

Abstract
Multi-drug resistance (MDR) is a major obstacle in cancer chemotherapy. There are contrasting data on a possible correlation between the level of expression of the drug transporter P-glycoprotein (P-gp) and susceptibility to complement-dependent cytotoxicity (CDC). We therefore investigated the sensitivity of human ovarian carcinoma cells and their P-gp expressing MDR variants to complement. Chemoselected P-gp expressing MDR cells showed increased resistance to CDC associated with overexpression of membrane-bound complement regulatory proteins (mCRP) and increased release of the soluble inhibitors C1 inhibitor and factor I. MDR1 gene transfection alone did not alter the susceptibility of P-gp expressing A2780-MDR and SKOV3-MDR cells to CDC. However, subsequent vincristine treatment conferred an even higher resistance to complement to these cells, again associated with increased expression of mCRP. Blocking the function of P-gp with verapamil, cyclosporine A or the anti-P-gp-antibody MRK16 had no impact on their complement resistance, whereas blocking of mCRP enhanced their susceptibility to complement. These results suggest that enhanced resistance of chemoselected MDR ovarian carcinoma cells to CDC is not conferred by P-gp, but is due at least partly to overexpression of mCRP, probably induced by treatment with the chemotherapeutic agents.
Keywords: cancer, complement regulators, complement resistance, P-glycoprotein, drug resistance
Introduction
The development of drug resistance is considered to be one of the major obstacles to efficient anti-tumour chemotherapy. Even after exposure to a single drug, tumour cells often develop co-resistance to several anti-tumour drugs, i.e. multi-drug resistance (MDR) [1]. The best-characterized MDR-promoting factor, the efflux transporter P-glycoprotein (P-gp), is a 170 kDa transmembrane glycoprotein encoded by the mdr1 gene on human chromosome 7p21 [2]. Overexpression of P-gp confers cross-resistance to several anti-tumour agents, including the anthracyclin doxorubicin and the vinca-alkaloid vincristine [3], varying in chemical structure, but all being lipophilic and positively charged [1]. P-gp actively removes lipophilic substrates from the intracellular compartment using hydrolysis of adenosine triphosphate as a source of energy [4]. P-gp may also act as a non-specific ion-channel, contributing to an increased intracellular pH and a decreased membrane potential, both of which reduce intracellular accumulation of positively charged molecules [5]. P-gp function can be blocked either with inhibitors such as verapamil [6] or cyclosporine A (CsA) [7] that compete with P-gp substrates for P-gp binding or with blocking monoclonal antibodies (mAb) such as MRK16 [8].
Immunotherapy of drug-resistant tumour cells with specific anti-tumour antibodies is a potential therapeutic alternative with an advantage of specific tumour targeting. As these antibodies may exert their anti-tumour effects through the action of complement [9,10], we investigated the effect of MDR on complement-mediated lysis. Activation of the complement cascade, either with antigen–antibody complexes that initiate the classical pathway or through the alternative and lectin pathways, results in deposition of opsonising complement proteins such as C3b and formation of the lytic C5b-9 transmembrane complex on the cell surface [11]. Nucleated cells use multiple mechanisms to prevent deposition of complement components and subsequent destruction by the cytolytic membrane attack complex. These mechanisms include the expression of membrane-bound complement regulatory proteins (mCRP) such as CD46, CD55 and CD59, which control the activation of the complement cascade by either affecting the generation of the C3/C5 convertase (CD46, CD55) [12,13] or interfering with the formation of the membrane attack complex (CD59) [14]. The therapeutic potential of anti-cancer antibodies is known to be reduced significantly because of complement resistance mechanisms exploited by tumour cells to evade complement-mediated destruction (for review see: [15]). For example, tumour cells often express higher quantities of mCRP than normal tissues, thereby limiting the efficacy of antibody immunotherapy [15–18].
The P-gp-positive MDR variants have been described previously to exert either enhanced susceptibility to complement attack [19] or enhanced complement resistance [20].
These conflicting findings on the correlation between P-gp-mediated MDR and complement resistance motivated us to analyse further the complement sensitivity of drug-selected and mdr1-gene transfected MDR variants of ovarian carcinoma cell lines, in comparison with parental cells.
Material and methods
Cancer cell lines
The MDR P-gp positive ovarian carcinoma cell variants OAW42-Dox and OAW42-Tax (kindly provided by Dr Volm and Dr Masanek, DKFZ, Heidelberg, Germany) were selected by cultivation of the parental cell line OAW42 in Dulbecco's modified Eagle's medium (DMEM) (BioWhittaker Europe, Verviers, Belgium) with 10% fetal calf serum (FCS) (Gibco, Paisley, Scotland, UK) supplemented with doxorubicin (Dox) (Pharmacia and Upjohn, Erlangen, Germany; final concentration: 19 µg/ml) or taxol (Tax) (Bristol Arzneimittel, Munich, Germany; final concentration: 30 µg/ml). Revertants were obtained by further cultivation of OAW42-Dox and OAW42-Tax cells in drug-free growth medium. Both the chemo-selected OAW42-Dox and OAW42-Tax cells have been described previously to over-express further drug resistance factors such as glutathione peroxidase, glutathione-S-transferase π and thymidylate synthase (OAW42-Dox) or protein kinase C β (OAW42-Tax) compared with their parental cell line OAW42 [21].
The MDR P-gp positive ovarian carcinoma cell lines, designated SKOV3-MDR/2 and A2780-MDR/2, were obtained by transduction of the parental cells with MDR1 gene containing SF-MDR vector [22] and further cultivation in a vincristine (Pharmacia and Upjohn)-containing DMEM (SKOV3-MDR/2: 4nM vincristine; A2780-MDR/2: 250nM vincristine). A second MDR1 gene transduced variant A2780-MDR was not exposed to drugs but sorted in a fluorescence activated cell sorter (FACScan; Becton-Dickinson, Heidelberg, Germany) using monoclonal anti-P-gp-antibody MRK16 (Alexis Biochemicals, Lausen, Switzerland).
Sera and antibodies
Normal human serum (NHS) was prepared from freshly collected blood from healthy donors and was used as a source for complement. Heat-inactivated serum was obtained by incubating the serum for 30 min at 56°C and was used as negative control.
To achieve optimal complement-dependent cytotoxicity (CDC) of the targeted tumour cells and because of the lack of broad reacting CDC-inducing mAb directed to the ovarian tumour cells under investigation, polyclonal rabbit anti-tumour antibodies to OAW42 and SKOV3 were used. The antibodies were generated by three subcutaneous injections of the respective human ovarian carcinoma cells (1 × 106) into rabbits and were used after heat inactivation (30 min; 56°C).
The following mAb directed to mCRP or directed to P-gp were used for flow cytometry: anti-CD59 (BRIC229, IgG2b), anti-CD55 (BRIC110, IgG1) (IBGRL, Bristol, UK), anti-CD46 (J4/48, IgG1, Dianova, Hamburg, Germany) and anti-P-gp (MRK16, IgG2a; Alexis Biochemicals).
For blocking the function of mCRP, we used the non-complement-activating mAb anti-CD59 (BRIC229), anti-CD55 (BRIC110), anti-CD55 (BRIC216, IgG1) (IBGRL) or anti-CD46 (GB24, IgG1, kindly provided by Dr. J. Atkinson, St Louis, MO, USA) [23].
Retroviral transduction of the MDR1 gene
The SF-MDR vector, based on a Friend mink cell focus-forming/murine embryonic stem-cell virus, contains the human MDR1 gene encoding for the P-gp transporter protein [24]. Retroviral supernatant used for MDR1 gene transduction was harvested from 90% confluent layers of GPenv+AM12/SF-MDR producer cells using a 0·45 µm filter after a 16-h cultivation period in DMEM cultivation medium supplemented with 10% FCS and 1% penicillin/streptomycin. For retroviral supernatant transduction of the MDR1 gene, ovarian carcinoma cells were incubated with DMEM containing this cell-free supernatant twice during 48 h.
RH 123-assay for P-pg activity
RH 123-efflux assay was used to assess the functional activity of P-gp by following the removal of the fluorescent agent RH 123 by the P-gp transporter pump out of cells. MRD cells (1 × 106 in 1 ml DMEM) were labelled for 30 min at 37°C with 10 µl lipophilic fluorescent agent RH 123 (Sigma-Aldrich, Steinheim, Germany; final concentration 200 ng/ml). Labelled cells were washed twice and resuspended in 1 ml DMEM. Cells were cultured for 1 h at 37°C to permit removal of RH 123 from MDR cells by P-gp. Again, the cells were washed twice and resuspended in 300 µl phosphate-buffered saline (PBS). Propidium iodide (30 µl) was added (Sigma; final concentration 10 µg/ml) to detect dead cells. Cells were then analysed by flow cytometry in a FACScan (Becton-Dickinson), as described previously [22]. Cells incubated with the P-gp blocker CsA (Sandoz, Basel, Switzerland; final concentration 1·5 µM) were used as negative control.
Complement-mediated cytotoxicity assay
Cells (1 × 106) were labelled for 90 min at 37°C with 51Cr (100 µCi) (Amersham Biosciences, Little Chalfont, Buckinghamshire, England). Labelled cells were washed three times, resuspended in DMEM and adjusted to 2 × 105 cells/ml. Cells (50 µl/well) were incubated for 30 min at 37°C with polyclonal rabbit anti-tumour antibodies and treated for 60 min with NHS (100 µl) as complement source or heat-inactivated serum as control, as described previously by Jurianz et al.[23]. In some experiments the assay was also performed in the presence of Mg2+ethylene glycol tetraacetic acid (EGTA) to analyse for alternative versus classical complement pathway activation. Cells were then centrifuged for 5 min at 300 gand 100 µl of cell-free supernatant were collected. Radioactivity within these aliquots was measured in a γ-counter (Berthold, Bad Wildbach, Germany). Maximal release was achieved by solubilizing cells with 1% Triton X-100 (Roche, Mannheim, Germany), and spontaneous release was determined by incubating the cells with DMEM instead of polyclonal antibodies and NHS. Specific 51Cr release was calculated as follows: specific release (%) = [sample/counts per minute (c.p.m.) − spontaneous c.p.m./max c.p.m. − spontaneous/c.p.m.)] × 100.
Because of large variation in the specific complement-mediated lysis among the different parental ovarian carcinoma cells (ranging from 40% in SKOV3 to 70% in OAW42 and 76% in A2780), the specific lysis of each of the parental cells was set at 100%. Lysis of MDR cells was calculated relative to parental cells lysis (adjusted lysis %). This permitted comparison of the impact of MDR on complement-mediated lysis among the individual cell lines.
Blocking of P-gp and mCRP
Tumour cells were incubated first with anti-tumour antibodies with or without P-gp inhibitors or anti-mCRP-antibodies. Human serum was then added as complement source as described above. To block P-gp function we used either the Ca2+-channel blocker verapamil (final concentration 10 µg/ml; Calbiochem, Schwalbach, Germany), CsA (final concentration 1 µM; Sandoz) or the monoclonal anti-P-gp antibody MRK16 (IgG2a, 10 µg/ml). For blocking mCRP function, tumour cells were preincubated with non-complement-activating neutralizing mAb to CD59 (BRIC229), CD55 (BRIC110, BRIC216) CD46 (GB24) or a combination of these antibodies at 10 µg/ml, as described previously in [23]. In the absence of the sensitizing polyclonal rabbit antibody, no complement-mediated lysis occurred under these conditions.
Flow cytometry analysis of mCRP and P-gp expression
Cells (105/well) were incubated for 30 min on ice with mouse mAb directed to CD59, CD55 or CD46 or to P-gp (MRK16). After washing with FACS buffer (1% bovine serum albumin, 0·1% NaN3 in PBS; PAA Laboratories, Coelbe, Germany), the cells were incubated for 30 min with fluorescein isothiocyanate-labelled F(ab)2 goat anti-mouse IgG (Dianova, Hamburg, Germany), washed again and analysed in a FACScan (Becton-Dickinson). To quantify antigen expression, we used the Dako-QIFI-Kit (Dako, Glostrup, Denmark), in which a series of beads coated with different well-defined quantities of mouse monoclonal IgG molecules allow the construction of the calibration curve (described previously in [23]).
Enzyme-linked immunosorbent assay for soluble complement inhibitors C1 inhibitor, factor H and factor
Levels of factor H (fH), factor I (fI) and C1 inhibitor (C1Inh) in cell culture supernatants were measured by enzyme-linked immunosorbent assay, as described previously [25]. In brief, 96-well microtitre plates (NUNC-MaxiSorb, Roskilde, Denmark) were coated with the respective specific antibodies in 50 mM Na2CO3/NaHCO3, pH 9·6 for 16 h at 4°C [fH: rabbit anti-human fH (Serotec, Morphosys, Duesseldorf, Germany), fI: goat anti-human fI (ICN Biomedical, Irvine, CA, USA), C1Inh: rabbit anti-human C1Inh (Dako)]. After blocking remaining unspecific binding sites with 1% gelatin/PBS, 50 µl per well of each supernatant was added in duplicate for 1 h at room temperature. The plates were washed three times and then incubated with the respective detection antibodies [fH: goat anti-human fH (Quidel, San Diego, CA, USA), fI: mouse anti-human fI (Quidel), C1Inh: goat anti-human C1Inh (Dianova, Hamburg, Germany)] for 1 h at room temperature followed by peroxidase-conjugated secondary antibody [F(ab)2 rabbit anti-goat (Dianova) or goat anti-mouse (Dianova) respectively] for 1 h at room temperature. The assay was developed using azino-bis(3-ethylbenzthiazoline-6-sulphonic acid) (Sigma)/H2O2 as substrate. After terminating the reaction with 0·2 M oxalic acid as stop solution, microtitre plates were analysed at λ = 405 nm/492 nm in an enzyme-linked immunosorbent assay reader (EAR 340; SLT Labinstruments, Overath, Germany). Purified C1Inh (CSL Behring, Marburg, Germany), fH (Advanced Research Technologies, San Diego, CA, USA) and fI (Calbiochem, Bad Soden, Germany) were used as standards.
Statistical analysis
Statistical analysis was performed using StatView software (Abacus Concepts, Inc., Berkeley, CA, USA). Student's t-test for independent data was used to determine statistical significance of differences in mCRP expression levels, soluble complement inhibitors as well as differences in complement-mediated lysis between parental and variant cell lines. Results are expressed as mean ± standard deviation. Statistical significance was assumed at P < 0·05.
Results
Chemo-selected MDR cells exert increased resistance to complement-mediated lysis
The P-gp-expressing MDR cell variants OAW42-Dox and OAW42-Tax, generated by incubation in drug containing medium (doxorubicin or taxol respectively), were significantly more resistant to complement-mediated lysis than parental drug-sensitive OAW42 cells. Both MDR variants showed a significantly reduced lysis in 51Cr-release assay (P < 0·001) (Fig. 1a). Fluorescence cytometry analyses excluded differences in binding of the polyclonal anti-tumour antibodies to OAW42 and to the drug-resistant variants OAW42-Dox and OAW42-Tax.
Fig 1.
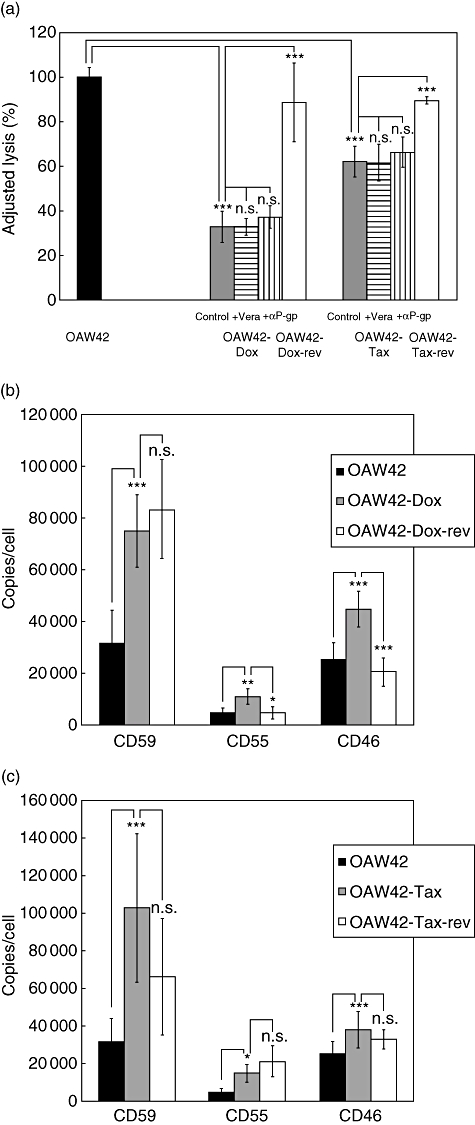
Analysis of complement sensitivity and membrane-bound complement regulatory protein (mCRP) level of OAW42 multi-drug-resistant (MDR) cells. (a) Susceptibility to complement-mediated lysis of OAW42 (black bars), OAW42-Dox (dark grey bars), OAW42-Dox-rev (open bars), OAW42-Tax (grey bars) and OAW42-Tax-rev (open bars) cells measured by 51Cr-release assay. Cells were labelled with 51Cr, incubated with anti-tumour antibody and human serum as complement source. Results are presented as mean % lysis ± standard deviation (s.d.), adjusted to lysis of OAW42 parental cells (set as 100%; triplicates of n = five independent experiments). Control = MDR cells without P-gp blockade, +Vera and +αP-glycoprotein (P-gp) = MDR cells incubated with verapamil or αP-gp antibody, respectively, to block P-gp. (b) Expression of membrane-bound complement regulators on OAW42 (black bars), OAW42-Dox (dark grey bars) and OAW42-Dox-rev (open bars) cells. Results are presented as mean mCRP copies/cell ± s.d. (CD59 n = 8; CD55 n = 5; CD46 n = 6). (c) Expression of membrane-bound complement regulators on OAW42 (black bars), OAW42-Tax (grey bars) and OAW42-Tax-rev (open bars) cells. Results are presented as mean mCRP copies/cell ± s.d. (n = six independent experiments). *P < 0·05; **P < 0·01; ***P < 0·001.
Revertant MDR variants became drug-sensitive upon incubation in drug-free medium, coinciding with a decrease in P-gp expression (Table 1) and accompanied by a decrease in complement resistance. The P-gp negative variants OAW42-Dox-rev and OAW42-Tax-rev regained a significantly increased susceptibility to complement-mediated lysis compared with the P-gp positive variants OAW42-Dox (P < 0·001) and OAW42-Tax (P < 0·001) (Fig. 1a). Blocking P-gp function, however, had no effect on complement susceptibility. Neither verapamil nor the monoclonal anti-P-gp-antibody MRK16 affected lysis level of OAW42-Dox or OAW42-Tax (Fig. 1a).
Table 1.
Expression of P-glycoprotein (P-gp) on ovarian carcinoma cells.
| Cell type | P-gp (copies/cell)* | Significance |
|---|---|---|
| OAW42 | 5 000 ± 7 000 | |
| OAW42-Dox | 326 000 ± 193 000 | P < 0·001 |
| OAW42-Dox-rev | 9 000 ± 6 800 | n.s. |
| OAW42-Tax | 168 000 ± 113 000 | P < 0·001 |
| OAW42-Tax-rev | 5 500 ± 6 300 | n.s. |
| A2780 | 800 ± 900 | |
| A2780MDR | 11 200 ± 3 800 | P < 0·05 |
| A2780MDR/2 | 357 000 ± 100 000 | P < 0·001 |
| SKOV3 | 2 600 ± 4 716 | |
| SKOV3MDR | 8 700 ± 3 100 | P < 0·001 |
| SKOV3MDR/2 | 19 200 ± 43 000 | P < 0·001 |
Cells were treated first with monoclonal antibody against P-gp (clone MRK16), followed by fluorescein isothiocyanate-labelled goat anti-mouse immunoglobulin G. Results are presented as mean copies/cell ± standard deviation (OAW42 n = eight; all other cells n = six independent experiments). Significance is determined relative to the parental cell line; n.s., not significant.
Analysis of mCRP expression on chemo-selected MDR variants
The P-gp positive chemo-selected variants OAW42-Dox and OAW42-Tax overexpressed the mCRP CD59 (P < 0·001), CD46 (P < 0·001) and CD55 (OAW42-Dox P = 0·002; OAW42-Tax P = 0·011) relative to parental OAW42 cells (Fig. 1b,c). Reversion of MDR correlated only partly with a decrease in mCRP expression levels on revertant P-gp negative variants. OAW42-Dox-rev cells showed significantly reduced levels of CD46 (P < 0·001) and CD55 (P = 0·017) compared with the MDR variant OAW42-Dox but slightly increased CD59 expression compared with OAW42-Dox, that was even significantly higher than the expression level on the initial parental cell line OAW42 (P < 0·001) (Fig. 1b). On the second revertant variant, OAW42-Tax-rev, only the expression of CD46 was slightly but non-significantly reduced compared with the MDR variant OAW42-Tax (P = 0·06), whereas the levels of CD59 and CD55 stayed on the same level as on the MDR variant OAW42-Tax. Moreover, CD55 remained significantly elevated compared with the parental cell line OAW42 (P < 0·05) (Fig. 1c).
The relevance of mCRP overexpression in conferring complement resistance to MDR tumour cells was established by blocking experiments. Neutralizing CD59 and CD55 with non-complement-activating mAb [23] markedly increased complement-mediated lysis of the MDR variants OAW42-Dox (anti-CD59 P < 0·001, anti-CD55 P < 0·001) and OAW42-Tax (anti-CD59 P < 0·001, anti-CD55 P = 0·01), whereas blocking the membrane-bound CD46 regulator only increased complement susceptibility of OAW42-Dox (P < 0·05) but had no effect on OAW42-Tax cells (Table 2). A combination of all neutralizing antibodies (anti-CD59, anti-CD55 and anti-CD46) revealed a further augmentation of lysis as exemplified for OAW42 cells and their taxol-treated variants OAW42-Tax, leading to similarly high complement susceptibility in the MDR variant OAW42-Tax and the revertant OAW42-Tax-rev compared with the parental OAW42 (Table 3). In the presence of Mg2+ EGTA cytotoxicity was abolished, indicating that complement-mediated lysis of the tumour cells depended on classical pathway activation (Table 3).
Table 2.
Effect of membrane-bound complement regulatory protein (mCRP) blockade on complement susceptibility of OAW42 cells and the multi-drug-resistant (MDR) variants OAW42-Dox, OAW42-Tax.
| Cell type | Adjusted lysis (%)* | Significance |
|---|---|---|
| OAW42 | 100 ± 4·8 | |
| OAW42-Dox | 33 ± 7·1 | P < 0·001 (versus OAW42) |
| OAW42-Dox + anti-CD59 | 91 ± 11·1 | P < 0·001 (versus OAW42-Dox) |
| OAW42-Dox + anti-CD55 | 84 ± 6·8 | P < 0·001 (versus OAW42-Dox) |
| OAW42-Dox + anti-CD46 | 42 ± 4·8 | P = 0·044 (versus OAW42-Dox) |
| OAW42 | 100 ± 4·8 | |
| OAW42-Tax | 68 ± 7·6 | P < 0·001 (versus OAW42) |
| OAW42-Tax + anti-CD59 | 99 ± 8 | P < 0·001 (versus OAW42-Tax) |
| OAW42-Tax + anti-CD55 | 88 ± 13 | P = 0·01 (versus OAW42-Tax) |
| OAW42-Tax + anti-CD46 | 77 ± 9·6 | n.s. (versus OAW42-Tax) |
Cells were incubated with polyclonal rabbit antitumour antibodies normal human serum as complement source. OAW42 cells, resistant against doxorubicine (OAW42-Dox) or taxol (OAW42-Tax), were preincubated with non-complement-activating monoclonal antibodies against CD59, CD55 or CD46 to block mCRP. Results are presented as mean percentage adjusted lysis ± standard deviation (as triplicates of n = five independent experiments). Specific complement-induced lysis of parental OAW42 (70%) was set as 100% and the specific lysis rates of the MDR-variants are presented relative to the parental cells.
Table 3.
Effect of combined membrane-bound complement regulatory protein (mCRP) blockade on complement susceptibility of OAW42, OAW42-Tax and the revertant OAW42-Tax-rev cells.
| Cell type | Adjusted lysis (%)* | Significance |
|---|---|---|
| OAW42 | 100 ± 1·3 | |
| OAW42 + Mg2+ EGTA | 1·4 ± 5·3 | P < 0·001 (versus OAW42) |
| OAW42 + α-mCRP* | 105·6 ± 2·9 | P < 0·05 (versus OAW42) |
| OAW42-Tax | 17 ± 1·5 | P < 0·001 (versus OAW42) |
| OAW42-Tax + Mg2+ EGTA | −3·8 ± 1·3 | P < 0·001 (versus OAW42-Tax) |
| OAW42-Tax + α-mCRP | 117·9 ± 4·2 | P < 0·001 (versus OAW42-Tax) |
| P < 0·05 (versus OAW42+αmCRP) | ||
| OAW42-Tax-rev | 32·4 ± 3·2 | P < 0·01 (versus OAW42-Tax) |
| P < 0·001 (versus OAW42) | ||
| OAW42-Tax-rev + Mg2+ EGTA | 1·2 ± 0·7 | P < 0·001 (versus OAW42-Tax-rev) |
| OAW42-Tax-rev + α-mCRP | 114 ± 2·5 | P < 0·001 (versus OAW42-Tax-rev) |
| P < 0·05 (versus OAW42+αmCRP) |
OAW42 cells and their variants OAW42-Tax and OAW42-Tax-rev were preincubated with a combination of non-complement-activating monoclonal antibodies against CD59, CD55 and CD46 to block mCRP. Results are presented as mean percentage adjusted lysis ± standard deviation (as triplicates). Mg2+-ethylene glycol tetraacetic acid (EGTA) was added in one set of experiments to test for classical versus alternative complement activation. Specific complement-induced lysis of parental OAW42 (85·3%) was set as 100% and the specific lysis rates of the MDR-variants and cells after mCRP blockade are presented relative to the parental cells.
Secretion of soluble complement inhibitors
To investigate further the discrepancy between an increased complement-susceptibility of the revertant cell lines compared with the MDR variants despite the still partly elevated mCRP levels on revertant cell lines, we analysed exemplarily the secretion of the complement regulators C1Inh, fI and fH by OAW42, OAW42-Tax and the revertant OAW42-Tax-rev (Table 4). Secretion of C1Inh and fI increased upon development of drug (and complement)-resistance in OAW42-Tax (OAW42 versus OAW42-Tax: C1Inh: 3·02 ± 0·96 ng/ml versus 6·15 ± 2·34 ng/ml, P < 0·05, fI: 5·49 ± 0·81 ng/ml versus 46·45 ± 8·35 ng/ml, P < 0·05) (Table 4). Revertant OAW42-Tax-rev released less fI compared with the MDR variant OAW42-Tax (Table 4), suggesting that this factor might – at least partly – account for the higher complement-susceptibility of the revertant variant compared with the MDR variant OAW42-Tax. In contrast, the secretion of C1Inh by the revertant variant remained on the same elevated level (Table 4); fH was never detected.
Table 4.
Secretion of soluble complement regulators by OAW42 ovarian carcinoma cells.
| Cell type | C1Inh (ng/ml) | Significance | Factor I (ng/ml) | Significance |
|---|---|---|---|---|
| OAW42 | 3·02 ± 1·0 | 5·49 ± 1·1 | ||
| OAW42-Tax | 6·18 ± 1·6 | P < 0·05 vs. OAW42 | 46·45 ± 11·8 | P < 0·01 versus OAW42 |
| OAW42-tax-rev | 6·15 ± 2·3 | P < 0·05 vs. OAW42 | 26·25±3·9 | P < 0·05 versus OAW42-Tax |
| P < 0·05 versus OAW42 |
Levels of C1 inhibitor (C1Inh) and factor I in cell culture supernatants were analysed with enzyme-linked immunosorbent assays. Results are presented as mean ± standard deviation (as duplicates of two independent experiments).
Complement susceptibility of MDR1 gene-transduced tumour cells
To focus on the impact of P-gp separately from the effect(s) mediated by selection in drug-containing medium, we generated a P-gp-positive MDR variant of A2780 by retroviral MDR1 gene transduction. These cells (A2780-MDR), FACS-sorted without further selection in drug-containing medium, were only slightly but not significantly less susceptible to complement-mediated lysis (P = 0·197, Fig. 2a) and did not express higher numbers of mCRP (Fig. 2b). However, when these cells were cultured subsequently in vincristine-containing selection medium (A2780-MDR/2) they became significantly more resistant to complement-mediated lysis than untreated chemo-sensitive parental A2780 cells (P < 0·001) (Fig. 2a). Again, this enhanced resistance was not affected by blocking P-gp function with verapamil (P = 0·42) or with CsA (P = 0·168). A2780-MDR/2 cells significantly overexpressed CD59 (P<0·001) and CD46 (P < 0·001) relative to A2780 cells, whereas only trace amounts of CD55 could be detected on parental A2780, A2780-MDR and A2780-MDR/2 cells (Fig. 2b).
Fig 2.
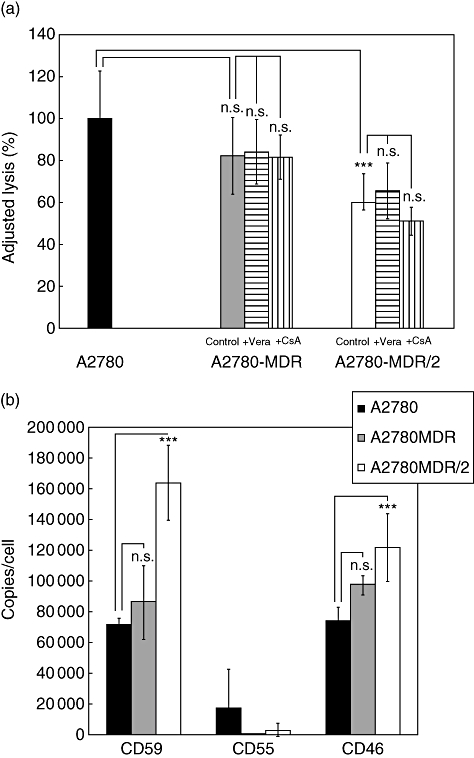
Analysis of complement sensitivity and membrane-bound complement regulatory protein (mCRP) level of A2780 cells and their multi-drug-resistant (MDR) variants. (a) Susceptibility to complement-mediated lysis of A2780 (black bars), A2780-MDR (grey bars) and A2780-MDR/2 (open bars) cells. Results are presented as mean % lysis ± standard deviation (s.d.), adjusted to lysis of A2780 parental cells (set as 100%; triplicates of n = six independent experiments). Control = MDR cells without P-glycoprotein (P-gp) blockade, +Vera and +cyclosporine A (CsA) = MDR cells incubated with verapamil or CsA, respectively, to block P-gp. (b) Expression of membrane-bound complement regulators on A2780 (black bars), A2780-MDR (grey bars) and A2780-MDR/2 (open bars) cells. Results are presented as mean mCRP copies/cell ± s.d. (n = six independent experiments). ***P < 0·001.
These observations were confirmed with a second ovarian carcinoma cell line, SKOV3, transduced with MDR1 gene (SKOV3-MDR), which initially did not show a significant difference in susceptibility to complement-mediated lysis compared with parental SKOV3 cells (Fig. 3a). At that time, mCRP expression on SKOV3-MDR cells remained at the same level as on parental SKOV3 cells (data not shown). Only after a long-term exposure (for several months) to vincristine, the cells (now designated SKOV3-MDR/2) developed resistance to complement (P < 0·001; Fig. 3a), now associated with elevated mCRP expression levels (Fig. 3b: CD59: P < 0·01, CD55: P < 0·001, CD46: P = 0·072). As observed already in OAW42-Dox, OAW42-Tax and A2780-MDR cells, treatment with verapamil or CsA did not reverse the enhanced resistance of SKOV-MDR/2 cells to complement-mediated lysis (Fig. 3a).
Fig 3.
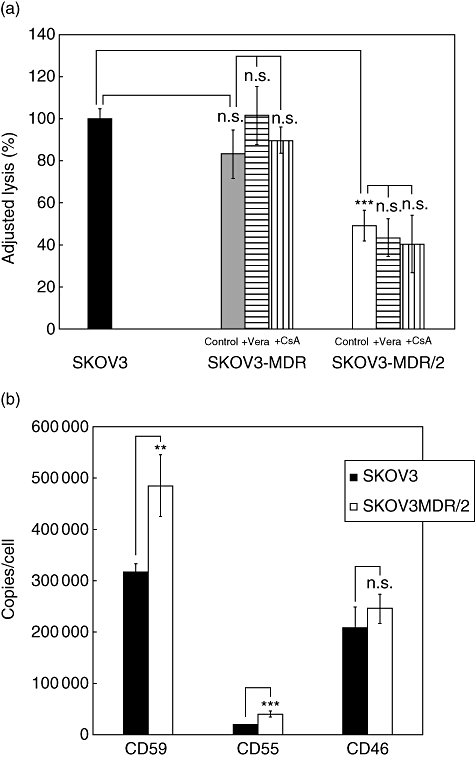
Analysis of complement sensitivity and membrane-bound complement regulatory protein (mCRP) level of SKOV3-multi-drug-resistant (MDR) cells. (a) Susceptibility to complement-mediated lysis of SKOV3 (black bars), SKOV3-MDR (grey bars) and SKOV3-MDR/2 (open bars) in 51Cr-release assay. Results are presented as mean % lysis ± standard deviation (s.d.), adjusted to lysis of SKOV3 parental cells (set as 100%; triplicates of n = six independent experiments). Control = MDR cells without P-glycoprotein (P-gp) blockade, +Vera and +cyclosporine A (CsA) = MDR cells incubated with verapamil (10 µg/ml) or CsA (1 µM), respectively, to block P-gp. (b) Expression of membrane-bound complement regulators on SKOV3 (black bars) and SKOV3-MDR/2 (open bars) cells (after long-term vincristine exposure). Results are presented as mean mCRP copies/cell ± s.d. (n = four independent experiments). **P < 0·01; ***P < 0·001.
Discussion
As MDR limits effective anti-cancer chemotherapy, antibody-based immunotherapy may provide an alternative treatment, also providing the advantage of tumour cell-specific targeting. Previous studies on the interaction of MDR tumour cells with the complement system aimed at defining a possible impact of P-gp on susceptibility of tumour cells to complement-mediated lysis has yielded conflicting results. LoVo human colon carcinoma [26] and KB-V1 human oral carcinoma [19] MDR cells were reported to be more sensitive to complement-mediated lysis compared with their P-gp-negative drug-sensitive parental cells. As P-gp is capable of altering the biophysical properties of the plasma membrane [27,28], it was speculated that P-gp might facilitate the insertion and stability of the C5b-9 MAC complex leading to enhanced lysis. In contrast, RV+ human myeloid leukaemia and K562 erythroleukaemia MDR cells showed enhanced resistance to complement lysis [20]. P-gp-mediated decrease of transmembrane potential and increase in intracellular pH have been proposed to interfere with the MAC pore formation leading to increased complement resistance in these cells [29], but the impact of any known complement-resistance factors on this observed complement resistance has not been tested.
We demonstrate here that MDR human ovarian carcinoma cells generated by drug selection are significantly more resistant to complement-mediated cytotoxicity than parental cells. Previous studies found no difference in membrane disruption by complement C5b-9 between P-gp-positive and -negative CEM cells [30]. This is in line with our observation that blocking P-gp function, either by CsA, verapamil or anti-P-gp mAb MRK16 does not alter complement susceptibility of MDR ovarian tumour cells. To further exclude a decisive role of P-pg in conferring complement resistance, we transduced two different MDR ovarian carcinoma cells with a retroviral MDR1 vector and highly purified them to homogeneity by subsequent FACS sorting. Upon exposure to antibody and complement, no alteration in susceptibilty to complement compared with parental control cells could be observed in these P-gp-positive MDR tumour cells. However, upon further cultivation in drug-containing media, in particular over a long time, these cells also became increasingly resistant to complement-mediated cytotoxicity.
Several extra- and intracellular factors regulate the rate and extent of complement deposition on the cells and their capability to escape from complement-mediated injury [25,31]. Alteration in any of these factors from the normal state will affect the sensitivity of the MDR cell to complement-mediated lysis. We and others could demonstrate clearly that enhanced resistance of tumour cells to complement is closely associated with overexpression of mCRP [15–17]. CD46 and CD59 are highly up-regulated on several ovarian carcinoma cell lines derived from patients [32] and recently high CD46 expression has been linked to a poorer prognosis in ovarian cancer patients [33]. We therefore compared the level of expression of CD46, CD55 and CD59 on MDR ovarian carcinoma variants and their parental cells. Transfected P-gp-positive MDR tumour cells which initially showed no difference in complement resistance compared with parental P-gp-negative controls expressed the same level of mCRP. However, when these cells, cultivated in drug-containing media, converted into a complement resistant phenotype, they expressed significantly higher levels of membrane regulators; but comparable levels of mCRP expressed by the two chemoselected MDR variants OAW42-Dox and OAW42-Tax did not result in a comparable level of resistance against complement attack. Furthermore, P-gp-negative revertant cells which regained susceptibility to complement-mediated lysis showed large variations in their mCRP expression, sometimes even exceeding that of the complement resistant variant (CD59, OAW42-Dox-rev; Fig. 1b). This points to additional resistance mechanisms beyond a high mCRP expression, as discussed previously [16]. For instance, the up-regulation of soluble complement regulatory factors such as C1Inh or fH [25] have been identified as additional important complement-resistance factors.
Indeed, as demonstrated here for the ovarian carcinoma line OAW42 and the MDR variant OAW42-Tax, these tumour cells secreted C1Inh and fI, but not fH. The latter is in contrast to previous findings [34,35] describing the release of fH and fH-like protein 1 by (other) ovarian cell lines. Neutralization of these soluble inhibitors has been shown to increase complement susceptibility significantly, even in the presence of highly up-regulated mCRP [36]. We show here an increased secretion of fI upon development of drug (and complement)-resistance in OAW42-Tax cells, which was significantly less pronounced in the revertant OAW42-Tax-rev, suggesting that this difference in fI might – at least partly – account for the increased complement-susceptibility of the revertant cell lines, despite their still partly elevated mCRP levels.
Bomstein and Fishelson [19] found an increased C3b deposition and a more efficient insertion of the C5b-9 MAC into plasma membrane of P-gp-positive KB-V1 cells compared with the parental cells. This increased sensitivity to complement could be explained partly by a reduced expression of CD55 on KB-V1. The claim that mCRP level of expression is one of the major factors determining the sensitivity of P-gp-positive MDR cells to complement is supported by the data showing that neutralization of CD59 and CD55 with non-complement-activating mAb abolishes complement resistance of OAW42-Dox and OAW42-Tax cells (Table 2) – which could be increased even further by simultaneously blocking CD59, CD55 and CD46, leading to the similarly high complement susceptibility in the MDR variant OAW42-Tax and the revertant OAW42-Tax-rev compared with the parental OAW42. This has been described previously with various tumour cell lines [15,23,32,37,38]. Although CD46 was highly overexpressed in the MDR-positive variants OAW42-Dox and OAW42-Tax, blocking CD46 with antibodies increased complement-susceptibility in OAW42-Dox only slightly and had no significant effect with OAW42-Tax variants. This agrees with previous findings [39], showing that in many carcinomas CD46 seems less important in conferring resistance to antibody-activated complement lysis. CD46 is postulated to play a more important role in regulation of the alternative complement pathway [12], which might contribute to its correlation with a poor clinical prognosis and reduced survival [33].
Those highly diverse observations suggest that development of MDR in tumour cells may lead to either complement resistance or to complement sensitivity, or may even have no impact on their original sensitivity to complement-mediated lysis – depending, to a large extent, on alterations in expression level of complement resistance factors such as mCRP, induced directly or indirectly by chemotherapy. Studies on the influence of chemotherapeutic agents on mCRP expression levels described alterations varying from drug-induced increased levels of CD59 and CD55 [38,39] to decreased levels of CD59 [40] and CD55 [41] under different chemotherapeutic regimens. In addition, some chemotherapeutic drugs even exerted different effects in different cell-types. While daunomycin did not affect CD59 on osteosarcoma cells [42], it increased CD59 expression on the ovarian carcinoma variant A2780-MDR [43]. In our different ovarian carcinoma cells we observed a drug-induced overexpression of mCRP in all cell lines varying from an increase in CD59 > CD46 > CD55 in OAW42 cells treated with doxorubicin or taxol to an increase in CD59 and CD46 in A2780 cells treated with vincristine and an overexpression of CD59 and CD55 in SKOV3 cells exposed to the same drug. In some cells, long-term drug exposure seems to be necessary to alter mCRP expression. Thus, our transfected and vincristine-treated SKOV-MDR cells initially showed no changes in mCRP expression levels, but converted into a complement-resistant variant overexpressing mCRPs after several months of vincristine treatment.
Thus, the level of sensitivity of each primary MDR tumour to complement and the resulting susceptibility to antibody-immunotherapy will have to be determined ad hoc. Each cell type has an innate threshold of resistance to complement that depends on numerous factors. We hypothesize that at a low level of expression (still sufficient to confer drug resistance), P-gp has no effect on complement sensitivity of the MDR cells. However, at a high level of expression it may induce secondary effects, not necessarily related to its drug efflux activity [44], that may also modify the sensitivity of the MDR cells to complement. Besides, exposure to chemotherapeutic drugs does not only induce P-gp overexpression but also other MDR conferring proteins such as MRP [45] and BCRP [46]. Their possible impact on complement resistance of chemoselected MDR tumour cells has not yet been investigated. Therefore, future studies on the interaction of MDR tumour cells with the complement system would require investigation of additional complement resistance and MDR conferring factors.
In summary, as shown in this series of experiments, the exposure of ovarian carcinoma cells to drug therapy may not only cause MDR but also induce enhanced complement resistance. Combined MDR and complement resistance may impede anti-tumour therapy. Complement resistance of MDR ovarian carcinoma cells appears to depend primarily on up-regulation of mCRP expression and cannot be linked to hitherto identified P-gp function. Nevertheless, as complement resistance seriously impairs the efficacy of antibody immunotherapy, the expression level of mCRP needs to be assessed when mAb therapy is considered in patients with MDR cancers.
Acknowledgments
This study was supported in part by the Cancer Cooperation Programme of the German Cancer Research Center (DKFZ), Heidelberg, Germany, by grant FR 1732/3-1 of the German Research Foundation (DFG) and the Israeli Ministry of Science.
References
- 1.Eytan GD, Kuchel PW. Mechanism of action of p-glycoprotein in relation to passive membrane permeation. Int Rev Cytol. 1999;190:175–250. doi: 10.1016/s0074-7696(08)62148-8. [DOI] [PubMed] [Google Scholar]
- 2.Ganapathi R, Hoeltge G, Casey G, Grabowski D, Neelon R, Ford J. Acquisition of doxorubicin resistance in human leukemia HL-60 cells is reproducibly associated with 7q21 chromosomal anomalies. Cancer Genet Cytogenet. 1996;86:116–9. doi: 10.1016/0165-4608(95)00207-3. [DOI] [PubMed] [Google Scholar]
- 3.Tsimberidou AM, Paterakis G, Androutsos G, et al. Evaluation of the clinical relevance of the expression and function of P-glycoprotein, multidrug resistance protein and lung resistance protein in patients with primary acute myelogenous leukaemia. Leuk Res. 2002;26:143–54. doi: 10.1016/s0145-2126(01)00106-0. [DOI] [PubMed] [Google Scholar]
- 4.Wigler PW, Patterson FK. Inhibition of the multidrug resistance efflux pump. Biochim Biophys Acta. 1993;1154:1173–81. doi: 10.1016/0304-4157(93)90010-l. [DOI] [PubMed] [Google Scholar]
- 5.Hoffman MM, Wei LY, Roepe PD. Are altered pHi and membrane potential in huMDR 1 transfectants sufficient to cause MDR protein-mediated multidrug resistance? J Gen Physiol. 1996;108:295–313. doi: 10.1085/jgp.108.4.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tsuruo T, Iida H, Tsukagoshi S, Sakurai Y. Overcoming of vincristine resistance in P388 leukemia in vivo and in vitro through enhanced cytotoxicity of vincristine and vinblastine by verapamil. Cancer Res. 1981;41:1967–72. [PubMed] [Google Scholar]
- 7.Wigler PW, Patterson FK. Reversal agent inhibition of the multidrug resistance pump in human leukemic lymphoblasts. Biochim Biophys Acta. 1994;1189:1–6. doi: 10.1016/0005-2736(94)90272-0. [DOI] [PubMed] [Google Scholar]
- 8.Hamada H, Tsuruo T. Functional role for the 170- to 180-kDa glycoprotein specific to drug-resistant tumour cells as revealed by monoclonal antibodies. Proc Natl Acad Sci USA. 1986;83:7785–9. doi: 10.1073/pnas.83.20.7785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Capone PM, Papsidero LD, Croghan GA, Chu TM. Experimental tumoricidal effects of monoclonal antibody against solid breast tumours. Proc Natl Acad Sci USA. 1983;80:7328–32. doi: 10.1073/pnas.80.23.7328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chapman PB, Lomberg M, Houghton AM. Light chain variants of an IgG3 anti-GD3 monoclonal antibody and the relationship among avidity, effector functions, tumour targeting, and antitumour activity. Cancer Res. 1990;50:1503–9. [PubMed] [Google Scholar]
- 11.Walport MJ. Complement. First of two parts. N Engl J Med. 2001;344:1058–66. doi: 10.1056/NEJM200104053441406. [DOI] [PubMed] [Google Scholar]
- 12.Kojima A, Iwata K, Seya T, et al. Membrane cofactor protein (CD46) protects cells predominantly from alternative complement pathway-mediated C3-fragment deposition and cytolysis. J Immunol. 1993;151:1519–27. [PubMed] [Google Scholar]
- 13.Medof ME, Kinoshita T, Nussenzweig V. Inhibition of complement activation on the surface of cells after incorporation of decay-accelerating factor (DAF) into their membranes. J Exp Med. 1984;160:1558–78. doi: 10.1084/jem.160.5.1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Meri S, Morgan BP, Davies A, et al. Human protectin (CD59), an 18,000–20,000 MW complement lysis restricting factor, inhibits C5b-8 catalysed insertion of C9 into lipid bilayers. Immunology. 1990;71:1–9. [PMC free article] [PubMed] [Google Scholar]
- 15.Fishelson Z, Donin N, Zell S, Schultz S, Kirschfink M. Obstacles to cancer immunotherapy: expression of membrane complement regulatory proteins (mCRPs) in tumours. Mol Immunol. 2003;40:109–23. doi: 10.1016/s0161-5890(03)00112-3. [DOI] [PubMed] [Google Scholar]
- 16.Jurianz K, Ziegler S, Garcia-Schuler H, et al. Complement resistance of tumour cells: basal and induced mechanisms. Mol Immunol. 1999;36:929–39. doi: 10.1016/s0161-5890(99)00115-7. [DOI] [PubMed] [Google Scholar]
- 17.Gorter A, Meri S. Immune evasion of tumour cells using membrane-bound complement regulatory proteins. Immunol Today. 1999;20:576–82. doi: 10.1016/s0167-5699(99)01537-6. [DOI] [PubMed] [Google Scholar]
- 18.Gelderman KA, Tomlinson S, Ross GD, Gorter A. Complement function in mAb mediated cancer immunotherapy. Trends Immunol. 2004;25:158–64. doi: 10.1016/j.it.2004.01.008. [DOI] [PubMed] [Google Scholar]
- 19.Bomstein Y, Fishelson Z. Enhanced sensitivity of P-glycoprotein-positive multidrug resistant tumour cells to complement-mediated lysis. Eur J Immunol. 1997;27:2204–11. doi: 10.1002/eji.1830270913. [DOI] [PubMed] [Google Scholar]
- 20.Weisburg JH, Curcio M, Caron PC, et al. The multidrug resistance phenotype confers immunological resistance. J Exp Med. 1996;183:2699–704. doi: 10.1084/jem.183.6.2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Masanek U, Stammler G, Volm M. Messenger RNA expression of resistance proteins and related factors in human ovarian carcinoma cell lines resistant to doxorubicin, taxol and cisplatin. Anticancer Drugs. 1997;8:189–98. doi: 10.1097/00001813-199702000-00010. [DOI] [PubMed] [Google Scholar]
- 22.Fruehauf S, Breems DA, Knaan-Shanzer S, et al. Frequent analysis of multidrug resistance-1 gene transfer into human primitive hematopietic progenitor cells using the cobblestone area-forming cell assay and detection of vector-mediated P-glycoprotein expression by rhodamine-123. Hum Gene Ther. 1996;7:1219–31. doi: 10.1089/hum.1996.7.10-1219. [DOI] [PubMed] [Google Scholar]
- 23.Jurianz K, Maslak S, Garcia-Schuler H, Fishelson Z, Kirschfink M. Neutralization of complement regulatory proteins augments lysis of breast carcinoma cells targeted with rhumAb anti-HER2. Immunopharmacology. 1999;42:209–18. doi: 10.1016/s0162-3109(99)00006-5. [DOI] [PubMed] [Google Scholar]
- 24.Hildinger M, Schilz A, Eckert HG, et al. Bicistronic retroviral vectors for combining myeloprotection with cell-surface marking. Gene Ther. 1999;6:1222–30. doi: 10.1038/sj.gt.3300942. [DOI] [PubMed] [Google Scholar]
- 25.Jurianz K, Ziegler S, Donin N, Reiter Y, Fishelson Z, Kirschfink M. K562 erythroleukemic cells are equipped with multiple mechanisms of resistance to lysis by complement. Int J Cancer. 2001;93:848–54. doi: 10.1002/ijc.1406. [DOI] [PubMed] [Google Scholar]
- 26.Gambacorti-Passerini C, Rivoltini L, Supino R, et al. Susceptibility of chemoresistant murine and human tumour cells to lysis by interleukin 2-activated lymphocytes. Cancer Res. 1988;48:2372–6. [PubMed] [Google Scholar]
- 27.Roepe PD, Wei LY, Cruz J, Carlson D. Lower electrical membrane potential and altered pHi homeostasis in multidrug-resistant (MDR) cells: further characterization of a series of MDR cell lines expressing different levels of P-glycoprotein. Biochemistry. 1993;32:11042–56. doi: 10.1021/bi00092a014. [DOI] [PubMed] [Google Scholar]
- 28.Rothnie A, Theron D, Soceneantu L, et al. The importance of cholesterol in maintenance of P-glycoprotein activity and its membrane perturbing influence. Eur Biophys J. 2001;30:430–42. doi: 10.1007/s002490100156. [DOI] [PubMed] [Google Scholar]
- 29.Weisburg JH, Roepe PD, Dzekunov S, Scheinberg DA. Intracellular pH and multidrug resistance regulate complement-mediated cytotoxicity of nucleated human cells. J Biol Chem. 1999;274:10877–88. doi: 10.1074/jbc.274.16.10877. [DOI] [PubMed] [Google Scholar]
- 30.Johnstone RW, Tainton KM, Ruefli AA, et al. P-glycoprotein does not protect cells against cytolysis induced by pore-forming proteins. J Biol Chem. 2001;276:16667–73. doi: 10.1074/jbc.M010774200. [DOI] [PubMed] [Google Scholar]
- 31.Donin N, Jurianz K, Ziporen L, Schultz S, Kirschfink M, Fishelson Z. Complement resistance of human carcinoma cells depends on membrane regulatory proteins, protein kinases and sialic acid. Clin Exp Immunol. 2003;131:254–63. doi: 10.1046/j.1365-2249.2003.02066.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bjorge L, Hakulinen J, Wahlstrom T, Matre R, Meri S. Complement-regulatory proteins in ovarian malignancies. Int J Cancer. 1997;70:14–25. doi: 10.1002/(sici)1097-0215(19970106)70:1<14::aid-ijc3>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 33.Surowiak P, Materna V, Maciejczyk A, et al. CD46 expression is indicative of shorter revival-free survival for ovarian cancer patients. Anticancer Res. 2006;26:4943–8. [PubMed] [Google Scholar]
- 34.Junnikkala S, Hakulinen J, Jarva H, et al. Secretion of soluble complement inhibitors factor H and factor H-like protein (FHL-1) by ovarian tumour cells. Br J Cancer. 2002;87:1119–27. doi: 10.1038/sj.bjc.6600614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bjorge L, Hakulinen J, Vintermyr OK, Jarva TS, Iversen OE, Meri S. Ascitic complement system in ovarian cancer. Br J Cancer. 2005;92:895–905. doi: 10.1038/sj.bjc.6602334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Junnikkala S, Jokiranta TS, Friese MA, Jarva H, Zipfel PF, Meri S. Exceptional resistance of human H2 glioblastoma cells to complement-mediated killing by expression and utilization of factor H and factor H-like protein 1. J Immunol. 2000;164:6075–81. doi: 10.4049/jimmunol.164.11.6075. [DOI] [PubMed] [Google Scholar]
- 37.Blok VT, Daha MR, Tijsma O, et al. A bispecific monoclonal antibody directed against both the membrane-bound complement regulator CD55 and the renal tumour-associated antigen G250 enhances C3 deposition and tumour cell lysis by complement. J Immunol. 1998;160:3437–43. [PubMed] [Google Scholar]
- 38.Gelderman KA, Blok VT, Fleuren GJ, Gorter A. The inhibitory effect of CD46, CD55, and CD59 on complement activation after immunotherapeutic treatment of cervical carcinoma cells with monoclonal antibodies or bispecific monoclonal antibodies. Lab Invest. 2002;82:483–93. doi: 10.1038/labinvest.3780441. [DOI] [PubMed] [Google Scholar]
- 39.Gorter A, Blok VT, Haasnoot WH, Ensink NG, Daha MR, Fleuren GJ. Expression of CD46, CD55, and CD59 on renal tumour cell lines and their role in preventing complement-mediated tumour cell lysis. Lab Invest. 1996;74:1039–49. [PubMed] [Google Scholar]
- 40.Kuraya M, Yefenof E, Klein G, Klein E. Expression of the complement regulatory proteins CD21, CD55 and CD59 on Burkitt lymphoma lines: their role in sensitivity to human serum-mediated lysis. Eur J Immunol. 1992;22:1871–6. doi: 10.1002/eji.1830220729. [DOI] [PubMed] [Google Scholar]
- 41.Maio M, Brasoveanu LI, Coral S, et al. Structure, distribution, and functional role of protectin (CD59) in complement-susceptibility and in immunotherapy of human malignancies. Int J Oncol. 1998;13:305–18. doi: 10.3892/ijo.13.2.305. [DOI] [PubMed] [Google Scholar]
- 42.Bjørge L, Matre R. Down-regulation of CD59 (protectin) expression on human colorectal adenocarcinoma cell lines by levamisole. Scand J Immunol. 1995;42:512–6. doi: 10.1111/j.1365-3083.1995.tb03688.x. [DOI] [PubMed] [Google Scholar]
- 43.Sedlak J, McGown A, Hrubisko M, Hunakova L, Chorvath B. Drug-resistance associated alterations of cell surface antigen expression in a human anthracycline-resistant ovarian carcinoma cell line. Neoplasma. 1994;41:259–62. [PubMed] [Google Scholar]
- 44.Ding S, Chamberlain M, McLaren A, Goh L, Duncan I, Wolf CR. Cross-talk between signalling pathways and the multidrug resistant protein MDR-1. Br J Cancer. 2001;85:1175–84. doi: 10.1054/bjoc.2001.2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zaman GJ, Flens MJ, van Leusden MR, et al. The human multidrug resistance-associated protein MRP is a plasma membrane drug-efflux pump. Proc Natl Acad Sci USA. 1994;91:8822–6. doi: 10.1073/pnas.91.19.8822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Doyle LA, Yang W, Abruzzo LV, et al. A multidrug resistance transporter from human MCF-7 breast cancer cells. Proc Natl Acad Sci USA. 1998;95:15665–70. doi: 10.1073/pnas.95.26.15665. [DOI] [PMC free article] [PubMed] [Google Scholar]