

Abstract
The aim of this work was to analyse C4 genotypes, C4 protein levels, phenotypes and genotypes in patients with the classical form of 21-hydroxylase deficiency. Fifty-four patients from 46 families (36 female, 18 male; mean age 10·8 years) with different clinical manifestations (31 salt-wasting; 23 simple-virilizing) were studied. Taq I Southern blotting was used to perform molecular analysis of the C4/CYP21 gene cluster and the genotypes were defined according to gene organization within RCCX modules. Serum C4 isotypes were assayed by enzyme-linked immunosorbent assay. The results revealed 12 different haplotypes of the C4/CYP21 gene cluster. Total functional activity of the classical pathway (CH50) was reduced in individuals carrying different genotypes because of low C4 concentrations (43% of all patients) to complete or partial C4 allotype deficiency. Thirteen of 54 patients presented recurrent infections affecting the respiratory and/or the urinary tracts, none of them with severe infections. Low C4A or C4B correlated well with RCCX monomodular gene organization, but no association between C4 haplotypes and recurrent infections or autoimmunity was observed. Considering this redundant gene cluster, C4 seems to be a well-protected gene segment along the evolutionary process.
Keywords: adrenal disease, complement, endocrine immunology disease, MHC, steroids
Introduction
The incidence of primary immunodeficiencies in the Caucasian population is approximately 1 : 3000, excluding cases of selective immunoglobulin (Ig)A deficiency [1]. Deficiencies of the complement system represent 2–6% of all primary immunodeficiency cases [2,3]. Complement plays a role in the innate and adaptative immune system. It interacts and modulates the specific immune response using specific receptors and molecular interactions [4,5].
C4 is an essential component of the classical and lectin activation pathways [6]. The genetics of human complement C4 is complex. The high polymorphic C4 proteins are categorized into two isotypes, C4A and C4B, with multiple allotypes [7,8].
An interindividual variation in the C4 gene copy number and dichotomies of C4 gene size and C4 protein isotypes were observed [9]. Theoretically, two to eight copies of C4 genes can be present in a diploid genome of an individual. The C4 gene size has two forms: long and short. The long and short genes are 21 kb and 14·6 kb in length respectively [10]. The duplication or multiplication of C4 genes also includes three neighbouring genes: the nuclear protein kinase RP1, the steroid cytochrome P450 21-hydroxylase (CYP21) and extracellular matrix protein tenascin-X (RCCX = RP, C4, CYP21, TNX genes) [11].
The C4-CYP21 complex is located about 400 kb from the DR locus of the major histocompatibility complex (MHC) class II region [8]. There are two copies of the 21-hydroxylase gene, a functional gene termed CYP21A2 and a pseudogene called CYP21A1P. The pseudogene is intercalated and arranged in tandem together with the C4A and C4B genes [12]. The CYP21A2 gene is located 3′ to C4B, and genetic alterations in the C4 genes are associated frequently with congenital adrenal hyperplasia (CAH) because of 21-hydroxylase deficiency (21-OHD) [13]. There is a continuous spectrum of manifestations of this disease, ranging from classical and severe forms such as ‘salt-losing’ and ‘simple-virilizing’ to non-classical forms which present with mild to moderate clinical manifestations (late onset form) or may occur even without phenotypic manifestations (cryptic form) [12–14].
Total C4 deficiency is rare but, on the other hand, heterozygous or partial deficiencies of C4A (C4AQ0) or C4B (C4QB0) affects approximately 35% of all individuals and about 1% express only a single C4 allele. C4A and C4B null alleles have been associated with systemic lupus erythematosus, insulin-dependent diabetes mellitus, IgA nephropathy, Schönlein–Henoch purpura, subacute sclerosing panencephalitis, autoimmune chronic active hepatitis, membranoproliferative glomerulonephritis, rapid progressive human immunodeficiency virus disease and other disorders [9,15–18]. These associations may be due to either C4 deficiency itself or to the linkage with other MHC III (6p21) genes or both. Individuals with total deficiency of C4B (homozygous C4BQ0) have a higher risk for bacterial meningitis [19]. On the contrary, excessive C4 or overactivation of C4 could aggravate an inflammatory response and render an individual more vulnerable to tissue injuries [9].
Considering the relationship of the genes encoding C4 and 21-hydroxylase, the aim of this study was to evaluate C4 isotype levels, the RCCX modules and the occurrence of recurrent infections and/or autoimmune diseases in Brazilian patients with 21-OHD.
Methods
Patients
Fifty-four patients from 46 families with established diagnosis of classical 21-OHD were included in the study. They have been followed at the Pediatric Endocrinology Outpatient Clinic at University of Campinas (UNICAMP) Medical School Hospital, Campinas, Brazil. All patients presented signs and symptoms of virilization, high plasma levels of 17-hydroxyprogesterone and androstenedione, associated or not with salt-losing history. The clinical and laboratory 21-OHD diagnosis were confirmed in all cases by the occurrence of two affected CYP21A2 alleles. The molecular evaluation included Southern blotting [20] of Taq I digested genomic DNA and allele-specific hybridization or allele-specific polymerase chain reaction (PCR) genotyping [21].
Thirty-six of the 54 patients were female, 31 (22 female, nine male) had the salt-wasting form and 23 (14 female, nine male) had the simple-virilizing form of the disease. The mean age was 10·8 years (ranging from 4 to 22 years). An eventual clinical history of recurrent infections and autoimmune disorders was evaluated. The definition for the recurrent infections was at least six upper respiratory infections per year [22] or two pneumonias in 1 year [23] or two urinary tract infections [24]. Autoimmunity was considered only for defined diseases and the isolated finding of anti-nuclear factor was not a diagnostic criteria. The study was approved by the UNICAMP Medical School Ethics Committee. All individuals gave written informed consent.
Analysis of the complement activation and C4 isotypes
Haemolytic assays for the classical (CH50, normal range: 53–110 IU/ml) and the alternative pathway (AH50, normal range: 34–78 IU/ml) were performed as described by Mayer [25] and Joiner et al.[26] respectively. Ethylenediamine tetraacetic acid (EDTA)-plasma samples were used for biochemical analysis of C4 isotypes. C4 isotypes (C4A and C4B) were evaluated by isotype-specific enzyme-linked immunosorbent assay (ELISA), basically described by Chrispeels et al.[27]. In brief, microtitre plates (Nunc, Wiesbade, Germany) were coated overnight at 4°C with rabbit anti-human C4 IgG (Dakopatts, Hamburg, Germany). Non-specific binding sites were blocked with phosphate-buffered saline, 1% bovine serum albumin, followed by the application of diluted samples or standards. After incubation with non-specific mouse anti-human IgG, C4A and C4B antibodies (Dianova, Hamburg, Germany) were added to the samples for 60 min respectively. The reaction was visualized by addition of 2·2-azino-bist (3-ethyleneben/thiazoline-6-sulphone) (Sigma, St Louis, MO, USA) and H2O2, and stopped with 0·2 M oxalic acid. The normal range (established previously in healthy volunteers) for C4A was 70–200 µg/ml, and for C4B from 200 to 400 µg/ml (M. K., Heidelberg).
Genotype analysis of the C4/CYP21 locus
Molecular analysis of the C4/CYP21 gene cluster was performed as described elsewhere [21,28]. Briefly, after Taq I digestion DNA samples were submitted to electrophoresis in 0·8% agarose gel in Tris-borate-EDTA buffer (0·09 M Tris-HCl; 0·09 M boric acid; 20 mM EDTA, pH 8·0). DNA samples in agarose gels were transferred to Hybond N+ membranes (Amersham Biosciences, Uppsala, Sweden) by Southern blotting and hybridized separately to radioactively labelled C4 (pC4B550) and CYP21A2 (pC21/3c) cDNA probes. On the basis of differences in both Taq I restriction fragment length polymorphism and band intensities, the C4/CYP21 gene organization in RCCX modules was revealed and used to define genotypes for each individual within a family. Band intensitites were obtained in a LKB 222-020 Ultroscan XL Laser Densitometer (Pharmacia, Uppsala, Sweden). Autoradiographies obtained with different exposition times were used to evaluate gene copy number. Figure 1 illustrates how C4 and CYP21 genes are organized in monomodular, bimodular and trimodular alleles and their Taq I variants. The absence of either C4 or CYP21 corresponding fragments indicated homozygous deletion, whereas decreased hybridization signals indicated a heterozygous condition. Increased hybridization signals were considered duplicated genes. Alleles carrying C4B long or short gene copies were recognized by 6·0 kb or 5·4 kb Taq I fragments respectively. To avoid misinterpretation between duplicated and deleted alleles, parents and non-affected siblings were analysed, allowing the verification of correct allelic segregation.
Fig. 1.

C4 and CYP21 genes organization in modular alleles and their Taq I variants.
Statistical analysis
Data were presented as mean, standard deviation, minimum and maximum values. Patients were grouped according to clinical history of recurrent infections or autoimmune diseases, the type of CAH and the genotype of C4 modules. The results of complement and C4 allotypes were compared among groups using the Mann–Whitney U-test. The P-value accepted for statistical significance was 0·05.
Results
Table 1 shows clinical features (sex, age, clinical form of 21-OHD, recurrent infections and autoimmune disorders), biochemical data of the complement pathway (CH50, APH50, C4A, C4B) and molecular data of C4 genotype (C4A and C4B genes) from the 54 patients evaluated.
Table 1.
Biochemical data of complement pathway and C4A and C4B haplotypes from 54 patients with classical 21-hydroxylase deficiency.
| C4 genotype |
C4 genotype |
||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Patient | 21-OHD | Sex | Age | CH50 | APH50 | C4A | C4B | Father | Mother | Patient | 21-OHD | Sex | Age | CH50 | APH50 | C4A | C4B | Father | Mother |
| 1 | SV | M | 16 | 27 | 0 | 54 | 129 | LL | LL | 28‡ | SW | M | 6 | 57 | 67 | 103 | 257 | LLL | LL |
| 2†‡ | SV | F | 9 | 46 | 27 | 56 | 138 | LL | LL | 29 | SW | M | 5 | 74 | 42 | 112 | 358 | LSS | L |
| 3 | SV | M | 13 | 44 | 31 | 101 | 228 | LL | LL | 30† | SW | M | 10 | 88 | 66 | 0 | 517 | LS | LS |
| 4 | SV | F | 9 | 41 | 48 | 81 | 338 | LSS | LL | 31† | SW | M | 12 | 61 | 54 | 0 | 245 | L | L |
| 5 | SV | F | 9 | 46 | 55 | 83 | 203 | LL | LL | 32 | SW | M | 7 | 76 | 52 | 122 | 506 | LSS | LLL |
| 6 | SV | F | 17 | 67 | 55 | 114 | 274 | LL | LL | 33 | SW | F | 11 | 36 | 35 | 84 | 255 | LL | LS |
| 7 | SV | F | 17 | 53 | 46 | 66 | 193 | LL | LL | 34 | SW | F | 10 | 52 | 50 | 133 | 394 | LL | LS |
| 8† | SV | F | 11 | 69 | 58 | 108 | 255 | LS | LS | 35 | SW | M | 6 | 51 | 51 | 43 | 126 | LL | LS |
| 9 | SV | M | 10 | 69 | 50 | 109 | 266 | LS | LS | 36†¶ | SW | F | 8 | 47 | 23 | 132 | 27 | S | S |
| 10 | SV | F | 9 | 77 | 75 | 129 | 24 | L | LL | 37 | SW | F | 7 | 49 | 47 | 91 | 220 | LL | LL |
| 11 | SV | F | 12 | 49 | 37 | 113 | 130 | LL | LL | 38 | SW | F | 15 | 45 | 47 | 80 | 480 | LL | LSS |
| 12 | SV | M | 7 | 58 | 36 | 97 | 243 | LS | LL | 39 | SW | F | 16 | 50 | 52 | 112 | 46 | L | LL |
| 13 | SV | F | 22 | 60 | 51 | 53 | 281 | LS | LL | 40 | SW | F | 10 | 38 | 34 | 40 | 238 | LS | LS |
| 14 | SV | M | 7 | 51 | 55 | 47 | 235 | LS | LL | 41 | SW | F | 12 | 53 | 54 | 123 | 481 | LLL | LS |
| 15 | SV | M | 11 | 47 | 42 | 46 | 244 | LL | S | 42 | SW | F | 16 | 72 | 34 | 131 | 400 | LS | LS |
| 16 | SV | F | 8 | 50 | 65 | 154 | 357 | LLL | LL | 43† | SW | M | 14 | 58 | 32 | 98 | 279 | LS | LS |
| 17 | SV | M | 12 | 48 | 36 | 61 | 140 | LL | LL | 44 | SW | F | 10 | 54 | 37 | 107 | 260 | LL | LS |
| 18 | SV | M | 14 | 0 | 40 | 23 | 44 | LL | LS | 45 | SW | M | 12 | 50 | 36 | 115 | 152 | LL | L |
| 19 | SV | F | 10 | 24 | 46 | 39 | 105 | LL | LS | 46 | SW | F | 7 | 56 | 43 | 105 | 295 | LS | LL |
| 20 | SV | M | 14 | 57 | 47 | 99 | 190 | LL | LL | 47†¶ | SW | F | 12 | 55 | 46 | 109 | 405 | LL | LSS |
| 21 | SV | F | 6 | 60 | 51 | 50 | 359 | LL | L | 48§ | SW | F | 6 | 59 | 53 | 106 | 295 | LL | LS |
| 22 | SV | F | 16 | 40 | 32 | 78 | 232 | LL | LL | 49§ | SW | F | 13 | 54 | 52 | 95 | 145 | LL | LL |
| 23 | SV | F | 5 | 31 | 35 | 42 | 237 | LS | LS | 50 | SW | F | 8 | 58 | 53 | 162 | 439 | LS | LS |
| 24 | SW | M | 15 | 64 | 40 | 162 | 413 | LS | LL | 51 | SW | F | 4 | 56 | 51 | 120 | 481 | LL | LL |
| 25 | SW | F | 9 | 55 | 58 | 139 | 441 | LS | LL | 52 | SW | F | 12 | 46 | 50 | 130 | 217 | LL | LLL |
| 26†‡¶ | SW | F | 14 | 64 | 53 | 39 | 260 | LS | LS | 53‡ | SW | F | 5 | 65 | 61 | 105 | 260 | LL | LSS |
| 27† | SW | F | 9 | 69 | 65 | 124 | 303 | LL | LR | 54 | SW | F | 15 | 43 | 37 | 99 | 331 | LL | LL |
Urinary tract infections;
upper airway infections;
lung infections;
autoimmune disorders (hypothyroidism). Normal ranges: CH50 = 53–110 IU/ml; APH50 = 34–78 IU/ml; C4A = 70–200 µg/ml; C4B = 200–400 µg/ml. SV, simple-virilizing form; SW, salt-wasting form; M, male; F, female; age, years.
Among the 46 families with 21-OHD evaluated, 12 different haplotypes were observed, according C4+CYP21 gene organization (Table 1, Fig. 2). Bimodular haplotypes were the most frequent and were combined in eight distinct genotypes, in 30 families: LL/LS (a/b; n = 7), where LL means C4A7·0/C4B6·0 and LS means C4A7·0/C4B5·4; LL/LL (a/a; n = 10); LS/LS (b/b; n = 4), LL/LR (a/c; n = 1), LR means C4A7·0/C4B3·9; LS/LS (b/f; n = 2); LS/LL (b/g; n = 1); LL/LL (a/g; n = 4; LL/LL (g/g; n = 1). Combinations of bi- and trimodular or tri- and trimodular units were observed in eight families: LS/LLL (b/d; n = 2); LL/LLL (a/d; n = 1); LL/LSS (a/e; n = 1); LLL/LSS (d/e; n = 1); LSS/LL (e/g; n = 1); and LL/LSS (a/h; n = 1). In eight families different genotypes with monomodular units were observed: LL/L (a/i; n = 3); L/L (i/i; n = 1); S/S (j/j; n = 1); LL/L (g/i; n = 1); LL/S (a/k; n = 1), S in this case is C4A6·4; L/LSS (i/l; n = 1). Distinct C4 gene organizations were dependent on CYP21 gene deletions, conversions or mutations.
Fig. 2.
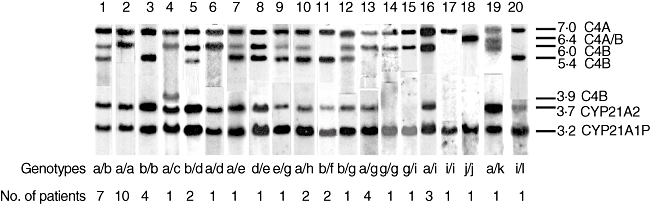
C4 and CYP21 genotypes and 21-hydroxylase deficiency patient numbers of each one.
Biochemical analysis of the complement pathway and C4 isotypes were expressed as the mean ± standard deviation, minimum and maximum values: CH50 = 53·1 ± 14·4 IU/ml (0–88), AH50 = 46·2 ± 12·7 IU/ml (0–75), C4A = 91·2 ± 38·5 µg/ml (0–162), and C4B = 266·1 ± 124·1 µg/ml (24–517) (Table 1).
Levels of CH50 (nadir 110 IU/ml) and APH50 (nadir 78 IU/ml) were not above the normal range in any patient, but CH50 (nadir 53 IU/ml) was below the normal range in 24 patients and undetectable in one (patient 18: Table 1), and APH50 (nadir 34 IU/ml) was below the normal range in only six patients and undetectable in one (patient 1: Table 1). Fourteen patients presented C4A values below the lower limit for the normal range (nadir 70 µg/ml). They carry in their genotypes either CYP21A2 gene conversion (LL/LL, n = 4; LS/LS, n = 3; LS/LL, n = 4) or C4/CYP21 gene deletion (LL/L, n = 2; L/L, n = 1). The C4B levels were elevated in nine patients (nadir 400 µg/ml) from whom five presented genotypes carrying three or four copies of C4B gene that combine either bi- and trimodular or tri- and trimodular haplotypes respectively (LS/LLL, n = 2; LLL/LSS, n = 1; LS/LLL, n = 1; LSS/LLL, n = 1); and four patients with bi and bimodular (LS/LL, n = 3; LS/LS, n = 1). One of these patients with high C4B expression and a genotype LS/LS had an undetectable C4A plasma level (patient 30: Table 1). Low levels for both variants C4A and C4B (nadir 200 µg/ml) were observed in six patients; all of them carry two bimodular haplotypes (LL/LL, n = 4; LL/LS, n = 2). Two patients (siblings) presented low levels of CH50, APH50, C4A and C4B (patients 1 and 2: Table 1). Two other siblings (patients 17 and 18: Table 1) and a non-related patient (patient 35: Table 1) had low levels of CH50, C4A and C4B.
Thirteen of 54 patients presented recurrent infections affecting the urinary tract (n = 8; Table 1: patients 2, 26, 27, 30, 31, 36, 43, 47), the upper airways (n = 5; Table 1: patients 2, 8, 26, 28, 53), the lungs (n = 2; Table 1: patients 48, 49) and urinary tract combined with upper airways infection (n = 2; Table 1: patients 2, 26). An additional three patients suffered from autoimmune disorders (hypothyroidism, n = 3; Table 1: patients 26, 36, 47), all associated with recurrent urinary infections, one of them combined with upper airway infections (Table 1: patient 26). None of them had severe infections.
Complement and C4 isotypes analysis showed a significant difference only for CH50 between groups with (60·9 ± 10·9 IU/ml) and without (50·7 ± 14·6 IU/ml) recurrent infections (Z = −2·45; P = 0·01). No statistical differences between groups with or without autoimmune diseases were detected.
Regarding the C4 modular genotype, significant differences were observed between LL/LL and LL/LLL for C4 (20·8 ± 5·2 mg/dl and 33·5 ± 0·7 mg/dl; Z = −2·24; P = 0·01) and for C4A (88·9 ± 21·6 µg/ml and 142 ± 17 µg/ml; Z = −2·23; P = 0·01). LL/LSS for C4 (20·8 ± 5·2 mg/dl and 30 ± 3·5 mg/dl; Z = −2·71; P = 0·004) and C4B (219 ± 93·3 µg/ml and 370·7 ± 93·9 µg/ml; Z = −2·40; P = 0·01) and LS/LS for CH50 (20·8 ± 5·2 IU/ml and 60·8 ± 17·4 IU/ml; Z = −2·71; P = 0·03) and C4B (219·4 ± 93·3 µg/ml and 321·2 ± 103·3 µg/ml; Z = −2·18; P = 0·004).
Discussion
Genetic recombination is a driving force for diversity and polymorphism. In general, recombination between misaligned homologous chromosomes can result in either deletion or duplication of genes. Furthermore, unequal cross-over between duplicated gene regions is responsible for a variety of human genetic diseases. Such duplications have been observed frequently in the human MHC class III. Located on chromosome 6p21, between class I and class II, the MHC class III encodes genes of various functions, including complement components C2, C4, factor B (Bf) and 21-hydroxylase genes (CYP21A2 and CYP21A1P). The order of the genes in the direction of transcription is C2, BF, C4A, CYP21A1P, C4B, CYP21A2 and they have mapped between human leucocyte antigen (HLA)-B and HLA DR. Because of the tight linkage, these genes are usually inherited as a single complotype. In some haplotypes, linkage disequilibrium includes the HLA-B and HLA-DR regions forming extended haplotypes [29]. The MHC class III region has been studied because of its association with many autoimmune and genetic diseases. A gene cluster containing the duplicated C4 and the CYP21 genes is of particular interest because of its linkage to diseases, such as systemic lupus erythematosous and CAH [30].
C4 is a highly polymorphic protein, which is reflected by a wide concentration range found in the population. There are two distinct classes of C4 protein, C4A and C4B, which have diversified in their predominant functions of opsonization/immune clearance (C4A) and the better-known killing of the pathogens by lyses and neutralization (C4B). C4 isotype levels were evaluated by ELISA using monoclonal antibodies to Rodgers (Rg1) and Chido (Ch1) determinants related to C4A and C4B respectively, so it was a specific binding [9,31].
Although we observed low C4A and C4B protein levels, no direct association with the recurrent respiratory and/or urinary tract infections was significant. Most of our patients had normal CH50 and C4 levels and no complete C4 deficiency. Twenty-five and six patients had, respectively, CH50 and AH50 levels below the normal range (53 IU/ml and 34 IU/ml respectively), reflecting either consumption because of inflammation or (partial) deficiency. Five patients had low levels of both CH50 and AH50.
Heterozygous deficiency of either C4A or C4B is common, with a frequency of approximately 20% each in the Caucasian population; complete deficiencies of both C4A and C4B proteins are extremely rare [31]. However, in patients suffering from infectious disorders and autoimmune diseases, homozygous or heterozygous deficiencies of C4A and/or C4B have been found with a significantly higher frequency. Probably because of the relatively low number of study patients and lack of complete C4 deficiency, we were unable to find a clear disease association with respect to C4 genotypes and C4 levels. Recurrent respiratory infections in our study population might be explained more clearly by the relative immaturity of the immune response system at this age. Urinary tract infections can also be justified by the presence of labial fusion in girls with CAH [32–34]. Although no significant correlation of C4 isotype deficiency was identified clearly in patients with hypothyroidism, it is relevant that three of our young patients presented this autoimmune manifestation. Female patients present hypothyroidism more frequently [35,36]. Recently, Demirbilek et al.[36] reported a mean age of 11·4 years for the hypothyroidism diagnosis in a paediatric population. The three patients with hypothyroidism reported by us were female, and were diagnosed at ages that varied from 8 to 14 years. Therefore, it could be concluded that thyroid impairment is relatively common in females at this age range; however, we evaluated a relatively low number of individuals, finding three of 54 patients affected.
Taq I Southern blot bears information on the dichotomous size variation of C4 genes and the number of RCCX modules – hence, the number of C4 genes – in a diploid genome. Chung et al.[9] tried to establish real-time PCR to determine the number of C4A and C4B genes, but the technique proved to be difficult. In 2000, Blanchong et al.[7] had already described the high variability and complexity of C4 gene size, number and modular variations in 150 healthy Caucasian females and 22 CAH patients. Our findings also indicate a high modular variability among 21-OHD patients.
The specific length variants and the number of RCCX modules are assigned based on presence and absence and relative band intensities. Variation in the number of RCCX modules and the size of the C4 genes leads to the presence of seven common RCCX length variants: monomodular L (long) and S (short); bimodular LL and LS; and trimodular LLL, LSS and LLS (or LSL) [7,37]. In addition, two rare length variants, bimodular SS and quadrimodular LLLL, have been implicated [38,39]. All the patients except one presented common RCCX length variants described previously. This patient (patient 36: Table 1, genotype j/j or S/S) presented a novel haplotype in which the C4A/B 6·4 kb fragment, associated frequently with the deletion of CYP21A1P 3·2 kb fragment, is associated with CYP21A2 3·7 kb deletion. This haplotype was observed for the first time in this patient and is probably a result of two different events of unequal cross-overs, the first resulting in the common haplotype bearing the the C4A/B 6·4 kb fragment and the CYP21A1P 3·2 kb fragment deletion and the second generating the CYP21A2 3·7 kb deletion haplotype. Those events must have occurred in at least three previous generations as the patient's parents, who are first cousins, and the maternal grandfather also carry the same haplotype.
The C4B levels were elevated in nine patients. Four presented genotypes carrying three or four copies of the C4B gene that combine either bi- and trimodular or tri- and trimodular haplotypes respectively. One of these patients with high C4B expression and homozygous for haplotype b had an undetectable C4A plasma level. Low levels for both variants C4A and C4B were observed in six patients; all of them carry two bimodular haplotypes. Fourteen patients presented C4A values below the lower limit for the normal range. They carry either CYP21A2 gene conversion or C4/CYP21 gene deletion. These data indicate that despite the fact that in some cases C4 gene deletion or duplication corresponded to low or high levels of C4 protein respectively, this association was not absolute.
We conclude that in a series of 46 families with 54 patients with CAH because of classical 21-OHD, in spite of the complexity of gene organization found in the cluster C4+CYP21 (12 different haplotypes), laboratory evaluation of complement activation and C4 allotypes were in the normal ranges, and the patients did not present significant recurrent infections or autoimmunity. Considering this redundant gene cluster, C4 seems to be a well-protected gene segment along the evolutionary process. The results show the importance of molecular medicine approach in diagnosing complex diseases and creating conditions for genetic counselling, treatment and prevention of diseases. Definitive and efficient techniques to elucidate the number of C4A and C4B genes and polymorphism of the protein products are important to determine the role that C4A and C4B play in disease associations in the prognosis and therapeutic intervention of autoimmune and inflammatory diseases.
Acknowledgments
This work was supported by grants from the FAPESP (92/03332-6 and 97/07622-2 to M.P.M.), CNPq (300357/01-0 to G. G. Jr and 302334/03-3 to M. P. M.) and FAEP-UNICAMP (0863/98 to G. G. Jr and 022/96 to M. P. M.). We thank Viviana Galimberti Arruk (LIM 56 – USP) for technical assistance in respective laboratory. Our special reference to Dr C. Yung Yu, who revised and contributed to the improvement of our manuscript.
References
- 1.Bonilla FA, Bernstein IL, Khan DA, et al. Practice parameter for the diagnosis and management of primary immunodeficiency. Ann Allergy Asthma Immunol. 2005;94(5)(1) Suppl.:S1–63. doi: 10.1016/s1081-1206(10)61142-8. [DOI] [PubMed] [Google Scholar]
- 2.Grumach AS, Duarte AJ, Bellinatti-Pires R, et al. Brazilian report on primary immunodeficiencies in children: 166 cases studied over a follow-up time of 15 years. J Clin Immunol. 1997;17:240–5. doi: 10.1023/a:1027335000994. [DOI] [PubMed] [Google Scholar]
- 3.Leiva LE, Zelazco M, Oleastro M, et al. Primary immunodeficiency diseases in Latin America: the second report of the LAGID registry. J Clin Immunol. 2007;27:101–8. doi: 10.1007/s10875-006-9052-0. [DOI] [PubMed] [Google Scholar]
- 4.Hawlisch H, Kohl J. Complement and Toll-like receptors: key regulators of adaptive immune responses. Mol Immunol. 2006;43:13–21. doi: 10.1016/j.molimm.2005.06.028. [DOI] [PubMed] [Google Scholar]
- 5.Lutz HU, Jelezarova E. Complement amplification revisited. Mol Immunol. 2006;43:2–12. doi: 10.1016/j.molimm.2005.06.020. [DOI] [PubMed] [Google Scholar]
- 6.Sjoholm AG, Jonsson G, Braconier JH, et al. Complement deficiency and disease: an update. Mol Immunol. 2006;43:78–85. doi: 10.1016/j.molimm.2005.06.025. [DOI] [PubMed] [Google Scholar]
- 7.Blanchong C, Zhou B, Rupert KL, et al. Deficiencies of human complement component C4A and C4B and heterozygosity in length variants of RP-C4-CYP21-TNX (CCX) modules in Caucasians: the load of RCCX genetic diversity on major histocompatibility complex-associated disease. J Exp Med. 2000;191:2183–96. doi: 10.1084/jem.191.12.2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yu CY. The complete exon–intron structure of a human complement C4A gene. J Immunol. 1991;146:1057–66. [PubMed] [Google Scholar]
- 9.Chung EK, Yang Y, Rupert KL, et al. Determining the one, two, three, or four and short loci of human complement C4 in a major histocompatibility complex haplotype encoding C4A or C4B proteins. Am J Hum Genet. 2002;71:810–22. doi: 10.1086/342778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang Y, Chung EK, Zhou B, et al. Diversity in intrinsic strengths of the human complement system: serum C4 protein concentrations correlate with C4 gene size and polygenic variations, hemolytic activities and body mass index. J Immunol. 2003;171:2734–45. doi: 10.4049/jimmunol.171.5.2734. [DOI] [PubMed] [Google Scholar]
- 11.Yang Z, Yu CY. Organizations and gene duplications of the human and mouse MHC complement gene clusters. Exp Clin Immunogenet. 2000;17:1–17. doi: 10.1159/000019119. [DOI] [PubMed] [Google Scholar]
- 12.Clayton PE, Miller WL, Oberfield SE, et al. Consensus statement on 21-hydroxylase deficiency from the European Society for Paediatric Endocrinology and the Lawson Wilkins Pediatric Endocrine Society. Horm Res. 2002;58:188–95. doi: 10.1159/000065490. [DOI] [PubMed] [Google Scholar]
- 13.Merke DP, Bornstein SR. Congenital adrenal hyperplasia. Lancet. 2005;365:2125–36. doi: 10.1016/S0140-6736(05)66736-0. [DOI] [PubMed] [Google Scholar]
- 14.White PC, Speiser PW. Congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Endocr Rev. 2000;21:245–91. doi: 10.1210/edrv.21.3.0398. [DOI] [PubMed] [Google Scholar]
- 15.Liszewski MK, Atkinson JP. The role of complement in autoimmunity. Immunol Ser. 1991;54:13–37. [PubMed] [Google Scholar]
- 16.Lokki ML, Circolo A, Ahokas P, et al. Deficiency of human complement protein C4 due to identical frameshift mutations in the C4A and C4B genes. J Immunol. 1999;162:3687–93. [PubMed] [Google Scholar]
- 17.Samano ET, Ribeiro LM, Gorescu RG, et al. Involvement of C4 allotypes in the pathogenesis of human diseases. Clinics. 2004;59:138–44. doi: 10.1590/s0041-87812004000300009. [DOI] [PubMed] [Google Scholar]
- 18.Yang Y, Chung EK, Wu YL, et al. Gene copy-number variation and associated polymorphisms of complement component C4 in human systemic lupus erythematosus (SLE): low copy number is a risk factor for and high copy number is a protective factor against SLE susceptibility in European Americans. Am J Hum Genet. 2007;80:1037–54. doi: 10.1086/518257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bishof NA, Welch TR, Beischel LS. C4B deficiency: a risk factor for bacteremia with encapsulated organisms. J Infect Dis. 1990;162:248–50. doi: 10.1093/infdis/162.1.248. [DOI] [PubMed] [Google Scholar]
- 20.Araujo M, Sanches MP, Suzuki LA, et al. Molecular analysis of CYP-21 and C4 genes in Brazilian families with the classical forms of the steroid 21-hydroxylase deficiency. Braz J Med Biol Res. 1996;29:1–13. [PubMed] [Google Scholar]
- 21.Paulino LC, Araujo M, Guerra-Junior G, et al. Mutation distribution and CYP21/C4 locus variability in Brazilian families with the classical form of the 21-hydroxylase deficiency. Acta Paediatr. 1999;88:275–83. doi: 10.1080/08035259950170024. [DOI] [PubMed] [Google Scholar]
- 22.Barata RCB, Waldman EA, Moraes JC, Guibu IA, Rozov T, Takimoto S. Gastroenteritis and acute respiratory infections among children up to 5 years old in area of Southeastern Brazil, 1986–1987. I. Acute respiratory infections. Rev Saude Publica. 1996;30:533–63. doi: 10.1590/s0034-89101996000600010. [DOI] [PubMed] [Google Scholar]
- 23.Heffelfinger JD, Davis TE, Gebrian B, Bordeau R, Schwartz B, Dowell SF. Evaluation of children with recurrent pneumonia diagnosed by World Health Organization criteria. Pediatr Infect Dis J. 2002;21:108–12. doi: 10.1097/00006454-200202000-00005. [DOI] [PubMed] [Google Scholar]
- 24.Le Saux N, Pham B, Moher D. Evaluating the benefits of antimicrobial prophylaxis to prevent urinary tract infections in children: a systematic review. Can Med Assoc J. 2000;163:523–9. [PMC free article] [PubMed] [Google Scholar]
- 25.Mayer MM. Complement and complement fixation. In: Kabat EA, Mayer MM, editors. Experimental immuonochemistry. Springfield: Thomas; 1961. pp. 133–240. [Google Scholar]
- 26.Joiner KA, Haiwinger A, Gelfand JA. A study of optimal reaction conditions for assay of the human alternative pathway. Am J Clin Pathol. 1983;79:65–72. doi: 10.1093/ajcp/79.1.65. [DOI] [PubMed] [Google Scholar]
- 27.Chrispeels J, Bank S, Rittner C, et al. Sandwich enzyme-linked immunosorbent assays for the quantification of the C4 isotypes (C4A and C4B) in human plasma. J Immunol Methods. 1989;125:5–12. doi: 10.1016/0022-1759(89)90071-9. [DOI] [PubMed] [Google Scholar]
- 28.Palsdottir A, Arnason A, Fossdal R, et al. Gene organization of haplotypes expressing two different C4A allotypes. Hum Genet. 1987;76:220–4. doi: 10.1007/BF00283611. [DOI] [PubMed] [Google Scholar]
- 29.Yu CY. Molecular genetics of the human MHC complement gene cluster. Exp Clin Immunogenet. 1998;15:212–29. doi: 10.1159/000019075. [DOI] [PubMed] [Google Scholar]
- 30.Rupert KL, Rennebohm RM, Yu CY. An unequal crossover between the RCCX modules of the human MHC leading to the presence of a CYP21B gene and a tenascin TNXB/TNXA-RP2 recombination between C4A and C4B genes in a patient with juvenile reumathoid arthritis. Exp Clin Immunogenet. 1998;16:81–97. doi: 10.1159/000019099. [DOI] [PubMed] [Google Scholar]
- 31.Schneider PM, Stradmann-Bellinghausen B, Rittner C. Genetic polymorphism of the fourth component of human complement: population study and proposal for a revised nomenclature based on genomic PCR typing of Rodgers and Chido determinants. Eur J Immunogenet. 1996;3:335–44. doi: 10.1111/j.1744-313x.1996.tb00006.x. [DOI] [PubMed] [Google Scholar]
- 32.Morton L, Podolsky MD. Labial fusion: a cause of recurrent urinary tract infections. Clin Pediatr (Phila) 1973;12:345–6. [PubMed] [Google Scholar]
- 33.Leung AKC, Robson WLM. Labial fusion and asymptomatic bacteriuria. Eur J Pediatr. 1993;152:250–1. doi: 10.1007/BF01956155. [DOI] [PubMed] [Google Scholar]
- 34.Elder JS. Urologic disorders in infants and children. In: Kliegman R, Behrman RE, Jenson HB, Stanton BF, editors. Nelson textbook of pediatrics. Philadelphia, PA: Saunders Elsevier; 2007. p. 2255. [Google Scholar]
- 35.Lorini R, Gastaldi R, Traggiai C, Perucchin PP. Hashimoto's thyroiditis. Pediatr Endocrinol Rev. 2003;1(Suppl.)(2):205–11. [PubMed] [Google Scholar]
- 36.Demirbilek H, Kandemir N, Gonc EN, Ozon A, Alikasifoglu A, Yordam N. Hashimoto's thyroiditis in children and adolescents: a retrospective study on clinical, epidemiological and laboratory properties of the disease. J Pediatr Endocrinol Metab. 2007;20:1199–205. doi: 10.1515/jpem.2007.20.11.1199. [DOI] [PubMed] [Google Scholar]
- 37.Yang Z, Mendoza AR, Welch TR, et al. Modular variations of the human major histocompatibility complex class III genes for serine/threonine kinase RP, complement component C4, steroid 21-hydroxylase CYP21, and tenascin TNX (the RCCX module). A mechanism for gene deletions and disease associations. J Biol Chem. 1999;274:12147–56. doi: 10.1074/jbc.274.17.12147. [DOI] [PubMed] [Google Scholar]
- 38.Collier S, Sinnott PJ, Dyer PA, et al. Pulsed field gel electrophoresis identifies a high degree of variability in the number of tandem 21-hydroxylase and complement C4 gene repeats in 21-hydroxylase deficiency haplotypes. EMBO J. 1989;8:1393–402. doi: 10.1002/j.1460-2075.1989.tb03520.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Weg-Remers S, Brenden M, Schwarz E, et al. Major histocompatibility complex (MHC) class III genetics in two Amerindian tribes from southern Brazil: the Kaingang and the Guarani. Hum Genet. 1997;100:548–56. doi: 10.1007/s004390050550. [DOI] [PubMed] [Google Scholar]