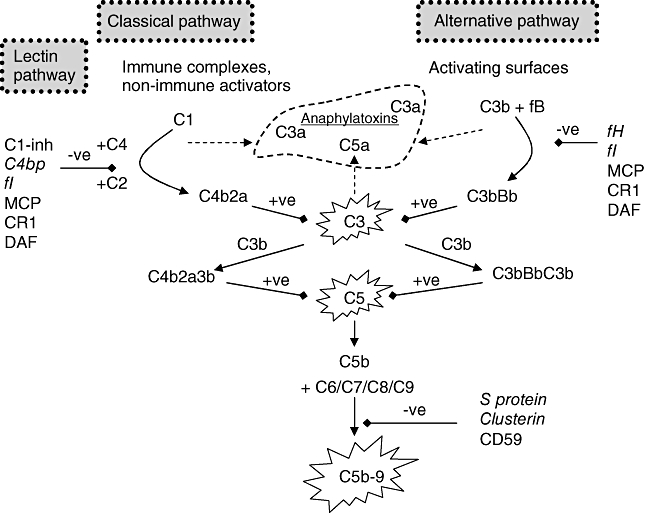
Activation and regulation of the complement (C) system: activation through both classical and alternative pathways results in the formation of C5 convertases, cleaving C5 and eventually forming the membrane attack complex (C5b–9). They also result in the formation of anaphylatoxins which cause inflammation and chemotaxis. The C system is under tight regulation by soluble (in italics) and membrane-bound (in bold) proteins (C1-inh, C1 inhibitor; C4bp, C4 binding protein; fI, factor I; MCP, membrane co-factor protein; CR1, C receptor 1; DAF, decay accelerating factor; fH, factor H; fB, factor B).
Humoral immunity in neuromyelitis optica and MS
Neuromyelitis optica (NMO) is an inflammatory demyelinating disease affecting predominantly the optic nerves and spinal cord. Traditionally seen as a variant of MS, it has been redefined recently according to new criteria using a combination of phenotypic subtyping along with a newly developed biomarker of disease, NMO-immunoglobulin G (IgG) (reported sensitivity of 58–76% and specificity of 85–99% for NMO) [15,16]. The clinical differences between NMO and MS are mirrored by pathological differences. NMO is typified by abundant Ig and C deposits surrounding blood vessels in a distinctive rim or rosette fashion (corresponding with aquaporin-4 expression) [17], and is thought to be driven by humoral processes [18]. Therapeutic plasma exchange, known to deplete plasma proteins non-selectively, has been reported as having a beneficial role in NMO [19,20] and is thought to exert its therapeutic effect by reducing circulating levels of relevant autoantibodies.
Until recently, studies of MS pathogenesis and treatment have focused on the role of T cells; however, it is now clear that humoral immune mechanisms also play an important role which has been reassessed recently [21,22]. Circumstantial evidence for a B cell contribution to disease came initially from the demonstration of central clonal expansion of B cells and production of oligoclonal cerebrospinal fluid IgG (used routinely as a diagnostic biomarker in MS) [23]. More recently, several studies have demonstrated antibodies to myelin and other CNS autoantigens in MS patients but without convincing disease specificity, and the specific antigens responsible for initiating the immune response in MS remain unidentified [24]. Although Ig and C deposits are described in MS lesions, the localization of infiltrate is different from that in NMO [25] and has been suggested to be present only in a histological subgroup of patients (pattern II lesions) [26].
Therapies targeting humoral mechanisms, such as plasma exchange [19,27,28] and rituximab [a B cell-depleting monoclonal antibody (mAb)][29], have been tested with variable success in acute demyelinating events and it has been suggested that patients with pattern II histological lesions, showing prominent Ig and C deposition, are more likely to have a favourable response to this intervention [30]. The recognition of specific biomarkers for MS patients with extensive C activation may therefore be important in identifying patients who may benefit most from humoral-mediated therapies [31].
Complement in MS – evidence from pathology
A role for C in MS was proposed initially following the demonstration of C component C3 deposited in the brains of MS patients [6]. Immunocytochemical localization of the TCC in five of seven MS patients was reported in 1989; TCC was demonstrated in association with capillary endothelial cells, predominantly within plaques and adjacent white matter [32]. Localization of C activation to areas of active myelin destruction was demonstrated in a case report showing TCC deposited exclusively in such areas [33]. In a study of 23 cases of secondary progressive MS, the opsonin C3d was shown deposited in association with short segments of disrupted myelin in plaques with low-grade active demyelination and provided evidence for a C contribution to disease progression as well as acute inflammation [34].
More recently a complex and heterogeneous pathological model for MS has been proposed by Luchinetti et al., who have described four distinct patterns of demyelination in a group of 83 patients, with specimens collected either at autopsy (n = 32) or biopsy (n = 51) [26,35]. The pathological classification was based mainly on the presence of Ig and activated C (pattern II only), remyelination (patterns I and II) and primary oligodendrocyte (OGD) damage (patterns III and IV). The authors suggested that the four different patterns of disease provided evidence for different mechanisms of immune-mediated demyelination, with C playing a role in a distinct but large subgroup of patients.
When considering the pathological subgroups and their relation to disease phenotype, it must noted that the frequencies of the different patterns seen may be skewed heavily by patient selection for biopsy (possibly more aggressive disease). Further, the specimens collected in the study by Luchinetti et al. were from a cohort of patients with very early disease (the mean disease duration before autopsy or biopsy was 39 and 9 months respectively) and therefore may not represent accurately a global picture of MS. In addition, the study showed C and Ig depositions only in pattern II lesions, represented exclusively in more than 50% of the patients and in all subgroups of disease (relapsing–remitting, secondary and primary progressive). However, a more recent study has failed to demonstrate this heterogeneity, showing C and Ig deposition present consistently in areas of demyelination [25]. These authors have suggested that the heterogeneity described by Luchinetti et al. may have been a phenomenon of early disease and was therefore not seen in their samples, in which the mean disease duration was 22 years.
Serum or CSF biomarkers that identify any pathological subgroups accurately would help considerably with clarifying these issues and would provide direction for future research.
Complement in MS – animal models
The EAE, most commonly in rodents, has long been used as an animal model for MS [36,37], but was constructed initially to examine the monophasic illness: acute disseminated encephalomyelitis [38]. However, since its introduction numerous variations to the initial concept have evolved. Disease induction with a known CNS antigen [such as myelin basic protein (MBP) [39], proteolipoprotein [40], myelin oligodendrocyte glycoprotein (MOG), myelin-associated glycoprotein and S-100 protein [41]] causes paralysis in animals with varying degrees of demyelination depending on the method of EAE induction and species used [42]. The two main models employed are the MBP model (resulting in a monophasic, non-demyelinating disease) and the antibody-mediated demyelinating EAE model (ADEAE) (where MBP immunization is followed by administration of anti-MOG antibodies). Although the MBP-induced model of EAE is thought by some to bear the closest resemblance to MS, it is likely that different models will have relevance in the understanding of different immunopathological subtypes or phases of disease. Information gained from these studies must be tempered by the knowledge that EAE, like most animal models of disease, is an incomplete and not entirely accurate reflection of the human situation. Models reflecting the axonal damage seen in MS have still to be used to analyse disruption of the CNS, but may yet provide a clearer understanding of progressive disease [43]. Despite all these problems, much information on C pathology, potential C biomarkers and therapeutics has been obtained from EAE studies in normal animals and transgenic mouse models [44], and these models have provided a test bed for studies of treatment with C suppressors [31].
Anti-C agents in EAE
The first studies to examine the role of C in EAE used the C activator cobra venom factor (CVF) as a treatment to reduce serum C, reducing the severity of EAE markedly [45–48]. Despite initial success, it was soon realized that response to this treatment was transient and repeated injections were highly immunogenic; further, CVF-induced C activation led to production of vast quantities of the proinflammatory anaphylatoxins C3a and C5a, sufficient to cause a shock syndrome in recipients. It was also reported that, although CVF treatment in MBP-induced EAE caused suppression of disease, in the ADEAE model CVF had no effect on disease severity despite C9 deposition being abolished [49].
To combat the problems of CVF, soluble C receptor 1 (sCR1) (which blocks the action of C3 and C5 convertases) was trialled as a treatment to decrease C activity and reduce the severity of EAE [50]. Treatment with sCR1 worked to some extent, but showed much less reduction in clinical severity scores when compared with treatment with CVF, perhaps reflecting an incomplete blockade of C activation [51].
Gene deletion models in EAE
Gene-deleted (knock-out) animal models have been used to enhance our understanding of the roles of the pathways of C activation in EAE. A study of C3 and factor B (fB) knock-out mice (the former unable to activate C, the latter alternative pathway-deficient) demonstrated normal incidence of MOG-induced EAE but attenuated severity of chronic disease in both mouse strains, with decreased numbers of infiltrating macrophages and T cells [52]. This work showed unequivocally that C contributed to the development of EAE, and also that the alternative pathway was vital in the development of disease. The role of C3 has since been questioned, with C3 knock-out mice showing similar disease to wild-type mice in one study [53], but attenuated disease in others [54,55]. The differences here may be explained by the use of different mouse models. Evidence that the alternative pathway is the main player is supported by studies in C4-deficient (classical pathway absent) animals, which showed EAE onset and progression virtually identical to wild-type animals [56,57].
Once activated, C has multiple roles in the EAE inflammatory lesion, causing demyelination [58], axonal damage and OGD injury [45]. The mechanisms by which this damage is mediated have been examined in detail. The membrane attack complex (MAC), the cytolytic end-product of the C cascade, is thought to play a significant role in demyelination, with evidence from pathological studies (as discussed), in vitro studies (deficiency of the MAC inhibitor CD59 was shown to be the cause of rat OGD susceptibility to lysis in culture [59] and in vivo studies. C9/MAC deposition was shown to correlate closely with demyelination in the spinal cord of rats in which EAE was induced with anti-MOG antibodies [58], while C6-deficient rats (unable to produce MAC), when immunized with MBP and given anti-MOG antibodies to induce ADEAE, failed to develop demyelination, axonal damage or paralysis, but when reconstituted with C6 went on to develop pathology and clinical disease similar to wild-type rats [60]. Despite these findings, the role of MAC has been questioned [54]; C5 knock-out mice were not protected from MBP-induced EAE, suggesting that the terminal pathway was not essential for disease [61,62]. The extent of the role of MAC in demyelination and remyelination in models and MS thus remains uncertain.
Anaphylatoxins C3a and C5a are important inflammatory mediators and were considered initially to play an important role in CNS inflammation, based predominantly on evidence showing the expression of their receptors (C3aR and C5aR) on neuronal cells [63]. C3aR expression on microglia and infiltrating macrophages was increased during EAE but neuronal expression was largely unchanged [64]. The expression of C5aR on microglia and hypertrophic astrocytes within the spinal cords of Lewis rats was highly up-regulated in EAE [65], leading to the hypothesis that C5a is an important inflammatory mediator in EAE. Since these initial studies, it has been demonstrated that C5a plays no role in demyelination in EAE; C5aR-deficient animals are fully susceptible to EAE [66], and treatment with a C5aR antagonist failed to protect against development of EAE [67]. In contrast to this, C3a-deficient mice in which EAE was induced exhibited an attenuated clinical course, and immunohistochemical analysis revealed reduced demyelination and infiltration of macrophages and T cells compared with control mice, indicating that anaphylatoxin C3a plays a role in inflammatory cell recruitment and demyelination [68]. Interestingly, these effects were more apparent in the chronic phase of disease, corresponding with results from C3-deficient mice [52].
Summary
Animal models have been useful in clarifying the role of C in EAE. It is clear that the alternative pathway contributes to disease while the roles of the classical and terminal pathways are less clear, with the latter reported to be vital for demyelination in some, but not all, EAE models. The anaphylatoxin C5a probably contributes little to disease, while C3a plays a larger part – predominantly through recruitment of inflammatory cells. The alternative pathway plays a dominant role in EAE, as has been shown in many other disease models [69,70], impacting particularly in chronic disease. While direct translation of these findings into human disease is not possible, they may suggest that the alternative pathway is important in disease perpetuation after initiation by a primary source. The roles of C in mediating axonal damage need to be investigated further and require animal models that represent axonal damage and chronic disease more clearly.
Complement as a serological or CSF biomarker of MS
In light of the evidence implicating C in MS, various C proteins have been considered as biomarkers of disease activity. These are summarized in Table 1 and discussed below.
Table 1.
Studies examining concentrations of complement (C) components in multiple sclerosis (MS) patients compared with control subjects.
| Reference | C component measured | Sample type | Significant alteration in concentration compared with controls |
|---|---|---|---|
| Kuwert et al. 1965 [127] | C3/C4 | CSF | Reduced |
| Link 1972 [128] | C3/C4 | CSF | No difference |
| Dube et al. 1973 [129] | C3/C4 | CSF | Increased |
| Yam et al. 1980 [130] | C3/C4 | CSF | C3: increased |
| C4: no difference | |||
| Price et al. 1980 [131] | C3/C4 | CSF | In relapse: no difference |
| In remission: decreased | |||
| Jans et al. 1984 [71] | C3/C4 | CSF/serum | Serum and CSF C3: no difference |
| Serum C4: increased (P = 0·05) | |||
| CSF C4: reduced (P < 0·01) | |||
| Jongen et al. 2000 [72] | C3/C4 | CSF/serum | Serum C3: reduced in RRMS and SPMS |
| CSF C3: reduced in RRMS | |||
| Serum and CSF C4: no difference | |||
| Sellebjerg et al. 1998 [76] | C3 and TCC | CSF/plasma | C3: no difference |
| CSF TCC: increased and levels correlated with disability (P = 0·003) | |||
| Morgan et al. 1984 [73] | C9 | CSF/plasma | Reduced in CSF |
| No difference in plasma | |||
| Compston et al. 1986 [132] | C9 | CSF/plasma | Reduced in CSF |
| No difference in plasma | |||
| Sanders et al. 1986 [78] | TCC | CSF | Increased |
| Mollnes et al. 1987 [77] | TCC | CSF/serum | Increased in CSF |
| No difference in serum | |||
| Rodriguez et al. 1990 [74] | C9 | CSF | No difference |
| Halawa et al. 1989 [75] | C9 | CSF | No difference |
CSF, cerebrospinal fluid; RRMS, relapsing–remitting multilpe sclerosis; SPMS, secondary progressive multiple sclerosis; TCC, terminal complement complex.
C3 and C4
C3 and C4 are the most abundant of the C proteins and are measured routinely in many laboratories. Like most of the C components, both C3 and C4 are acute-phase reactants and increased synthesis in response to inflammation can mask even quite marked consumption. Perhaps, as a consequence, studies of C3 and C4 levels in MS serum and CSF have shown inconsistent and conflicting results [71,72]. Levels in serum and CSF from patients with neuroinflammatory diseases are likely to be influenced both by the presence of active inflammation and the integrity of the blood–brain barrier, both highly fluctuant in MS. Indeed, positive correlation of C3 and C4 CSF : serum ratios with albumin CSF : serum ratios has been identified in controls but not in MS patients [72].
Terminal pathway components and the TCC
Measurements of the terminal component, C9, and of the TCC have been performed in MS, although results have again been inconsistent. Low levels of C9 in MS CSF have been demonstrated in two studies, including one in which patients were subclassified into acute relapsing, remission and progressive with lowered C9 levels in all groups, implicating prior activation of the terminal C pathway [32,73]. Other studies have shown no difference in C9 levels compared with controls [74,75]. Measurement of TCC in CSF in three studies has consistently shown raised levels in MS patients [76–78], one study showing good correlation of CSF TCC concentration with disability [76]. There were no changes in C9 or C5b–9 levels in plasma in any of these studies, suggesting that CSF changes were a much better indicator of CNS disease.
Complement regulators
To date, little work has been conducted looking specifically at C regulators in MS. A recent study using proteomic analyses to examine C proteins in MS CSF found significant reduction in one unspecified isoform of fB, and of the alternative splice product of the fH gene, fHL-1, in MS CSF [79]. The authors suggested that measurement of specific C component and regulator protein isoforms would be most informative as biomarkers. Although there have been no follow-up studies, this work highlights the potential of C measurements as biomarkers in MS.
In one study, antibodies to two membrane C regulators (CD46 and CD59) were identified in MS serum, present in the acute phase of relapsing–remitting MS but not in chronic MS or control groups [80]. The antibodies were directed against the active site of the C regulator, inactivating their regulatory function and leading to excess activation of C. The authors suggest this as a mechanism for damage caused during acute relapse, but so far no one has either replicated this very small study, performed with less than 20 patients in each subgroup, or investigated these antibodies as possible biomarkers of disease. Nevertheless, the study highlights the need for the subclassification of MS on clinical grounds at the point of study design.
Summary
Although showing some variation in MS, C components and activation products have not been established as biomarkers of disease, in part because results obtained from different studies measuring the same C component have often been conflicting. These differences could be due to lack of standard techniques of measurement or to low patient numbers leading to underpowered studies; however, universally there has been poor stratification of study groups according to phenotype. In a relapsing immunologically heterogeneous inflammatory disease, clinical phenotype (such as aggressive or benign disease or intercurrent relapse) is likely to have a large effect on levels of C components. It is the view of the authors that much information can be gained from further analysis of C components and regulators in serum, plasma and CSF employing correlation of levels with clinical phenotype and other modalities of disease activity and severity such as magnetic resonance imaging.
Role of C in neuroprotection in MS
Neuroprotection from sublytic MAC
The neurotoxic effects of MAC are concentration-dependent and it has been suggested that partial deficiency of terminal components has evolutionary benefits [81–83]. Several groups have shown that a sublytic dose of MAC can be cytoprotective, with prior exposure leading to accumulated resistance to a subsequent lytic attack [84,85].
In one study in EAE, C5-deficient mice showed greater inflammatory demyelination and axonal loss [61], with more OGD apoptosis compared with controls [62]. Studies in vitro suggested strongly that this was a consequence of sublytic levels of MAC conferring protection from OGD apoptosis [86–89].
It is known that OGD apoptosis is associated with activation of caspases 3, 8, 9 and 11 [88–91] and inhibition with caspase-3 and -8 inhibitors can prevent OGD apoptosis in vitro[89]. Sublytic MAC has been shown in vitro to inhibit cytochrome c activation of caspase-3 and -9 and induce phosphorylation of BAD which, when phosphorylated, can bind to cytoplasmic 14-3-3 protein and increase cell survival [86]. Sublytic MAC has also be shown to inhibit Fas ligand and tumour necrosis factor-α-induced OGD apoptosis by inhibition of caspase-8 processing [87,89]. This provides in vitro and in vivo evidence of neuroprotection mediated by low levels of MAC, acting primarily via regulation of caspases [92,93].
Neuroprotection by anaphylatoxins
In vitro studies of anaphylatoxins C3a and C5a have shown potential neuroprotective effects. Pretreatment of primary murine corticohippocampal neurones with human or mouse recombinant C5a reduced glutamate neurotoxicity and prevented glutamate-induced neuronal apoptosis [94]; these events were signalled through mitogen-activated protein kinase-mediated regulation of caspase cascades [95]. C3a was neuroprotective against N-methyl-d-aspartate toxicity in the presence of astrocytes, but did not protect against serum deprivation-induced apoptotic neuronal death, or α-amino-3-hydroxy-5-methyl-4-isoxazolepropionate/kainate-mediated excitotoxicity [96]. In animal models, C5a receptor-deficient mice were more susceptible to apoptotic injury in vivo because of increased excitotoxicity [97].
Genetic findings implicating C in MS
Epidemiological studies have long recognized the important role of genetic factors in susceptibility to MS, with a number of largely unidentified genes contributing to disease risk [98–100]. More recently, genome-wide association studies have confirmed that single nucleotide polymorphisms (SNPs) in inflammatory cytokine receptors interleukin (IL)-2Rα and IL-7R are associated with susceptibility to MS [101–103].
A more significant role for genetic variability influencing C in MS may be signalled by recent findings in other CNS disorders with a significant inflammatory component. Studies in age-related macular degeneration (AMD) have recently identified a SNP in the locus encoding the C regulator fH (Y402H); homozygosity for the minor allele conferred a sixfold increased risk of disease [104–106]. This common polymorphism is located in short consensus repeat 7, a region implicated in binding multiple ligands, and has been suggested to alter affinity of fH for surfaces, thereby influencing capacity to inhibit C [107]. Other identified polymorphisms in the alternative pathway component fB and the classical pathway component C2 may contribute further to disease risk [108]. These minor changes in the C proteins causing subtle dysregulation in C have been shown to be associated strongly with the disease and are suggested to be global markers for AMD [109]. The Y402H polymorphism in fH described in AMD has also been identified as a risk allele for Alzheimer's disease (AD) in some, but not all, studies [110,111].
Given the role of C in neuroinflammation and the reports of C polymorphisms contributing to other inflammatory conditions, a candidate gene approach in MS to identify association with C components and regulators would seem logical. Although no studies have yet identified susceptibility genes within C components or receptors (Table 2), evidence for linkage in MS in the region around the centromere of chromosome 5 (containing genes for C components C6, C7 and C9) has been identified in four independent linkage studies [112–115].
Table 2.
Studies examining complement (C) component and regulator single nucleotide polymorphisms (SNPs) in relation to multiple sclerosis (MS) disease susceptibility.
| Study | Gene tested | No. SNPs tested from gene | Cohort of patients | Result |
|---|---|---|---|---|
| Chataway et al. 1999 [133] | C6 | 1 | 227 multiplex families | No association |
| C7 | 3 | 466 affected siblings | No distortion of transmission | |
| 112 unaffected siblings | ||||
| Simon et al. 2007 [134] | CR2 | 9 | Not disclosed | No association |
| Bulman et al. 1991 [135] | C3 | 1 allele | 129 patients | No association |
| 69 controls | ||||
| Unpublished | FH | 3 | 480 patients | No association |
All the studies conducted so far looking at candidate genes have been underpowered to detect significant differences where the risk proportioned to each identified allele is very small (odds ratios < 1·3) [116], therefore negative studies can not be deemed reliable. Also, work to date has focused on disease susceptibility risk and has not examined phenotype, which may be more likely to be altered by C dysregulation. Further studies with large groups of patients are needed in order to uncover genetic biomarkers for MS within the C system, and to direct functional studies.
Complement activation and dysregulation in neurological disorders
Complement has been implicated firmly in the pathology of a number of diseases of the CNS and its role in NMO and AMD has been discussed. In addition, there have been reports of the alternative pathway regulator fH being a marker in AD, elevated significantly in AD patients compared with controls [117]. Further evidence of a role for fH in AD pathology has been provided by Honda and co-workers, who developed a mAb that specifically stained senile plaques and demonstrated subsequently that the mAb bound fH, present both in the senile plaques and in CSF in AD [118]. The Y402H polymorphism in fH described in AMD has also been identified as a risk allele for AD in some, but not all, studies [110,111]. Further genetic and functional work is being conducted to establish this C regulator as a clinically useful biomarker in AD.
Complement activation is also described in several other CNS conditions, including Guillain–Barré syndromes in which antibody and C-mediated mechanisms have been shown to be important in pathogenesis [119], and ischaemia–reperfusion injuries such as trauma and stroke in which C inhibition has been shown to be neuroprotective [120,121] (reviewed in [10]. These disorders demonstrate how, although not being the primary pathogenic factor, C dysregulation can contribute significantly to disease in the presence of other disease-initiating mechanisms.
Conclusion
This review summarizes compelling evidence from pathological, animal model and functional studies that C plays an important role in the immunopathogenesis of MS. Prior understanding of the immunology of MS and knowledge gained from animal studies leads us to conclude that C does not initiate disease, but propagates ongoing disease with increased contribution over the course of the illness. Severity may vary between individuals according to the extent of C dysregulation, contributed to possibly by functional polymorphisms in C components and regulators. Despite detailed information from animal model studies regarding the activation and regulation of C in demyelination, questions still remain as to the role of the terminal pathway, the extent of the neuroprotective function of C in disease, the role of C in axonal damage and the translation of these studies into the more complex human disease of MS.
This review has highlighted the importance of clinical classification of disease subgroups at the point of study design for functional and genetic work. In particular, studies looking at concentrations of C in serum and CSF have often been complicated by the disease heterogeneity in which acute relapse, stable and chronic disease are highly likely to have distinctly varying C contributions. In recent years advances in technology, in particular the use of proteomic and genetic analytical techniques, have led to better opportunities for finding sensitive and specific biomarkers of disease, as proven in other complex disorders such as AD, AMD and NMO. Use of these techniques, along with better clinical stratification, will bring us closer to finding useful biomarkers for MS; however, given the genetic, pathological, immunological and clinical heterogeneity of MS, it is highly unlikely there will be a single effective surrogate biomarker for disease and a combination of markers, possibly including C components and polymorphisms, are more likely to be informative.
References
- 1.Compston A, Coles A. Multiple sclerosis. Lancet. 2002;359:1221–31. doi: 10.1016/S0140-6736(02)08220-X. [DOI] [PubMed] [Google Scholar]
- 2.McFarland HF, Martin R. Multiple sclerosis: a complicated picture of autoimmunity. Nat Immunol. 2007;8:913–19. doi: 10.1038/ni1507. [DOI] [PubMed] [Google Scholar]
- 3.Hirst C, Ingram G, Swingler R, et al. Change in disability in patients with multiple sclerosis: a 20-year prospective population-based analysis. J Neurol Neurosurg Psychiatry. 2008;79:1137–43. doi: 10.1136/jnnp.2007.133785. [DOI] [PubMed] [Google Scholar]
- 4.Compston A. The pathogenesis and basis for treatment in multiple sclerosis. Clin Neurol Neurosurg. 2004;106:246–8. doi: 10.1016/j.clineuro.2004.02.007. [DOI] [PubMed] [Google Scholar]
- 5.Dutta R, Trapp BD. Pathogenesis of axonal and neuronal damage in multiple sclerosis. Neurology. 2007;68(Suppl.)(3):S22–31. doi: 10.1212/01.wnl.0000275229.13012.32. discussion S43–54. [DOI] [PubMed] [Google Scholar]
- 6.Lumsden CE. The immunogenesis of the multiple sclerosis plaque. Brain Res. 1971;28:365–90. doi: 10.1016/0006-8993(71)90052-7. [DOI] [PubMed] [Google Scholar]
- 7.Barnum SR, Szalai AJ. Complement as a biomarker in multiple sclerosis. J Neuropathol Exp Neurol. 2005;64:741. doi: 10.1097/01.jnen.0000175643.57245.08. [DOI] [PubMed] [Google Scholar]
- 8.Walport MJ. Complement. Second of two parts. N Engl J Med. 2001;344:1140–4. doi: 10.1056/NEJM200104123441506. [DOI] [PubMed] [Google Scholar]
- 9.Walport MJ. Complement. First of two parts. N Engl J Med. 2001;344:1058–66. doi: 10.1056/NEJM200104053441406. [DOI] [PubMed] [Google Scholar]
- 10.Gasque P, Dean YD, McGreal EP, et al. Complement components of the innate immune system in health and disease in the CNS. Immunopharmacology. 2000;49:171–86. doi: 10.1016/s0162-3109(00)80302-1. [DOI] [PubMed] [Google Scholar]
- 11.Levi-Strauss M, Mallat M. Primary cultures of murine astrocytes produce C3 and factor B, two components of the alternative pathway of complement activation. J Immunol. 1987;139:2361–6. [PubMed] [Google Scholar]
- 12.Morgan BP, Gasque P. Expression of complement in the brain: role in health and disease. Immunol Today. 1996;17:461–6. doi: 10.1016/0167-5699(96)20028-f. [DOI] [PubMed] [Google Scholar]
- 13.Morgan BP, Meri S. Membrane proteins that protect against complement lysis. Springer Semin Immunopathol. 1994;15:369–96. doi: 10.1007/BF01837366. [DOI] [PubMed] [Google Scholar]
- 14.Morgan BP, Harris CL. Regulation in the complement system. Complement Regul Proteins. 1999:32–40. [Google Scholar]
- 15.Giovannoni G. Neuromyelitis optica and anti-aquaporin-4 antibodies: widening the clinical phenotype. J Neurol Neurosurg Psychiatry. 2006;77:1001–2. doi: 10.1136/jnnp.2006.090944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wingerchuk DM, Lennon VA, Pittock SJ, et al. Revised diagnostic criteria for neuromyelitis optica. Neurology. 2006;66:1485–9. doi: 10.1212/01.wnl.0000216139.44259.74. [DOI] [PubMed] [Google Scholar]
- 17.Jarius S, Paul F, Franciotta D, et al. Mechanisms of disease: aquaporin-4 antibodies in neuromyelitis optica. Nat Clin Pract Neurol. 2008;4:202–14. doi: 10.1038/ncpneuro0764. [DOI] [PubMed] [Google Scholar]
- 18.Wingerchuk DM. Evidence for humoral autoimmunity in neuromyelitis optica. Neurol Res. 2006;28:348–53. doi: 10.1179/016164106X98260. [DOI] [PubMed] [Google Scholar]
- 19.Keegan M, Pineda AA, McClelland RL, et al. Plasma exchange for severe attacks of CNS demyelination: predictors of response. Neurology. 2002;58:143–6. doi: 10.1212/wnl.58.1.143. [DOI] [PubMed] [Google Scholar]
- 20.Watanabe S, Nakashima I, Misu T, et al. Therapeutic efficacy of plasma exchange in NMO-IgG-positive patients with neuromyelitis optica. Mult Scler. 2007;13:128–32. doi: 10.1177/1352458506071174. [DOI] [PubMed] [Google Scholar]
- 21.Franciotta D, Salvetti M, Lolli F, et al. B cells and multiple sclerosis. Lancet Neurol. 2008;7:852–8. doi: 10.1016/S1474-4422(08)70192-3. [DOI] [PubMed] [Google Scholar]
- 22.Racke MK. The role of B cells in multiple sclerosis: rationale for B-cell-targeted therapies. Curr Opin Neurol. 2008;21(Suppl)(1):S9–18. doi: 10.1097/01.wco.0000313359.61176.15. [DOI] [PubMed] [Google Scholar]
- 23.Link H, Huang YM. Oligoclonal bands in multiple sclerosis cerebrospinal fluid: an update on methodology and clinical usefulness. J Neuroimmunol. 2006;180:17–28. doi: 10.1016/j.jneuroim.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 24.Berger T, Reindl M. Multiple sclerosis: disease biomarkers as indicated by pathophysiology. J Neurol Sci. 2007;259:21–6. doi: 10.1016/j.jns.2006.05.070. [DOI] [PubMed] [Google Scholar]
- 25.Breij EC, Brink BP, Veerhuis R, et al. Homogeneity of active demyelinating lesions in established multiple sclerosis. Ann Neurol. 2008;63:16–25. doi: 10.1002/ana.21311. [DOI] [PubMed] [Google Scholar]
- 26.Lucchinetti CF, Bruck W, Rodriguez M, et al. Distinct pattern s of multiple sclerosis pathology indicates heterogeneity on pathogenesis. Brain Pathol. 1996;6:259–74. doi: 10.1111/j.1750-3639.1996.tb00854.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Weinshenker BG. Plasma exchange for severe attacks of inflammatory demyelinating diseases of the central nervous system. J Clin Apher. 2001;16:39–42. doi: 10.1002/jca.1010. [DOI] [PubMed] [Google Scholar]
- 28.Weinshenker BG, O'Brien PC, Petterson TM, et al. A randomized trial of plasma exchange in acute central nervous system inflammatory demyelinating disease. Ann Neurol. 1999;46:878–86. doi: 10.1002/1531-8249(199912)46:6<878::aid-ana10>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 29.Hauser SL, Waubant E, Arnold DL, et al. B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. N Engl J Med. 2008;358:676–88. doi: 10.1056/NEJMoa0706383. [DOI] [PubMed] [Google Scholar]
- 30.Keegan M, Konig F, McClelland R, et al. Relation between humoral pathological changes in multiple sclerosis and response to therapeutic plasma exchange. Lancet. 2005;366:579–82. doi: 10.1016/S0140-6736(05)67102-4. [DOI] [PubMed] [Google Scholar]
- 31.Oh S, Cudrici C, Ito T, et al. B-cells and humoral immunity in multiple sclerosis. Implications for therapy. Immunol Res. 2008;40:224–34. doi: 10.1007/s12026-007-8009-6. [DOI] [PubMed] [Google Scholar]
- 32.Compston DA, Morgan BP, Campbell AK, et al. Immunocytochemical localization of the terminal complement complex in multiple sclerosis. Neuropathol Appl Neurobiol. 1989;15:307–16. doi: 10.1111/j.1365-2990.1989.tb01231.x. [DOI] [PubMed] [Google Scholar]
- 33.Storch MK, Piddlesden S, Haltia M, et al. Multiple sclerosis: in situ evidence for antibody- and complement-mediated demyelination. Ann Neurol. 1998;43:465–71. doi: 10.1002/ana.410430409. [DOI] [PubMed] [Google Scholar]
- 34.Prineas JW, Kwon EE, Cho ES, et al. Immunopathology of secondary-progressive multiple sclerosis. Ann Neurol. 2001;50:646–57. doi: 10.1002/ana.1255. [DOI] [PubMed] [Google Scholar]
- 35.Lucchinetti C, Bruck W, Parisi J, et al. Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyelination. Ann Neurol. 2000;47:707–17. doi: 10.1002/1531-8249(200006)47:6<707::aid-ana3>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 36.Lassmann H. Models of multiple sclerosis: new insights into pathophysiology and repair. Curr Opin Neurol. 2008;21:242–7. doi: 10.1097/WCO.0b013e3282fee94a. [DOI] [PubMed] [Google Scholar]
- 37.Mackay IR, Carnegie PR, Coates AS. Immunopathological comparisons between experimental autoimmune encephalomyelitis and multiple sclerosis. Clin Exp Immunol. 1973;15:471–82. [PMC free article] [PubMed] [Google Scholar]
- 38.Kabat EA, Wolf A, Bezer AE. Studies on acute disseminated encephalomyelitis produced experimentally in rhesus monkeys; disseminated encephalomyelitis produced in monkeys with their own brain tissue. J Exp Med. 1949;89:395–8. doi: 10.1084/jem.89.4.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zamvil S, Nelson P, Trotter J, et al. T-cell clones specific for myelin basic protein induce chronic relapsing paralysis and demyelination. Nature. 1985;317:355–8. doi: 10.1038/317355a0. [DOI] [PubMed] [Google Scholar]
- 40.Yamamura T, Namikawa T, Endoh M, et al. Experimental allergic encephalomyelitis induced by proteolipid apoprotein in Lewis rats. J Neuroimmunol. 1986;12:143–53. doi: 10.1016/0165-5728(86)90027-5. [DOI] [PubMed] [Google Scholar]
- 41.Kojima K, Wekerle H, Lassmann H, et al. Induction of experimental autoimmune encephalomyelitis by CD4+ T cells specific for an astrocyte protein, S100 beta. J Neural Transm Suppl. 1997;49:43–51. doi: 10.1007/978-3-7091-6844-8_5. [DOI] [PubMed] [Google Scholar]
- 42.Lassmann H. Experimental models of multiple sclerosis. Rev Neurol (Paris) 2007;163:651–5. doi: 10.1016/s0035-3787(07)90474-9. [DOI] [PubMed] [Google Scholar]
- 43.Bjartmar C, Trapp BD. Axonal degeneration and progressive neurologic disability in multiple sclerosis. Neurotox Res. 2003;5:157–64. doi: 10.1007/BF03033380. [DOI] [PubMed] [Google Scholar]
- 44.Steinman L, Zamvil SS. How to successfully apply animal studies in experimental allergic encephalomyelitis to research on multiple sclerosis. Ann Neurol. 2006;60:12–21. doi: 10.1002/ana.20913. [DOI] [PubMed] [Google Scholar]
- 45.Linington C, Morgan BP, Scolding NJ, et al. The role of complement in the pathogenesis of experimental allergic encephalomyelitis. Brain. 1989;112:895–911. doi: 10.1093/brain/112.4.895. [DOI] [PubMed] [Google Scholar]
- 46.Hinman CL, Stevens-Truss R, Schwarz C, et al. Sequence determinants of modified cobra venom neurotoxin which induce immune resistance to experimental allergic encephalomyelitis: molecular mechanisms for immunologic action. Immunopharmacol Immunotoxicol. 1999;21:483–506. doi: 10.3109/08923979909007122. [DOI] [PubMed] [Google Scholar]
- 47.Pabst H, Day NK, Gewurz H, et al. Prevention of experimental allergic encephalomyelitis with cobra venom factor. Proc Soc Exp Biol Med. 1971;136:555–60. doi: 10.3181/00379727-136-35310. [DOI] [PubMed] [Google Scholar]
- 48.Abrahamson HA. Prevention of experimental allergic encephalomyelitis with cobra venom factor. J Asthma Res. 1971;8:151–2. doi: 10.3109/02770907109108369. [DOI] [PubMed] [Google Scholar]
- 49.Piddlesden S, Lassmann H, Laffafian I, et al. Antibody-mediated demyelination in experimental allergic encephalomyelitis is independent of complement membrane attack complex formation. Clin Exp Immunol. 1991;83:245–50. doi: 10.1111/j.1365-2249.1991.tb05622.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Piddlesden SJ, Storch MK, Hibbs M, et al. Soluble recombinant complement receptor 1 inhibits inflammation and demyelination in antibody-mediated demyelinating experimental allergic encephalomyelitis. J Immunol. 1994;152:5477–84. [PubMed] [Google Scholar]
- 51.Vriesendorp FJ, Flynn RE, Pappolla MA, et al. Soluble complement receptor 1 (sCR1) is not as effective as cobra venom factor in the treatment of experimental allergic neuritis. Int J Neurosci. 1997;92:287–98. doi: 10.3109/00207459708986406. [DOI] [PubMed] [Google Scholar]
- 52.Nataf S, Carroll SL, Wetsel RA, et al. Attenuation of experimental autoimmune demyelination in complement-deficient mice. J Immunol. 2000;165:5867–73. doi: 10.4049/jimmunol.165.10.5867. [DOI] [PubMed] [Google Scholar]
- 53.Calida DM, Constantinescu C, Purev E, et al. Cutting edge: C3, a key component of complement activation, is not required for the development of myelin oligodendrocyte glycoprotein peptide-induced experimental autoimmune encephalomyelitis in mice. J Immunol. 2001;166:723–6. doi: 10.4049/jimmunol.166.2.723. [DOI] [PubMed] [Google Scholar]
- 54.Barnum SR, Szalai AJ. Complement and demyelinating disease: no MAC needed? Brain Res Rev. 2006;52:58–68. doi: 10.1016/j.brainresrev.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 55.Szalai AJ, Hu X, Adams JE, et al. Complement in experimental autoimmune encephalomyelitis revisited: C3 is required for development of maximal disease. Mol Immunol. 2007;44:3132–6. doi: 10.1016/j.molimm.2007.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Morariu MA, Dalmasso AP. Experimental allergic encephalomyelitis in cobra venom factor-treated and C4-deficient guinea pigs. Ann Neurol. 1978;4:427–30. doi: 10.1002/ana.410040507. [DOI] [PubMed] [Google Scholar]
- 57.Boos LA, Szalai AJ, Barnum SR. Murine complement C4 is not required for experimental autoimmune encephalomyelitis. Glia. 2005;49:158–60. doi: 10.1002/glia.20093. [DOI] [PubMed] [Google Scholar]
- 58.Piddlesden SJ, Lassmann H, Zimprich F, et al. The demyelinating potential of antibodies to myelin oligodendrocyte glycoprotein is related to their ability to fix complement. Am J Pathol. 1993;143:555–64. [PMC free article] [PubMed] [Google Scholar]
- 59.Piddlesden SJ, Morgan BP. Killing of rat glial cells by complement: deficiency of the rat analogue of CD59 is the cause of oligodendrocyte susceptibility to lysis. J Neuroimmunol. 1993;48:169–75. doi: 10.1016/0165-5728(93)90189-6. [DOI] [PubMed] [Google Scholar]
- 60.Mead RJ, Singhrao SK, Neal JW, et al. The membrane attack complex of complement causes severe demyelination associated with acute axonal injury. J Immunol. 2002;168:458–65. doi: 10.4049/jimmunol.168.1.458. [DOI] [PubMed] [Google Scholar]
- 61.Weerth SH, Rus H, Shin ML, et al. Complement C5 in experimental autoimmune encephalomyelitis (EAE) facilitates remyelination and prevents gliosis. Am J Pathol. 2003;163:1069–80. doi: 10.1016/S0002-9440(10)63466-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Niculescu T, Weerth S, Niculescu F, et al. Effects of complement C5 on apoptosis in experimental autoimmune encephalomyelitis. J Immunol. 2004;172:5702–6. doi: 10.4049/jimmunol.172.9.5702. [DOI] [PubMed] [Google Scholar]
- 63.Gasque P, Neal JW, Singhrao SK, et al. Roles of the complement system in human neurodegenerative disorders: pro-inflammatory and tissue remodeling activities. Mol Neurobiol. 2002;25:1–17. doi: 10.1385/mn:25:1:001. [DOI] [PubMed] [Google Scholar]
- 64.Davoust N, Jones J, Stahel PF, et al. Receptor for the C3a anaphylatoxin is expressed by neurons and glial cells. Glia. 1999;26:201–11. doi: 10.1002/(sici)1098-1136(199905)26:3<201::aid-glia2>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 65.Nataf S, Davoust N, Barnum SR. Kinetics of anaphylatoxin C5a receptor expression during experimental allergic encephalomyelitis. J Neuroimmunol. 1998;91:147–55. doi: 10.1016/s0165-5728(98)00169-6. [DOI] [PubMed] [Google Scholar]
- 66.Reiman R, Campos Torres A, Martin BK, et al. Expression of C5a in the brain does not exacerbate experimental autoimmune encephalomyelitis. Neurosci Lett. 2005;390:134–8. doi: 10.1016/j.neulet.2005.08.022. [DOI] [PubMed] [Google Scholar]
- 67.Morgan BP, Griffiths M, Khanom H, et al. Blockade of the C5a receptor fails to protect against experimental autoimmune encephalomyelitis in rats. Clin Exp Immunol. 2004;138:430–8. doi: 10.1111/j.1365-2249.2004.02646.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Boos L, Campbell IL, Ames R, et al. Deletion of the complement anaphylatoxin C3a receptor attenuates, whereas ectopic expression of C3a in the brain exacerbates, experimental autoimmune encephalomyelitis. J Immunol. 2004;173:4708–14. doi: 10.4049/jimmunol.173.7.4708. [DOI] [PubMed] [Google Scholar]
- 69.Fang CJ, Richards A, Liszewski MK, et al. Advances in understanding of pathogenesis of aHUS and HELLP. Br J Haematol. 2008;143:336–48. doi: 10.1111/j.1365-2141.2008.07324.x. [DOI] [PubMed] [Google Scholar]
- 70.Abarrategui-Garrido C, Melgosa M, Pena-Carrion A, et al. Mutations in proteins of the alternative pathway of complement and the pathogenesis of atypical hemolytic uremic syndrome. Am J Kidney Dis. 2008;52:171–80. doi: 10.1053/j.ajkd.2008.01.026. [DOI] [PubMed] [Google Scholar]
- 71.Jans H, Heltberg A, Zeeberg I, et al. Immune complexes and the complement factors C4 and C3 in cerebrospinal fluid and serum from patients with chronic progressive multiple sclerosis. Acta Neurol Scand. 1984;69:34–8. doi: 10.1111/j.1600-0404.1984.tb07777.x. [DOI] [PubMed] [Google Scholar]
- 72.Jongen PJ, Doesburg WH, Ibrahim-Stappers JL, et al. Cerebrospinal fluid C3 and C4 indexes in immunological disorders of the central nervous system. Acta Neurol Scand. 2000;101:116–21. doi: 10.1034/j.1600-0404.2000.101002116.x. [DOI] [PubMed] [Google Scholar]
- 73.Morgan BP, Campbell AK, Compston DA. Terminal component of complement (C9) in cerebrospinal fluid of patients with multiple sclerosis. Lancet. 1984;2:251–4. doi: 10.1016/s0140-6736(84)90298-8. [DOI] [PubMed] [Google Scholar]
- 74.Rodriguez M, Wynn DR, Kimlinger TK, et al. Terminal component of complement (C9) in the cerebrospinal fluid of patients with multiple sclerosis and neurologic controls. Neurology. 1990;40:855–7. doi: 10.1212/wnl.40.5.855. [DOI] [PubMed] [Google Scholar]
- 75.Halawa I, Lolli F, Link H. Terminal component of complement C9 in CSF and plasma of patients with MS and aseptic meningitis. Acta Neurol Scand. 1989;80:130–5. doi: 10.1111/j.1600-0404.1989.tb03854.x. [DOI] [PubMed] [Google Scholar]
- 76.Sellebjerg F, Jaliashvili I, Christiansen M, et al. Intrathecal activation of the complement system and disability in multiple sclerosis. J Neurol Sci. 1998;157:168–74. doi: 10.1016/s0022-510x(98)00086-0. [DOI] [PubMed] [Google Scholar]
- 77.Mollnes TE, Vandvik B, Lea T, et al. Intrathecal complement activation in neurological diseases evaluated by analysis of the terminal complement complex. J Neurol Sci. 1987;78:17–28. doi: 10.1016/0022-510x(87)90074-8. [DOI] [PubMed] [Google Scholar]
- 78.Sanders ME, Koski CL, Robbins D, et al. Activated terminal complement in cerebrospinal fluid in Guillain–Barré syndrome and multiple sclerosis. J Immunol. 1986;136:4456–9. [PubMed] [Google Scholar]
- 79.Finehout EJ, Franck Z, Lee KH. Complement protein isoforms in CSF as possible biomarkers for neurodegenerative disease. Dis Markers. 2005;21:93–101. doi: 10.1155/2005/806573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Pinter C, Beltrami S, Caputo D, et al. Presence of autoantibodies against complement regulatory proteins in relapsing-remitting multiple sclerosis. J Neurovirol. 2000;6(Suppl.)(2):S42–6. [PubMed] [Google Scholar]
- 81.Tanhehco EJ, Lee H, Lucchesi BR. Sublytic complement attack reduces infarct size in rabbit isolated hearts: evidence for C5a-mediated cardioprotection. Immunopharmacology. 2000;49:391–9. doi: 10.1016/s0162-3109(00)00258-7. [DOI] [PubMed] [Google Scholar]
- 82.Wurzner R, Orren A, Lachmann PJ. Inherited deficiencies of the terminal components of human complement. Immunodefic Rev. 1992;3:123–47. [PubMed] [Google Scholar]
- 83.Ross SC, Densen P. Complement deficiency states and infection: epidemiology pathogenesis and consequences of neisserial and other infections in an immune deficiency. Medicine (Balt) 1984;63:243–73. [PubMed] [Google Scholar]
- 84.Wurzner R. Deficiencies of the complement MAC II gene cluster (C6, C7, C9): is subtotal C6 deficiency of particular evolutionary benefit? Clin Exp Immunol. 2003;133:156–9. doi: 10.1046/j.1365-2249.2003.02230.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Rus H, Cudrici C, Niculescu F. The role of the complement system in innate immunity. Immunol Res. 2005;33:103–12. doi: 10.1385/IR:33:2:103. [DOI] [PubMed] [Google Scholar]
- 86.Soane L, Cho HJ, Niculescu F, et al. C5b-9 terminal complement complex protects oligodendrocytes from death by regulating Bad through phosphatidylinositol 3-kinase/Akt pathway. J Immunol. 2001;167:2305–11. doi: 10.4049/jimmunol.167.4.2305. [DOI] [PubMed] [Google Scholar]
- 87.Cudrici C, Niculescu F, Jensen T, et al. C5b-9 terminal complex protects oligodendrocytes from apoptotic cell death by inhibiting caspase-8 processing and up-regulating FLIP. J Immunol. 2006;176:3173–80. doi: 10.4049/jimmunol.176.5.3173. [DOI] [PubMed] [Google Scholar]
- 88.Cudrici C, Niculescu T, Niculescu F, et al. Oligodendrocyte cell death in pathogenesis of multiple sclerosis: protection of oligodendrocytes from apoptosis by complement. J Rehabil Res Dev. 2006;43:123–32. doi: 10.1682/jrrd.2004.08.0111. [DOI] [PubMed] [Google Scholar]
- 89.Soane L, Rus H, Niculescu F, et al. Inhibition of oligodendrocyte apoptosis by sublytic C5b-9 is associated with enhanced synthesis of bcl-2 and mediated by inhibition of caspase-3 activation. J Immunol. 1999;163:6132–8. [PubMed] [Google Scholar]
- 90.Hisahara S, Yuan J, Momoi T, et al. Caspase-11 mediates oligodendrocyte cell death and pathogenesis of autoimmune-mediated demyelination. J Exp Med. 2001;193:111–22. doi: 10.1084/jem.193.1.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Shibata M, Hisahara S, Hara H, et al. Caspases determine the vulnerability of oligodendrocytes in the ischemic brain. J Clin Invest. 2000;106:643–53. doi: 10.1172/JCI10203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Rus H, Cudrici C, Niculescu F, et al. Complement activation in autoimmune demyelination: dual role in neuroinflammation and neuroprotection. J Neuroimmunol. 2006;180:9–16. doi: 10.1016/j.jneuroim.2006.07.009. [DOI] [PubMed] [Google Scholar]
- 93.Rus H, Cudrici C, Niculescu F. C5b-9 complement complex in autoimmune demyelination: dual role in neuroinflammation and neuroprotection. Adv Exp Med Biol. 2006;586:139–51. doi: 10.1007/0-387-34134-X_10. [DOI] [PubMed] [Google Scholar]
- 94.Osaka H, Mukherjee P, Aisen PS, et al. Complement-derived anaphylatoxin C5a protects against glutamate-mediated neurotoxicity. J Cell Biochem. 1999;73:303–11. [PubMed] [Google Scholar]
- 95.Mukherjee P, Pasinetti GM. Complement anaphylatoxin C5a neuroprotects through mitogen-activated protein kinase-dependent inhibition of caspase 3. J Neurochem. 2001;77:43–9. doi: 10.1046/j.1471-4159.2001.00167.x. [DOI] [PubMed] [Google Scholar]
- 96.van Beek J, Nicole O, Ali C, et al. Complement anaphylatoxin C3a is selectively protective against NMDA-induced neuronal cell death. Neuroreport. 2001;12:289–93. doi: 10.1097/00001756-200102120-00022. [DOI] [PubMed] [Google Scholar]
- 97.Mukherjee P, Thomas S, Pasinetti GM. Complement anaphylatoxin C5a neuroprotects through regulation of glutamate receptor subunit 2 in vitro and in vivo. J Neuroinflammation. 2008;5:5. doi: 10.1186/1742-2094-5-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sadovnick AD, Ebers GC, Dyment DA, et al. Evidence for genetic basis of multiple sclerosis. The Canadian Collaborative Study Group. Lancet. 1996;347:1728–30. doi: 10.1016/s0140-6736(96)90807-7. [DOI] [PubMed] [Google Scholar]
- 99.Ebers GC, Bulman DE, Sadovnick AD, et al. A population-based study of multiple sclerosis in twins. N Engl J Med. 1986;315:1638–42. doi: 10.1056/NEJM198612253152603. [DOI] [PubMed] [Google Scholar]
- 100.Robertson NP, Clayton D, Fraser M, et al. Clinical concordance in sibling pairs with multiple sclerosis. Neurology. 1996;47:347–52. doi: 10.1212/wnl.47.2.347. [DOI] [PubMed] [Google Scholar]
- 101.Gregory SG, Schmidt S, Seth P, et al. Interleukin 7 receptor alpha chain (IL7R) shows allelic and functional association with multiple sclerosis. Nat Genet. 2007;39:1083–91. doi: 10.1038/ng2103. [DOI] [PubMed] [Google Scholar]
- 102.Hafler DA, Compston A, Sawcer S, et al. Risk alleles for multiple sclerosis identified by a genomewide study. N Engl J Med. 2007;357:851–62. doi: 10.1056/NEJMoa073493. [DOI] [PubMed] [Google Scholar]
- 103.International Multiple Sclerosis Genetic Consortium. Refining genetic associations in multiple sclerosis. Lancet Neurol. 2008;7:567–9. doi: 10.1016/S1474-4422(08)70122-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Edwards AO, Ritter R, 3rd, Abel KJ, et al. Complement factor H polymorphism and age-related macular degeneration. Science. 2005;308:421–4. doi: 10.1126/science.1110189. [DOI] [PubMed] [Google Scholar]
- 105.Hageman GS, Anderson DH, Johnson LV, et al. A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age-related macular degeneration. Proc Natl Acad Sci USA. 2005;102:7227–32. doi: 10.1073/pnas.0501536102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Haines JL, Hauser MA, Schmidt S, et al. Complement factor H variant increases the risk of age-related macular degeneration. Science. 2005;308:419–21. doi: 10.1126/science.1110359. [DOI] [PubMed] [Google Scholar]
- 107.Skerka C, Lauer N, Weinberger AA, et al. Defective complement control of factor H (Y402H) and FHL-1 in age-related macular degeneration. Mol Immunol. 2007;44:3398–406. doi: 10.1016/j.molimm.2007.02.012. [DOI] [PubMed] [Google Scholar]
- 108.Gold B, Merriam JE, Zernant J, et al. Variation in factor B (BF) and complement component 2 (C2) genes is associated with age-related macular degeneration. Nat Genet. 2006;38:458–62. doi: 10.1038/ng1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kaur I, Hussain A, Hussain N, et al. Analysis of CFH, TLR4, and APOE polymorphism in India suggests the Tyr402His variant of CFH to be a global marker for age-related macular degeneration. Invest Ophthalmol Vis Sci. 2006;47:3729–35. doi: 10.1167/iovs.05-1430. [DOI] [PubMed] [Google Scholar]
- 110.Hamilton G, Proitsi P, Williams J, et al. Complement factor H Y402H polymorphism is not associated with late-onset Alzheimer's disease. Neuromolecular Med. 2007;9:331–4. doi: 10.1007/s12017-007-8013-y. [DOI] [PubMed] [Google Scholar]
- 111.Zetterberg M, Landgren S, Andersson ME, et al. Association of complement factor H Y402H gene polymorphism with Alzheimer's disease. Am J Med Genet B Neuropsychiatr Genet. 2008;147B:720–6. doi: 10.1002/ajmg.b.30668. [DOI] [PubMed] [Google Scholar]
- 112.Kuokkanen S, Gschwend M, Rioux JD, et al. Genomewide scan of multiple sclerosis in Finnish multiplex families. Am J Hum Genet. 1997;61:1379–87. doi: 10.1086/301637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ebers GC, Kukay K, Bulman DE, et al. A full genome search in multiple sclerosis. Nat Genet. 1996;13:472–6. doi: 10.1038/ng0896-472. [DOI] [PubMed] [Google Scholar]
- 114.Sawcer S, Jones HB, Feakes R, et al. A genome screen in multiple sclerosis reveals susceptibility loci on chromosome 6p21 and 17q22. Nat Genet. 1996;13:464–8. doi: 10.1038/ng0896-464. [DOI] [PubMed] [Google Scholar]
- 115.Haines JL, Ter-Minassian M, Bazyk A, et al. A complete genomic screen for multiple sclerosis underscores a role for the major histocompatability complex. The Multiple Sclerosis Genetics Group. Nat Genet. 1996;13:469–71. doi: 10.1038/ng0896-469. [DOI] [PubMed] [Google Scholar]
- 116.Sawcer S, Ban M, Maranian M, et al. A high-density screen for linkage in multiple sclerosis. Am J Hum Genet. 2005;77:454–67. doi: 10.1086/444547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Hye A, Lynham S, Thambisetty M, et al. Proteome-based plasma biomarkers for Alzheimer's disease. Brain. 2006;129:3042–50. doi: 10.1093/brain/awl279. [DOI] [PubMed] [Google Scholar]
- 118.Honda S, Itoh F, Yoshimoto M, et al. Association between complement regulatory protein factor H and AM34 antigen, detected in senile plaques. J Gerontol A Biol Sci Med Sci. 2000;55:M265–9. doi: 10.1093/gerona/55.5.m265. [DOI] [PubMed] [Google Scholar]
- 119.Willison HJ, Halstead SK, Beveridge E, et al. The role of complement and complement regulators in mediating motor nerve terminal injury in murine models of Guillain–Barre syndrome. J Neuroimmunol. 2008;201(202):172–82. doi: 10.1016/j.jneuroim.2008.05.028. [DOI] [PubMed] [Google Scholar]
- 120.Arumugam TV, Shiels IA, Woodruff TM, et al. The role of the complement system in ischemia–reperfusion injury. Shock. 2004;21:401–9. doi: 10.1097/00024382-200405000-00002. [DOI] [PubMed] [Google Scholar]
- 121.Arumugam TV, Woodruff TM, Lathia JD, et al. Neuroprotection in stroke by complement inhibition and immunoglobulin therapy. Neuroscience. 2008 doi: 10.1016/j.neuroscience.2008.07.015. PMID: 18691639 (Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Cuzner ML, Opdenakker G. Plasminogen activators and matrix metalloproteases, mediators of extracellular proteolysis in inflammatory demyelination of the central nervous system. J Neuroimmunol. 1999;94:1–14. doi: 10.1016/s0165-5728(98)00241-0. [DOI] [PubMed] [Google Scholar]
- 123.Selmaj KW, Raine CS. Tumor necrosis factor mediates myelin and oligodendrocyte damage in vitro. Ann Neurol. 1988;23:339–46. doi: 10.1002/ana.410230405. [DOI] [PubMed] [Google Scholar]
- 124.Smith KJ, Kapoor R, Felts PA. Demyelination: the role of reactive oxygen and nitrogen species. Brain Pathol. 1999;9:69–92. doi: 10.1111/j.1750-3639.1999.tb00212.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Smith KJ, Lassmann H. The role of nitric oxide in multiple sclerosis. Lancet Neurol. 2002;1:232–41. doi: 10.1016/s1474-4422(02)00102-3. [DOI] [PubMed] [Google Scholar]
- 126.Morgan BP, Gasque P, Singhrao SK, et al. Role of complement in inflammation and injury in the nervous system. Exp Clin Immunogenet. 1997;14:19–23. [PubMed] [Google Scholar]
- 127.Kuwert E, Pette E, Firnhaber W, et al. Demonstration of complement in spinal fluid in multiple sclerosis. Ann NY Acad Sci. 1965;122:429–38. doi: 10.1111/j.1749-6632.1965.tb20225.x. [DOI] [PubMed] [Google Scholar]
- 128.Link H. Complement factors in multiple sclerosis. Acta Neurol Scand. 1972;48:521–8. doi: 10.1111/j.1600-0404.1972.tb07572.x. [DOI] [PubMed] [Google Scholar]
- 129.Dube VE, McDuffie FC, Burton RC, et al. Cerebrospinal fluid complement in multiple sclerosis. J Lab Clin Med. 1973;81:530–7. [PubMed] [Google Scholar]
- 130.Yam P, Petz LD, Tourtellotte WW, et al. Measurement of complement components in cerebral spinal fluid by radioimmunoassay in patients with multiple sclerosis. Clin Immunol Immunopathol. 1980;17:492–505. doi: 10.1016/0090-1229(80)90145-2. [DOI] [PubMed] [Google Scholar]
- 131.Price P, Cuzner ML. Cerebrospinal fluid complement proteins in neurological disease. J Neurol Sci. 1980;46:49–54. doi: 10.1016/0022-510x(80)90042-8. [DOI] [PubMed] [Google Scholar]
- 132.Compston DA, Morgan BP, Oleesky D, et al. Cerebrospinal fluid C9 in demyelinating disease. Neurology. 1986;36:1503–6. doi: 10.1212/wnl.36.11.1503. [DOI] [PubMed] [Google Scholar]
- 133.Chataway J, Sawcer S, Sherman D, et al. No evidence for association of multiple sclerosis with the complement factors C6 and C7. J Neuroimmunol. 1999;99:150–6. doi: 10.1016/s0165-5728(99)00054-5. [DOI] [PubMed] [Google Scholar]
- 134.Simon K, Yang X, Munger K, et al. Variation in the Epstein–Barr virus receptor, CR2, and risk of multiple sclerosis. Mult Scler. 2007;13:947–8. doi: 10.1177/1352458506072983. [DOI] [PubMed] [Google Scholar]
- 135.Bulman DE, Armstrong H, Ebers GC. Allele frequencies of the third component of complement (C3) in MS patients. J Neurol Neurosurg Psychiatry. 1991;54:554–5. doi: 10.1136/jnnp.54.6.554. [DOI] [PMC free article] [PubMed] [Google Scholar]