

Abstract
The immunopathology of type 1 diabetes (T1D) has proved difficult to study in man because of the limited availability of appropriate samples, but we now report a detailed study charting the evolution of insulitis in human T1D. Pancreas samples removed post-mortem from 29 patients (mean age 11·7 years) with recent-onset T1D were analysed by immunohistochemistry. The cell types constituting the inflammatory infiltrate within islets (insulitis) were determined in parallel with islet insulin content. CD8+ cytotoxic T cells were the most abundant population during insulitis. Macrophages (CD68+) were also present during both early and later insulitis, although in fewer numbers. CD20+ cells were present in only small numbers in early insulitis but were recruited to islets as beta cell death progressed. CD138+ plasma cells were infrequent at all stages of insulitis. CD4+ cells were present in the islet infiltrate in all patients but were less abundant than CD8+ or CD68+ cells. Forkhead box protein P3+ regulatory T cells were detected in the islets of only a single patient. Natural killer cells were detected rarely, even in heavily inflamed islets. The results suggest a defined sequence of immune cell recruitment in human T1D. They imply that both CD8+ cytotoxic cells and macrophages may contribute to beta cell death during early insulitis. CD20+ cells are recruited in greatest numbers during late insulitis, suggesting an increasing role for these cells as insulitis develops. Natural killer cells and forkhead box protein P3+ T cells do not appear to be required for beta cell death.
Keywords: beta cell, insulitis, T cell, type 1 diabetes
Introduction
Type 1 diabetes (T1D) in man is an immune-mediated disease originating from the selective destruction of pancreatic beta cells following infiltration of the islets of Langerhans by cells of the immune system (insulitis). However, the cellular and molecular basis of disease progression remains unclear because, although these processes have been documented in animal models [1–3], only limited data are available in man. This reflects the relative lack of availability of pancreatic specimens recovered from individuals in whom ongoing beta cell destruction is present.
In an attempt to address this deficiency, a single time-point pancreatic biopsy method was employed in an earlier study [4] to study the immunopathology of T1D in man. However, the samples obtained contained only small numbers of islets per section and very little insulitis was seen. In the present work, we have adopted an alternative approach by analysing larger pancreas samples removed post-mortem from patients who died within a maximum of 18 months of diagnosis of T1D. As reported previously [5], these contain islets at various stages of disease progression. Islets appear to progress from insulin-containing islets with no inflammation to islets with a near normal complement of beta cells and a light infiltrate of inflammatory cells, then to islets with pronounced inflammation and reduced numbers of beta cells, and finally to islets devoid of beta cells and inflammation. These pancreas samples are thus well suited for analysis of the temporal sequence of insulitis. Accordingly, we provide the first detailed mapping of the evolution of insulitis in man.
Materials and methods
Subjects
The investigations reported in this study were performed with full ethical approval. Twenty-nine human pancreases recovered from patients with recent-onset T1D were employed in the study; 28 of these were removed at autopsy and one pancreas was recovered by surgical resection at the time of organ donation (Table 1). The cases were selected randomly from within a collection of 70 cases of recent-onset T1D in which the stored pancreatic sections contain residual insulin-positive islets. A detailed description of these cases has been given previously [6]. Random selection was achieved without prior examination of the sections by arbitrary choice from a numbered list. The cohort consisted of 21 female and eight male diabetic patients. Twenty-eight of these were in the age range 1–23 years (mean 10·6 ± 1·2), while the additional patient (female) was 42 years old (overall mean: 11·7 ± 1·6 years). Controls were eight non-diabetic paediatric pancreases (age range 2–9 years; six male, two female), four non-diabetic adult, two type 2 diabetes (T2D) pancreases and two surgical resections of chronic pancreatitis (Table 2). Pancreatic specimens had been fixed previously in buffered p-formaldehyde, unbuffered formol saline or Bouin's fixative, and they were all paraffin-embedded.
Table 1.
Clinical information and cause of death for the cohort of patients with type 1 diabetes.
| Case | Age (years) | Sex | Duration of diabetes symptoms | Cause of death |
|---|---|---|---|---|
| D1 | 2 | F | 2 weeks | Ketoacidosis |
| D2 | 19 | F | 18 months | Pneumonia |
| D3 | 3 | F | < 1 week | Ketoacidosis |
| D4 | 5 | M | 1 week | Ketoacidosis |
| D5 | 1 | F | 3 days | Cerebral oedema |
| D6 | 4 | F | 2 weeks | Cerebral oedema |
| D7 | 15 | M | 6 months | Ketoacidosis |
| D8 | 18 | F | 4 days | Ketoacidosis |
| D9 | 6 | F | < 1 week | Ketoacidosis |
| D10 | 11 | F | 5 days | Ketoacidosis |
| D11 | 13 | F | < 1 week | Ketoacidosis |
| D12 | 7 | F | 1 week | Ketoacidosis |
| D13 | 18 | F | 3 weeks | Ketoacidosis |
| D14 | 7 | F | Unknown | Ketoacidosis |
| D15 | 18 | M | Unknown | Addison's adrenal insufficiency |
| D16 | 12 | F | 2 months | Ketoacidosis |
| D17 | 6 | M | Unknown | Ketoacidosis |
| D18 | 3 | F | 3 weeks | Ketoacidosis |
| D19 | 14 | F | 1 day | Ketoacidosis |
| D20 | 7 | F | Unknown | Ketoacidosis |
| D21 | 8 | F | < 1 week | Ketoacidosis |
| D22 | 14 | F | < 1 week | Ketoacidosis |
| D23 | 12 | F | 2 months | Ketoacidosis |
| D24 | 17 | M | < 1 week | Ketoacidosis |
| D25 | 23 | M | 2 weeks | Ketoacidosis |
| D26 | 4 | F | 3 weeks | Ketoacidosis |
| D27 | 13 | M | 2 days | Ketoacidosis |
| D28 | 18 | M | 4 months | Hyperglycaemia |
| D29 | 42 | F | 18 months | Surgical resection |
Table 2.
Clinical information and cause of death for the control groups of patients.
| Case | Age (years) | Sex | Disease (if known) | Cause of death if autopsy |
|---|---|---|---|---|
| C1 | 50 | F | Insulinoma | Surgical specimen |
| C2 | 40 | M | Insulinoma | Surgical |
| C3 | 38 | M | Carcinoma ampulla of Vater | Surgical |
| C4 | 53 | F | Insulinoma | Surgical |
| C5 | 5 | M | Mental disability | Unknown |
| C6 | 9 | M | Intussusception | |
| C7 | 6 | M | Meningitis following head injury | |
| C8 | 2 | F | Bronchopneumonia and epilepsy | |
| C9 | 3 | F | Road traffic accident | |
| C10 | 3 | M | Meningococcal septicaemia | |
| C11 | 2 | M | Haemophilus influenzae meningitis | |
| C12 | 6 | M | Porencephaly and pneumonia | |
| C13 | 70 | F | Type 2 diabetes | Uncertain at post mortem |
| post-mortem C14 | 72 | F | Type 2 diabetes | Peritonitis following surgery |
| C15 | 65 | M | Chronic pancreatitis | Surgical |
| C16 | 34 | M | Chronic pancreatitis | Surgical |
Immunohistochemistry
Serial sections (4 µm) were cut from each case and mounted on glass slides coated in (3-aminopropyl)-triethoxysilane (Sigma, Dorset, UK). Sections were processed and labelled using a standard immunoperoxidase technique for paraffin sections. With the exception of glucagon and insulin, antigens were unmasked by heat-induced epitope retrieval. Sections to be labelled with anti-CD45+ and anti-CD20+ anti-sera were heated in 10 mM citrate buffer, pH 6·0 while those to be labelled with anti-CD3+, anti-CD4+, anti-CD8+, anti-CD68+ and anti-forkhead box protein P3 (FoxP3)+ anti-sera were heated in 10 mM Tris, 1 mM ethylenediamine tetraacetic acid, pH 9·0, and those to be labelled with anti-CD138+ anti-sera were heated in 1 mM ethylenediamine tetraacetic acid, pH 8·0. The antibody clone (236A/E7), utilized for study of FoxP3 expression, was shown to be optimal for detection of this antigen in paraffin-embedded tissues in an antibody comparison study [7]. Primary antibodies were applied for 1 h at room temperature, with the exception of anti-CD4, which was incubated overnight at 4°C (see Supplementary material at end, Table S1). The Dako REAL™ Envision™ Detection System was used for antigen detection. Some slides were processed in the absence of primary antibody or with isotype control anti-sera to confirm the specificity of labelling.
Morphometry and cell counting
Slides were analysed by light microscopy. Initially, the total number of islets on each section was determined and these were subdivided into insulin-containing or insulin-deficient islets. Each islet was then examined for the presence of insulitis. For the analysis of CD4+, CD8+, CD20+ and CD68+ cells, approximately 10 islets per case were chosen at random and analysed in serial sections. The percentage of insulin-positive area per islet section was measured by morphometry using image analysis software (ImageJ, NIH, Bethesda, MD, USA). The number of individual immune cells was then counted within the islet section area. Any stained cells in the surrounding exocrine tissue were not included unless these were in direct contact with immune cells within the islet and appeared to be in the process of either entering or leaving the islet. For the analysis of FoxP3 and CD138 staining, every islet section in all of the 29 cases was examined and the number of positively stained cells counted. These were correlated with insulin-positivity.
Statistical analysis
Each islet was assigned to one of four groups according to its insulin-positive area (taken to represent the preservation of beta cells). The mean number of each type of immune cell per islet section was then calculated for each of these groups. Results are expressed as the mean number of cells ± standard error of the mean. Statistical significance was calculated by analysis of variance with Tukey's post hoc test using spss software.
Results
Defining insulitis
An important issue initially was to define ‘insulitis’, as it was noted that islets from individuals without autoimmune diabetes contained occasional immune cells. To assess this, more than 3800 islets in relevant control cases were studied. Examination of 853 islets in the normal adult pancreases revealed a total of only five islet sections containing either lymphocytes or macrophages and for each cell type there was never more than a single immunopositive cell per islet section. Normal paediatric cases had a total of six islet sections among 2399 islet sections containing a single lymphocyte and 14 islet sections each with one to three macrophages. In three of the cases, a single islet section contained more than five macrophages, but these were never accompanied by CD45+ cells. One of the T2D cases contained three islet sections among 286 that were immunopositive for CD45+ in the peri-islet area (never more than three per islet section). Both cases of chronic pancreatitis displayed large-scale infiltration of the organ with lymphocytes and macrophages. However, these were localized almost entirely to the fibrous tissue that had replaced the normal exocrine tissue, and only five islet sections among 271 were found to contain immune cells (lymphocytes) which never exceeded two per islet section. Therefore, in the patients with T1D, insulitis was defined as the presence of a minimum of five immune cells (stained positively for CD45+ and/or CD68+) within an islet section.
One potential limitation of the present study is the inevitable presence of varying degrees of autolysis in some pancreatic specimens recovered at autopsy. Therefore, each of the antibodies was tested in surgically removed control human pancreases and duodenum that were deliberately allowed to autolyse for increasing periods of time (between 0 and 42 h) prior to fixation. This preliminary work confirmed that antibody staining was affected minimally by the degree of autolysis.
Insulitis in islets with varying degrees of insulin-positivity
Twenty-nine T1D patients having a mean age of 11·7 ± 1·6 years were then analysed. Among these, the sections contained a total of 3075 islets (average of 106 ± 9·6 islets/section), of which 732 contained insulin (23·8%). A total of 255 (34·8%) of the insulin-containing islets were inflamed, as were 125 (5%) insulin-deficient islets. Within our cohort of patients, there was no clear correlation between the age and duration of diabetes and either the percentage of insulin-containing islets or the percentage of inflamed islets.
A range of islets with significant insulitis (equivalent to that shown in Fig. 1) were examined first to confirm that the sum of each individual lymphocyte subtype stained with anti-CD4 or anti-CD8 was approximately equal to the total number of cells stained with anti-CD3. Further, it was determined that the summated number of lymphocytes stained with anti-CD3 and anti-CD20 was approximately equal to those which were immunopositive for anti-CD45. Because these values correlated appropriately, this implies that the majority of immune cells were detected and that no subset of cells was underestimated systematically.
Fig. 1.
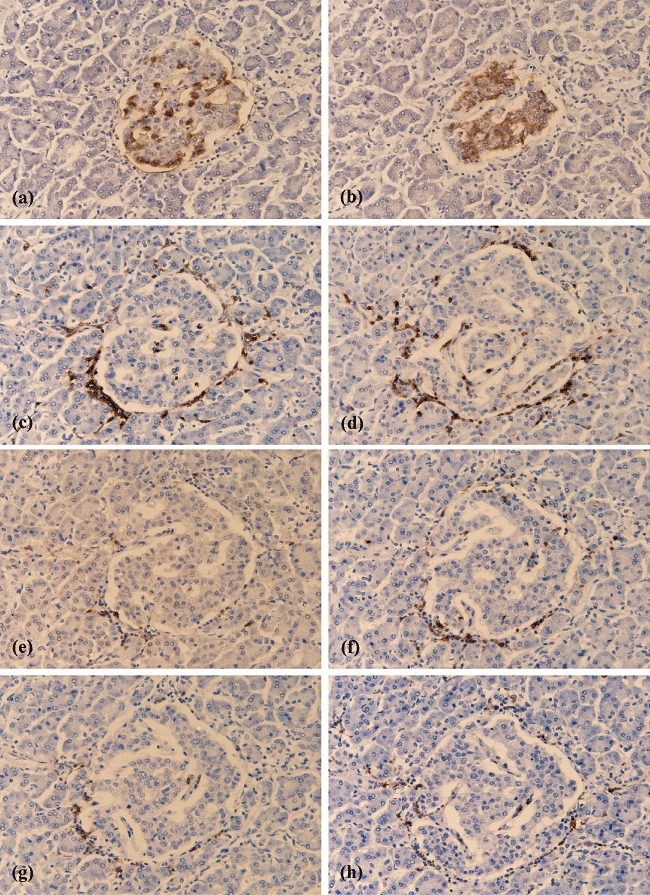
Immunostaining of individual immune cells in serial sections of a single islet from a patient with recent-onset type 1 diabetes (T1D). The micrographs reveal the extent of endocrine cell immunostaining and the immune cell complement in serial sections of one representative islet. Sections were stained with anti-sera raised against glucagon (a), insulin (b), CD45 (c), CD3 (d), CD4 (e), CD8 (f), CD20 (g) and CD68 (h) respectively. All images were taken at ×200 magnification.
Figure 1(a–h) provides an illustrative example of the selective staining of each immune cell subtype in a single islet in serial sections taken from one patient with T1D. In order to analyse the course of insulitis more fully, a total of 279 islets across the 29 cases were selected randomly and examined in detail. The percentage of insulin-positive area was measured for each islet section and taken to represent an index of beta cell mass. The mean insulin-positive area of 50 islet sections from non-diabetic control paediatric and adult pancreases was 73 ± 1%. On this basis, it was concluded that islet sections in which the insulin-positive area was 35% of the total islet section area had lost approximately half their beta cells. A total of 114 of the 279 T1D islets were insulin-negative (recorded as 0% and defined hereafter as category 1). The remaining 165 islet sections ranged between 0·5% and 68·2% insulin-positive area (mean: 29 ± 1%). These were categorized into four additional groups according to their percentage insulin-positivity [> 0–9% (category 2), 10–29% (category 3), 30–49% (category 4) or 50–69% (category 5)]. The individual immune cell numbers (CD4+, CD8+, CD20+ and CD68+) present within the islet section area were then recorded for each category of islets (Fig. 2 and see Supplementary material at end, Table S2).
Fig. 2.
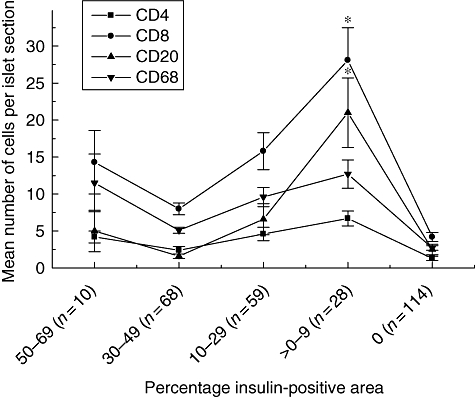
Relationship between insulin immunopositivity and the complement of defined subsets of immune cells in the islets of patients with recent-onset type 1 diabetes (T1D). The number of immunopositive CD4+, CD8+, CD20+ and CD68+ cells were counted in a total of 279 islets across 29 patients with T1D. These were then correlated with insulin immunopositivity, as quantified by morphometry. The islets were subdivided according to their percentage insulin-positive area, which was taken to be representative of the residual beta cell mass. Each point is the mean ± standard error of the mean for the number of relevant immune cells detected in each islet section. The number of islets analysed in each insulin-positive category is given in parenthesis.
It was found that at all stages of beta cell decline, CD8+ T cells represented the predominant cellular component of the insulitic infiltrate. These cells increased in number as insulin-positivity decreased – from a mean minimum of 8 ± 0·68 cells per islet section (in category 4) to reach a peak of 28·1 ± 4·4 cells/islet section when few beta cells remained (category 2; P < 0·01). Their numbers then declined dramatically once insulin-positivity was lost completely. This profile was mirrored by each of the other major immune cell subtypes (Fig. 2), although all were present in fewer numbers. CD4+ T cells were the least abundant population in all islet categories, with a mean minimum of 2·4 ± 0·5 cells/islet section and maximum of 6·7 ± 1·0 cells/islet section. CD20+ B cells were present in many of the inflamed islets and represented the second most abundant population (their numbers increased from a mean minimum of 1·6 ± 0·3 (category 4) to a maximum 21 ± 4·7 cells/islet section (category 2); P < 0·001). It was also noted that, with the exception of a single islet in one case, CD20+ B cells were detected only when CD8+ T cells were also present. Macrophages were present in the majority of insulin-positive islet sections examined and at relatively constant numbers irrespective of the insulin-positive area (mean minimum 5·1 ± 0·4, maximum 12·7 ± 1·9 cells/islet section). They were also the most frequent cell type detected in insulin-deficient islets (see Supplementary material at end).
All immune cell subtypes declined in number to a mean of fewer than five cells per islet section in the insulin-negative (category 1) islets (CD4+ 1·3 ± 0·3 cells/islet section; CD8+ 4·2 ± 0·6; CD20+ 2·5 ± 0·7; CD68+ 2·7 ± 0·3).
Regulatory T cells in human insulitis
To examine the frequency at which regulatory T cells (Tregs) were found within the CD4+ population of inflamed islets, 16 of the 29 cases were selected at random and stained with an anti-FoxP3 antibody. In humans, FoxP3 is expressed abundantly in Treg cells but it may also be expressed transiently at low levels in activated effector T (Teff) cells [8]. Irrespective of this, FoxP3+ cells were detected only rarely and were present in the infiltrate of only two adjacent islets in one section of the 16 T1D patients analysed (Fig. 3a and b). They were not found in the islets of any of the non-diabetic control pancreases.
Fig. 3.
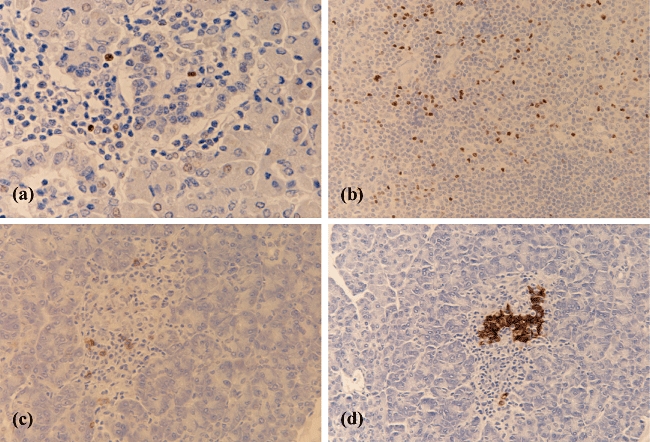
Immunodetection of forkhead box P3 (FoxP3)+ or CD138+ cells in inflamed islets from patients with recent-onset type 1 diabetes (T1D). The micrographs reveal the presence of FoxP3+ T cells (a) within a single islet from a T1D patient. (b) FoxP3+ staining of a section of human tonsil as a positive control. CD138+ (c) and insulin (d) immunostaining respectively, on serial sections of a single representative islet from a second patient with T1D. (a) ×400 magnification; all other images taken at ×200 magnification.
Plasma cells in human insulitis
In view of the observation that CD20+ B cells are present at high frequency in inflamed islets, the possibility was considered that plasma cells (CD138+CD20−) might also be present. Accordingly, sections from all 29 T1D patients were stained with an anti-CD138 antibody. CD138+ cells were detected in five of the 29 patients and were observed in a total of 40 islet sections. Twenty-four of these were from one patient, while a further two patients each had five CD138+-positive islet sections and two additional patients each had three CD138+ islet sections. All but one of these islets were insulin-positive (Fig. 3c and d). In those islets where CD138+ cells were present, between 1 and 20 such cells were detected (mean 5·25 ± 0·81 cells/islet section). CD138+ cells were present only in islet sections that were also positive for CD20+ B cells. CD138+ cells were not detected in any islets of the non-diabetic controls.
Natural killer cells are detected infrequently during insulitis
Unequivocal detection of natural killer (NK) cells in human tissue sections is notoriously difficult, as there are no NK cell-specific markers that are expressed invariably by all subtypes of NK cells [9,10]. Therefore, in this study we utilized antibodies raised against a range of potential NK cell markers including CD56 (neural cell adhesion molecule), CD57, CD94, NKp46 and Pen5, all of which are expressed to varying degrees by subsets of NK cells in man. The anti-CD94 and NKp46 antibodies proved unsuitable for use on paraffin-embedded tissue sections. By contrast, CD56+ cells could be detected in paraffin-embedded sections of pancreas, but this antigen is not ideal because, as shown in Fig. 4a, CD56 is not specific for NK cells but is also expressed on islet endocrine cells and by nerve cells. Similarly, CD57 is also expressed by islet endocrine cells (Fig. 4b), and this lack of specificity made the selective detection of NK cells expressing either CD56+ or CD57+ very difficult within an islet. Nevertheless, we were able to study a series of islets in which the number of beta cells was reduced but which contained an abundance of infiltrating immune cells. Examples are shown in Fig. 4(a and b), and it is clear that, in these islet sections, hardly any of the immune cells stained positively for CD56 or CD57. To confirm this, numbers of CD56+ immune cells were counted in the 29 T1D patients. Twelve patients contained CD56+ cells, but these amounted to a combined total of only 20 cells in 17 different islet sections. To verify the conclusion that NK cells were not an abundant population within the infiltrate, an antibody raised against Pen5 was also used. Pen5 is a glycoprotein expressed specifically by activated NK cells [11]. This antibody generated a ‘wash’ of non-specific background staining in the majority of islets from both control patients and those with T1D, which again made unequivocal identification of Pen5+ immune cells troublesome. However, in spite of these difficulties, it was possible to determine that very few of the immune cells were immunopositive for Pen5, even in islet sections that contained very few insulin-positive cells but an abundance of immune cells (Fig. 4c). Therefore, when considered together, these results lead to the conclusion that NK cells do not form a major component of the insulitic infiltrate in T1D in man.
Fig. 4.
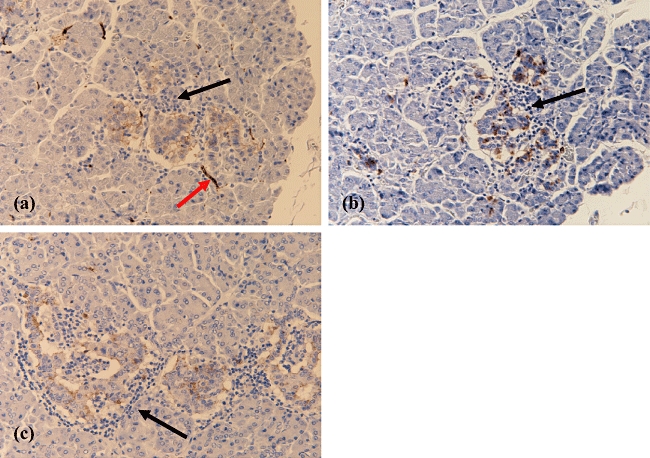
Immunodetection of natural killer (NK) cell markers in inflamed islets from patients with recent-onset type 1 diabetes (T1D). The micrographs demonstrate the immunostaining patterns for anti-CD56+ (a), CD57+ (b) and Pen5+ (c), respectively, in heavily inflamed islets from a single patient with recent-onset T1D. An example of a nerve that is stained positively for CD56 is highlighted by a red arrow in (a). While endocrine cells are stained by all these NK cell antibodies, lymphocytes which are clearly visible within each islet by virtue of their dense nuclei and small size do not stain (examples are indicated by black arrows in a, b and c). All images were taken at ×200 magnification.
Discussion
Type 1 diabetes in man is characterized by infiltration of pancreatic islets by cells of the immune system, but very little is known about the temporal progression of insulitis [5,12] nor of the subtypes of immune cells recruited during the process. A previous study [4] analysed insulitis in human pancreas biopsy samples, but the development of insulitis in relation to the residual beta cell area in individual islets was not studied. Moreover, insulitis was defined as a minimum of two mononuclear cells per islet section. Our observations reveal that control paediatric pancreases contain occasional immune cells and we noted that the number of immune cells could reach as many as five per islet in these cases, although it rarely exceeded this number. Indeed, in more than 3800 control islets examined, only three contained more than five immune cells (< 0·2%) and, even in these cases, the cells were identified as CD68+ (macrophages) which were not accompanied by CD45+ lymphocytes. In this context it is interesting to note that a recent study [13] has described the presence of small numbers of macrophages in the islets of patients with T2D. In a separate unpublished analysis of a larger number of patients with T2D, we also observed an overall increase in CD68+ cells compared with non-diabetic adult controls but this still did not exceed an average of five cells per islet. Accordingly, we adopted a higher threshold than that used previously to define insulitis in the T1D pancreases (a minimum of five mononuclear cells per islet) and, on this basis, we recorded mononuclear cell infiltration in 380 islets (255 insulin-positive and 125 insulin-negative islets) from a total of 3075 examined in the T1D patients (12%). Evidence of insulitis was observed in all 29 cases.
Another difference between the earlier biopsy procedure [4] and the samples employed in the present work is that the former yielded sections containing a maximum of 21 islets for analysis. By contrast, the mean figure in our samples was > 100 islets/section (with a maximum of 274). Thus, it is possible that the biopsy sampling method may have underestimated the prevalence of insulitis. Alternatively, it is also possible that insulitis may occur to a greater extent in younger patients as has been suggested previously [6], as the mean age of our cohort was 11·7 ± 1·6 years, whereas the patients studied by Itoh et al.[4] had an average age of 30 years. Additionally, we cannot exclude the possibility that the extent of insulitis may have been greater prior to disease onset as our samples were all taken from patients after diagnosis of T1D.
It is believed widely that the demise of beta cells occurs over a protracted time-course (possibly extending to several years) in T1D [14,15]. It might be expected therefore that insulitis, being the mediator of beta cell destruction, would be relatively infrequent in a diabetic pancreas at clinical presentation. In the present study, 732 islets contained insulin and 255 of these had insulitis.
Analysis of the immune cell infiltrate in T1D islets revealed that, during the initial phase of beta cell death within a given islet (i.e. those defined as categories 4 and 5 in terms of their insulin content), CD8+ cytotoxic T cells comprised the most abundant immune cell population. The presence of CD8+ cells in inflamed islets concurs with that in biopsied pancreases [4] and with the first ever autopsy case report [16]. This finding is also in agreement with studies in the LEW.1AR1/Ztm-iddm (insulin-dependent diabetes mellitus: IDDM) model of T1D, where CD8+ T cells form the predominant immune cell type in inflamed islets at disease onset and during later stages of infiltration [3]. Similarly, in the non-obese diabetic mouse, T cells are present in high numbers during both the pre-diabetic and diabetic stages [17]. Hence, proteins secreted by CD8+ T cells (e.g. perforin and granzymes [18]) might be expected to play an important role in mediating the early demise of beta cells within an islet. However, in contrast to earlier work [16], we also noted the presence of significant numbers of CD68+ cells (macrophages) within the early infiltrate which might contribute to the initial phase of beta cell death (e.g. by secretion of proinflammatory cytokines such as interleukin-1β[19]). CD68+ cells were seen in the islets of control paediatric patients only occasionally and, therefore, we conclude that they are likely to be recruited actively during the process of insulitis. Their numbers remained fairly constant during all phases of insulitis, suggesting that beta cells may be exposed to elevated levels of proinflammatory cytokines throughout the period of inflammation. As a result, the stimulus for increased Fas expression (which is mediated by interleukin-1β and has been seen in the beta cells of patients with T1D previously [20]) could be maintained throughout the evolution of insulitis. These results are consistent with observations in the LEW.1AR1/Ztm-iddm (IDDM) rat, where macrophages were present in the infiltrate at all stages of disease [3]. In the rat model, these cells were predominant during early stages of insulitis but increasing numbers of CD8+ cells were seen during disease development. Because the present human samples were all analysed after disease onset it is not possible to assess whether macrophages were predominant during the pre-diabetic phase, although it is interesting to note that CD8+ T cells still outnumbered macrophages in those islets which retained high levels of insulin immunopositivity.
During the period when few residual beta cells remain within islets (category 2) CD8+ T cells increased in number (and remained the predominant cell type), but the largest proportionate increase was in the population of CD20+ cells. Thus, these cells appear to be recruited to the islets in greatest numbers once the decline in beta cells is established. A similar situation has been reported in both the non-obese diabetic mouse and the BB rat, where B lymphocytes are either low in numbers or undetectable during early stages of insulitis, but increase as insulitis proceeds [17,21]. By contrast, in the IDDM rat model B cells did not form a major component of the inflammatory infiltrate of islets at any stage [3]. The role of CD20+ cells remains to be established in man but a recent study in mice has suggested that these cells are necessary for the continued survival of cytotoxic T cells within inflamed islets, as B cell deficiency resulted in increased apoptosis of cytotoxic T cells [22]. It is possible, therefore, that they may fulfil a similar role in man. It is unlikely that the B cells detected with anti-CD20 anti-serum within inflamed islets were secreting antibody actively, because CD20 expression is lost during the differentiation to antibody-secreting plasma cells [23]. In order to confirm this conclusion, a second B cell marker (CD138) was examined in a subset of the patient samples, as plasma cells are known to express this protein. This analysis revealed that CD138+ cells were found only very infrequently in inflamed islets, confirming that antibody secretion is unlikely to represent a major function of the B cell population. Nevertheless, it cannot be excluded that modest autoantibody secretion within the islet milieu might lead to exacerbation of beta cell loss in some patients. It is interesting to note that CD138+ cells were not detected within inflamed islets that lacked CD20+ B cells, suggesting the possibility that a common stimulus may induce the migration of both of these cell types in a subset of T1D patients.
At no stage did the complement of CD4+ cells form a major component of the immune infiltrate, an observation similar to that in the IDDM rat model of T1D [3]. Having said this, it is important to note that the functional activity of CD4+ cells varies according to the ratio of T helper cell (Th1 and Th2) subtypes [24]. Therefore, it is conceivable that the lack of efficiency with which beta cells are killed during insulitis might reflect the opposing activities of two populations of immune cells, one of which is protective to beta cells. In this context, a number of recent studies have suggested that, within the CD4+ T cell population, Treg cells may play an important role in regulating the inflammatory process. Specific markers of Treg cells have been difficult to define in man although, in mice, FoxP3 is often utilized as a selective marker of the Treg population. By contrast, in humans, FoxP3 is also expressed transiently during the activation of Teff cells [7], although it is present in these cells at a much lower level than that found in Tregs. Thus, while FoxP3 is not entirely specific for Treg cells in man, the fact that only very few FoxP3+ cells were observed within the islet infiltrate suggests that Treg cells do not form a major component of the inflammatory population in human T1D. Moreover, the transient and low level of FoxP3 expression in Teff cells may not be sufficient to allow its accurate detection in these cells using standard immunohistochemical techniques in post-mortem samples. Thus, we consider it likely that the small number of FoxP3+ cells observed in the T1D islets in the present work were probably Treg cells. Whatever their precise identity, FoxP3+ cells were not found commonly in inflamed islets.
Natural killer cells were also detected only rarely in inflamed islets in the present study, a result which differs from a previous report implying the presence of large numbers of NK cells at the site of insulitis in humans [25]. In drawing this conclusion, it must be accepted that the unequivocal detection of NK cells in man is fraught with difficulty because of the lack of highly specific markers and the probable existence of several NK subsets having different phenotypes. However, we used a range of different markers in an attempt to define NK cells and were not able to detect the presence of large numbers of these cells in inflamed islets. This result concurs with our observation that the total number of CD45+ cells was approximately equal to the summated number of CD8+, CD4+ and CD20+ cells within any given islet, suggesting that no additional CD45+ immune cell type was present in large numbers. This conclusion can be drawn because NK cells are immunopositive for CD45 but do not express CD4, CD8 or CD20. The relative lack of NK cells is also consistent with previous findings that major histocompatibilty complex (MHC) class 1 antigens are overexpressed by the endocrine cells during islet inflammation in man [16,26], and the understanding that engagement of MHC class 1 ligands is inhibitory to NK cell function [9,10]. Thus, even if NK cells were present in inflamed islets, it is unlikely that these could be involved functionally in beta cell cytotoxicity as they would be inhibited by the overexpression of MHC class 1 antigens. Therefore, we conclude that the detection of CD56+ or Pen5+ NK cells in the inflamed islets of patients with T1D only rarely is consistent with the likelihood that these cells play, at best, a very minor role in the disease process.
Once beta cell destruction was complete and the islets became insulin-deficient (category 1), the number of all subsets of immune cells declined dramatically, suggesting that immune cells are retained within islets only when viable beta cells are present. Glucagon staining was normal in such islets, consistent with the failure of infiltrating immune cells to induce the death of α-cells. This implies that the principal chemokine gradients promoting immune cell recruitment and retention probably originate from within the beta cells themselves and that, once these cells are lost, the stimulus for insulitis declines [27,28].
In conclusion, the present study has revealed, in detail, the immune cell subsets present within the islets at different stages of beta cell destruction in recent-onset human T1D. The results imply that specific immune cells are recruited in a defined temporal manner during insulitis to mediate beta cell death in man.
Acknowledgments
We thank Diabetes UK and a Coordinated Action of the European Union (TONECA) for financial support of this study. We also thank Eric Vivier from the Centre d'Immunologie Marseille-Luminy for the kind gift of the Pen5 antibody.
Declarations of interest
The authors have no declarations of interest to declare.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Table S1. List of antibodies used in this study.
Table S2. Immune cell numbers in each islet with varying levels of insulin-positive staining (percentage insulinpositive area/total islet section ranging from low to high).
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- 1.Yang Y, Santamaria P. Lessons on autoimmune diabetes from animal models. Clin Sci (Lond) 2006;110:627–39. doi: 10.1042/CS20050330. [DOI] [PubMed] [Google Scholar]
- 2.Lally FJ, Ratcliff H, Bone AJ. Apoptosis and disease progression in the spontaneously diabetic BB/S rat. Diabetologia. 2001;44:320–24. doi: 10.1007/s001250051621. [DOI] [PubMed] [Google Scholar]
- 3.Jorns A, Gunther A, Hedrich HJ, Wedekind D, Tiedge M, Lenzen S. Immune cell infiltration, cytokine expression, and beta-cell apoptosis during the development of type 1 diabetes in the spontaneously diabetic LEW.1AR1/Ztm-iddm rat. Diabetes. 2005;54:2041–52. doi: 10.2337/diabetes.54.7.2041. [DOI] [PubMed] [Google Scholar]
- 4.Itoh N, Hanafusa T, Miyazaki A, et al. Mononuclear cell infiltration and its relation to the expression of major histocompatibility complex antigens and adhesion molecules in pancreas biopsy specimens from newly diagnosed insulin-dependent diabetes mellitus patients. J Clin Invest. 1993;92:2313–22. doi: 10.1172/JCI116835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Foulis AK. The pathogenesis of beta cell destruction in type I (insulin-dependent) diabetes mellitus. J Pathol. 1987;152:141–8. doi: 10.1002/path.1711520302. [DOI] [PubMed] [Google Scholar]
- 6.Foulis AK, Liddle CN, Farquharson MA, Richmond JA, Weir RS. The histopathology of the pancreas in type 1 (insulin-dependent) diabetes mellitus: a 25-year review of deaths in patients under 20 years of age in the United Kingdom. Diabetologia. 1986;29:267–74. doi: 10.1007/BF00452061. [DOI] [PubMed] [Google Scholar]
- 7.Roncador G, Brown PJ, Maestre L, et al. Analysis of FOXP3 protein expression in human CD4+CD25+ regulatory T cells at the single-cell level. Eur J Immunol. 2005;35:1681–91. doi: 10.1002/eji.200526189. [DOI] [PubMed] [Google Scholar]
- 8.Allan SE, Crome SQ, Crellin NK, et al. FOXP3 in human T effector cells does not suppress proliferation or cytokine production. Int Immunol. 2007;19:345–54. doi: 10.1093/intimm/dxm014. [DOI] [PubMed] [Google Scholar]
- 9.Caligiuri MA. Human natural killer cells. Blood. 2008;112:461–9. doi: 10.1182/blood-2007-09-077438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vivier E, Nunès JA, Vély F. Natural killer cell signalling pathways. Science. 2004;306:1517–19. doi: 10.1126/science.1103478. [DOI] [PubMed] [Google Scholar]
- 11.Vivier E, Munroe M, Ariniello P, Anderson P. Identification of tissue-infiltrating lymphocytes expressing PEN5, a mucin-like glycoprotein selectively expressed on natural killer cells. Am J Pathol. 1995;146:409–18. [PMC free article] [PubMed] [Google Scholar]
- 12.Jahromi MM, Eisenbarth GS. Cellular and molecular pathogenesis of type 1A diabetes. Cell Mol Life Sci. 2007;64:865–72. doi: 10.1007/s00018-007-6469-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ehses JA, Perren A, Eppler E, et al. Increased number of islet-associated macrophages in type 2 diabetes. Diabetes. 2007;56:2356–70. doi: 10.2337/db06-1650. [DOI] [PubMed] [Google Scholar]
- 14.Sherry NA, Tsai EB, Herold KC. Natural history of beta-cell function in type 1 diabetes. Diabetes. 2005;54(Suppl. 2):S32–9. doi: 10.2337/diabetes.54.suppl_2.s32. [DOI] [PubMed] [Google Scholar]
- 15.Achenbach P, Bonifacio E, Koczwara K, Ziegler AG. Natural history of type 1 diabetes. Diabetes. 2005;54(Suppl. 2):S25–31. doi: 10.2337/diabetes.54.suppl_2.s25. [DOI] [PubMed] [Google Scholar]
- 16.Bottazzo GF, Dean BM, McNally JM, MacKay EH, Swift PG, Gamble DR. In situ characterization of autoimmune phenomena and expression of HLA molecules in the pancreas in diabetic insulitis. N Engl J Med. 1985;313:353–60. doi: 10.1056/NEJM198508083130604. [DOI] [PubMed] [Google Scholar]
- 17.Signore A, Pozzilli P, Gale EA, Andreani D, Beverley D. The natural history of lymphocyte subsets infiltrating the pancreas of NOD mice. Diabetologia. 1989;32:282–9. doi: 10.1007/BF00265543. [DOI] [PubMed] [Google Scholar]
- 18.Pearl-Yafe M, Kaminitz A, Yolcu ES, Yaniv I, Stein J, Askenasy N. Pancreatic islets under attack: cellular and molecular effectors. Curr Pharm Des. 2007;13:749–60. doi: 10.2174/138161207780249155. [DOI] [PubMed] [Google Scholar]
- 19.Cnop M, Welsh N, Jonas JC, Jorns A, Lenzen S, Eizirik DL. Mechanisms of pancreatic beta-cell death in type 1 and type 2 diabetes: many differences, few similarities. Diabetes. 2005;54(Suppl. 2):S97–107. doi: 10.2337/diabetes.54.suppl_2.s97. [DOI] [PubMed] [Google Scholar]
- 20.Moriwaki M, Itoh N, Miyagawa J, et al. Fas and Fas ligand expression in inflamed islets in pancreas sections of patients with recent-onset Type I diabetes mellitus. Diabetologia. 1999;42:1332–40. doi: 10.1007/s001250051446. [DOI] [PubMed] [Google Scholar]
- 21.Hanenberg H, Kolb-Bachofen V, Kantwerk-Funk G, Kolb H. Macrophage infiltration precedes and is a prerequisite for lymphocytic insulitis in pancreatic islets of pre-diabetic BB rats. Diabetologia. 1989;32:126–34. doi: 10.1007/BF00505185. [DOI] [PubMed] [Google Scholar]
- 22.Brodie GM, Wallberg M, Santamaria P, Wong FS, Green EA. B-cells promote intra-islet CD8+ cytotoxic T-cell survival to enhance type 1 diabetes. Diabetes. 2008;57:909–17. doi: 10.2337/db07-1256. [DOI] [PubMed] [Google Scholar]
- 23.Harada H, Kawano MM, Huang N, et al. Phenotypic difference of normal plasma-cells from mature myeloma cells. Blood. 1993;81:2658–63. [PubMed] [Google Scholar]
- 24.Sia C. Imbalance in Th cell polarization and its relevance in type 1 diabetes mellitus. Rev Diabet Stud. 2005;2:182–6. doi: 10.1900/RDS.2005.2.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dotta F, Censini S, van Halteren AGS, et al. Coxsackie B4 virus infection of beta cells and natural killer cell insulitis in recent-onset type 1 diabetic patients. Proc Natl Acad Sci USA. 2007;104:5115–20. doi: 10.1073/pnas.0700442104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Foulis AK, Farquharson MA, Hardman R. Aberrant expression of class II major histocompatibility complex molecules by B cells and hyperexpression of class I major histocompatibility complex molecules by insulin containing islets in type 1 (insulin-dependent) diabetes mellitus. Diabetologia. 1987;30:333–43. doi: 10.1007/BF00299027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Frigerio S, Junt T, Lu B, et al. Beta cells are responsible for CXCR3-mediated T-cell infiltration in insulitis. Nat Med. 2002;8:1414–20. doi: 10.1038/nm1202-792. [DOI] [PubMed] [Google Scholar]
- 28.Hill NJ, Hultcrantz M, Sarvetnick N, Flodstrom-Tullberg M. The target tissue in autoimmunity – an influential niche. Eur J Immunol. 2007;37:589–97. doi: 10.1002/eji.200636368. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.