

Abstract
Cellular adjuvants such as dendritic cells (DC) are in the focus of tumour immunotherapy. In DC-vaccine trials, induction of tumour antigen-specific immunity is observed frequently and well-documented clinical responses have been reported. However, the overall response rate is less than 3%, therefore alternative strategies are being investigated. CD40-activated B cells (CD40-B) have been characterized previously as an interesting alternative because they present antigen efficiently and can be expanded by several logs from small amounts of peripheral blood. To determine the central technical challenges of cell-based vaccines we performed a single-patient analysis of 502 patients from DC-based tumour vaccine trials and identified at least three factors contributing to their limited efficiency: (1) lack of cell numbers; (2) lack of documented purity thus high contamination of bystander cells; and (3) lack of quality control and thus heterogeneous or unknown expression of important surface molecules such as major histocompatibility complex (MHC) and chemokine receptors. Based on these findings we re-evaluated the CD40-B approach in cancer patients. Here, we show that proliferation of B cells from cancer patients is equivalent to that observed in healthy donors. Purity is always > 90% after 2 weeks and remains stable for several weeks. They have comparable antigen-presenting capability determined phenotypically and by allogeneic mixed lymphocyte reaction. Expression of CCR7 and CD62L was detected in all samples and B cells migrated towards the relevant homing chemokines. Taken together, CD40-B cells from cancer patients can be expanded in virtually unlimited numbers at high purity and full function concerning antigen-presentation and migratory properties.
Keywords: antigen-presenting cells, CD40-activated B cells, tumour immunothrapy
Introduction
Cancer immunotherapy using dendritic cells (DC) as cellular adjuvants represents a promising modality for the treatment of malignant disease. Despite some successful experimental data, which were gathered mainly in mice, successful and standardized clinical applications for the treatment of human cancers are still lacking. Many clinical trials have been conducted evaluating the use of such cancer vaccines for the treatment of cancer. Even though measurable immunological reactions can be induced, relevant clinical responses are rare and the outcome has generally been disappointing [1]. Many questions regarding the optimal composition of tumour vaccines remain to be solved. Tumour-induced immunosuppression is considered a major barrier to successful therapeutic vaccination against cancer. Prostaglandin E2[2–5] and other inhibitory cytokines such as interleukin-10 [6,7], transforming growth factor-β[8,9] and vascular endothelial growth factor [10] have been shown to inhibit the differentiation and immunostimulatory function of DC. Furthermore, monocyte-derived DC (MoDC), as applied currently in cancer vaccine trials, lack CD62L, a key molecule implicated in the initial phase of migration through high endothelial venules. As shown in animal and human studies, homing of these cells can be very limited. Accordingly, transfection of CD62L into DC has been demonstrated to improve homing [11]. Nevertheless, this approach is hardly feasible for clinical application because of cost and regulatory obstacles.
However, while the biological criteria are certainly crucial for a successful induction of anti-tumour immunity it remains uncertain whether the currently used cellular vaccines meet basic requirements relevant for any therapeutic application and whether these can be used to address the above issues. In the first part of this work, we therefore address whether DC-based vaccines can be considered biologicals that are of standard quality, free of contaminants and can be generated to obtain relevant dose levels.
In recent years B cells have been recognized increasingly as important antigen-presenting cells (APC) capable of inducing antigen-specific CD4 and CD8 T cell responses under physiological and pathological conditions [12–14].
While it has been shown that antigen presentation by resting B cells and B cells activated by lipopolysaccharide induce tolerance in vivo[15,16], B cells activated via CD40 receptor can act as immunogenic APC [17,18]. CD40-activated B cells are being studied currently as an alternative type of APC for cellular vaccines [19–23]. Contrary to DC, little is known about the regulation of antigen presentation by B cells in cancer patients. We therefore studied the antigen presentation function and the migratory potential of activated B cells from cancer patients. In light of the above considerations, we also focused on the purity, standard phenotype, homing properties and cell numbers.
Material and methods
Systematic analysis of DC-based clinical trials in renal cell cancer and prostate cancer
A Medline–Ovid search was performed to identify articles reporting on clinical immunotherapy trials utilizing DC in tumour patients. For this first step the tumour entities renal cell cancer and prostate cancer were chosen for a detailed DC subanalysis, as they have a long-standing history and thus a high standard of DC-based vaccinations. Original papers published no earlier than 1 January 1 2000 in international, peer-reviewed journals, investigating at least six patients with the primary end-points of clinical or immune response, were included. Trials using allogeneic DC, describing mixed tumour entities or follow-up reports were excluded. Data obtained were collected and analysed in an Access database. DC subanalysis addressed the following items: DC type, DC purity and number of DC per vaccination.
Donor and patient samples
Peripheral blood from healthy volunteers and cancer patients was obtained by phlebotomy following informed consent and approval by the Institute's Review Board. Peripheral blood mononuclear cells (PBMC) were purified by Ficoll density centrifugation.
CD40-B cell generation
CD40-B cells were generated as described previously [24] In brief, whole PBMC were cultured on irradiated NIH3T3 cells transfected with CD154 (tCD40L) in the presence of recombinant human interleukin (rhIL)-4 (2 ng/ml; R&D Systems, Minneapolis, MN, USA) and clinical-grade cyclosporin A (CsA) (5·5 × 10−7 M; Novartis, Basel, Switzerland) in 4 ml of Iscove's modified Dulbecco's medium (IMEM; Invitrogen, Karlsruhe, Germany) supplemented with 10% pooled human serum. The expanding cells were transferred onto freshly prepared t-CD40L cells and fed with cytokine-replenished medium without CsA every 3–4 days.
Allogeneic mixed lymphocyte reaction assay
CD4+ lymphocytes were separated from peripheral blood by using a RosetteSep CD4+ T cell enrichment kit (StemCell Technologies, Vancouver, Canada) and stained with carboxyfluorescein succinimidyl ester (CFSE; Sigma, St Louis, MO, USA). CFSE-labelled CD4+ T cells were co-cultured for 4 days with CD40-B cells at various effector: target ratios and assessed by flow cytometric analysis.
Flow cytometry
The expression of cell surface molecules was determined by flow cytometry using standard procedures. Antibodies were purchased from BD Biosciences (San Jose, CA, USA) with the exception of CCR7 (R&D Systems).
Generation of cytotoxic T lymphocyte lines and cytotoxicity assay
Cytotoxic T lymphocyte lines were generated as described and tested for cytotoxicity using 2-h Europium (Eu3+) release assays. In brief, targets were labelled with Eu3+ by electoroporation, and 5 × 103 labelled cells per well were plated with various concentrations of effector cells. The percentage of cytotoxicity was calculated as follows: (experimental release – spontaneous release)/(maximum release – spontaneous release) × 100.
Chemotaxis assay
For B cell migration, 5 × 105 CD40-B cells were transferred into the upper chamber of 5-µm pore size transwell plates (Costar, Cambridge, MA, USA). Varying amounts of chemokines (R&D Systems) were added to the lower chamber. Migration of cells was assessed after 2 and 3 h using a Coulter Z2 counter (Beckman Coulter, Fullerton, CA, USA).
Results
Systematic analysis of DC as ‘biological’ in clinical trials
The systematic search identified 20 clinical trials fulfilling the described inclusion criteria [25–44]. A total of 502 patients had been enrolled in these trials. Individual data with regard to DC type and number of DC were published for only 51% of the patients (256). In most of the trials DC purity of cells assembled for therapeutic vaccinations was not specified. Only in one of the trials was purity indicated to be above 50% (Fig. 1a). MoDC were used in the majority (85%) of the investigated trials. Surprisingly, the portion of studies using mature MoDC was only 40%. In these eight trials 25·5% (128 of 502) of the entire population were treated (Fig. 1b). In three (15%) of the trials peripheral blood-derived DC enriched after sequential density centrifugation were used. Thus, in these the maturation status was not specified.
Fig. 1.
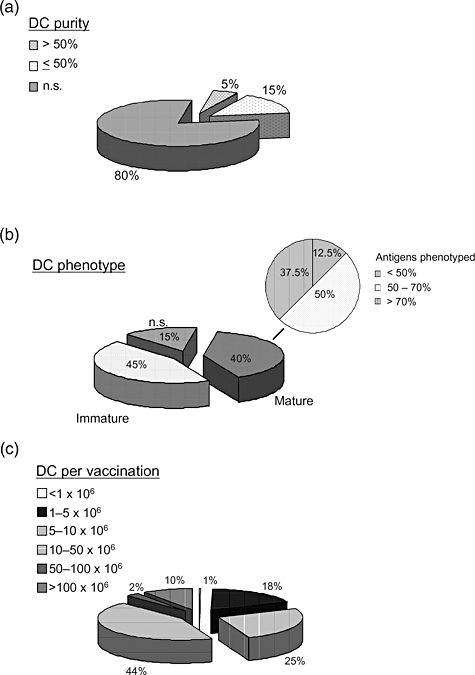
Quality of dendritic cells (DC) used in clinical vaccination trials. Exemplarily, publications of DC-based vaccination trials in patients with renal cell cancer (RCC) and prostate cancer were analysed. (a) Studies were categorized as to whether or not to include information about DC purity, and whether the purity was above or below 50%. (b) The proportion of studies based on the use of mature or immature DC was analysed. Additionally, focusing on the studies using mature DC, the completeness of DC phenotype analyses was investigated with regard to quality control criteria provided by Figdor et al.[26]. (c) Number of DC used per vaccination was classified in six dose groups for those 256 patients with individual information.
Focusing on studies using mature DC, quality control criteria as defined by Figdor and colleagues [45] were met by none of the trials concerning phenotype analysis. In 50% of the trials, less than half of the suggested analyses were performed (Fig. 1b). DC numbers used for individual vaccinations were marked by significant variability (between < 1 × 106 and > 100 × 106). Most patients were vaccinated with 10–50 × 106 DC (Fig. 1c). This equals 1·25 × 104 cells per kg to 6·25 × 104 cells per kg in a 80-kg patient and is in striking contrast to DC vaccinations in mice that use between 4 × 107/kg to 4 × 108/kg per vaccination or 3–4-log more cells. With regard to the individual patient information provided, the median number of vaccinations was three and median total ‘dose’ of vaccinated DC (number of vaccinations × number of DC per vaccination) was 4·5 × 107 DC per patient.
Generation of CD40-B cells from cancer patients
To determine whether CD40-activated B cells can be generated from cancer patients we tested several tumour entities (see below), among which colon cancer was most available and thus chosen as a primary model for solid tumours. Ficoll density separated PBMC were cultured with tCD40-L in the presence of rhIL-4, and proliferation of the cells and their phenotype was assessed. As shown in Fig. 2a, the cultured cells expanded significantly and the efficiency was comparable to that of CD40-B cells from healthy donors. Similar to CD40-B from healthy donors, for whom long-term expansion has been described [46,47], proliferation remained strong to day 21, when culture was discontinued for functional characterization. Cell numbers were calculated to reach more than 1 × 1010 after 5 weeks of culture when starting with two draws of 125 ml of blood; e.g. in an 80-kg patient this would be sufficient for > 10 vaccinations of 1·25 × 108. Fluorescence activated cell sorter analysis revealed that more than 90% of the cultured cells were B cells and histocompatibility and co-stimulatory molecules were expressed highly on the cells, which are consistent with CD40-activated B cells from healthy donors (Fig. 3). While preliminary observations in other tumours, such as prostate cancer, ovarian cancer, multiple myeloma, breast cancer, mantle cell lymphoma and acute myeloid leukaemia, confirm our observations (Kondo, unpublished results), the increasing use of anti-CD20 monoclonal antibodies might reduce the applicability of the above approach in the case of B cell malignancies.
Fig. 2.
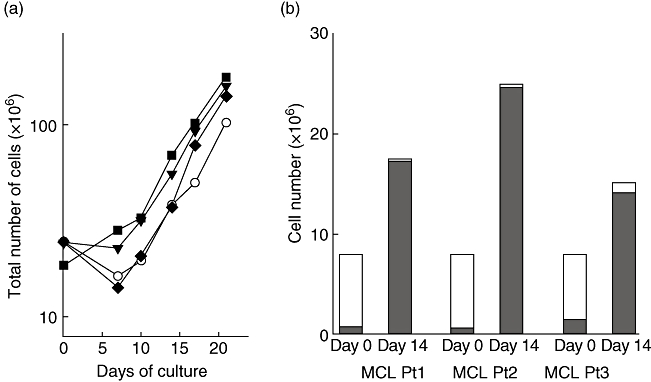
Expansion of CD40-B in cancer patients. CD40-B cells were generated using CD40L-transfected NIH3T3 cells and recombinant human interleukin-4. (a) CD40-B cells were generated from peripheral blood mononuclear cells of colon cancer patients (filled squares, triangles and diamonds). One representative healthy donor is included as reference (open circles). Total cell numbers of CD40-B cell cultures are shown. (b) CD40-B cell generation from mantle cell lymphoma patients. Total cell numbers (open bars) and purity of B cells (CD19+ cells) (closed bars) on days 0 and 14 are shown.
Fig. 3.
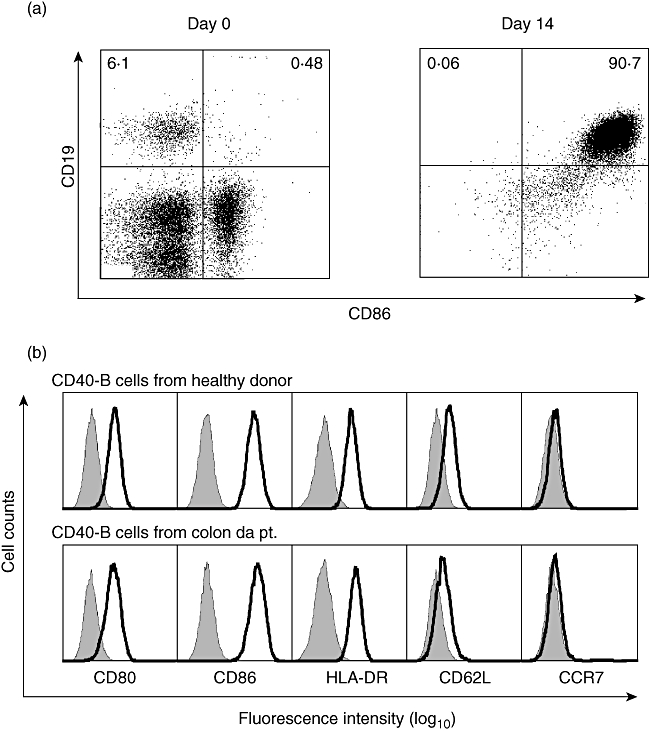
Expression of immunostimulatory molecules on CD40-B cells in cancer patients. (a) Two-colour immunophenotypic analysis, including CD19 and co-stimulatory molecule (CD86) on peripheral blood mononuclear cells on day 0 (left panel) and day 14 (right panel). Representative data are shown. (b) Surface expression of co-stimulatory molecules (CD80, CD86), human leucocyte antigen D-related, lymph node homing molecules (CD62L, CCR7) on CD40-B cells from healthy donor and colon cancer patient. The shaded histogram represents isotype control of each antibody.
Choosing mantle cell lymphoma as a complementary haematological tumour, CD40-B-activated B cells could be generated from four of six mantle cell lymphoma patients (Fig. 2b). In those four patients, there was no difference in expansion, purity or the phenotype (co-stimulatory molecules expression) compared with CD40-B cells from healthy donors. However, two patients had recently received Rituximab therapy (data not shown). The percentage of CD19+ cells in peripheral blood of the Rituximab-treated patients were 0·1% and 1·4%, meaning that almost all B cells were killed or currently undergoing apoptosis.
T cell stimulation by CD40-B cells from cancer patients
Primary allogeneic mixed lymphocyte reaction (MLR) with purified CD4+ T cell subsets were used to characterize antigen presentation of CD40-B cells from cancer patients. As shown in Fig. 4a,b, CFSE staining showed 20–30% of CD4+ T cells had proliferated after 4 days of co-culture with CD40-B cells from colon cancer patients. The results of MLR with colon cancer patients’ CD40-B cells were comparable to those with CD40-B cells from healthy donors (data not shown), indicating that CD40-B cells from cancer patients have sufficient functional ability to present alloantigens to T cells.
Fig. 4.
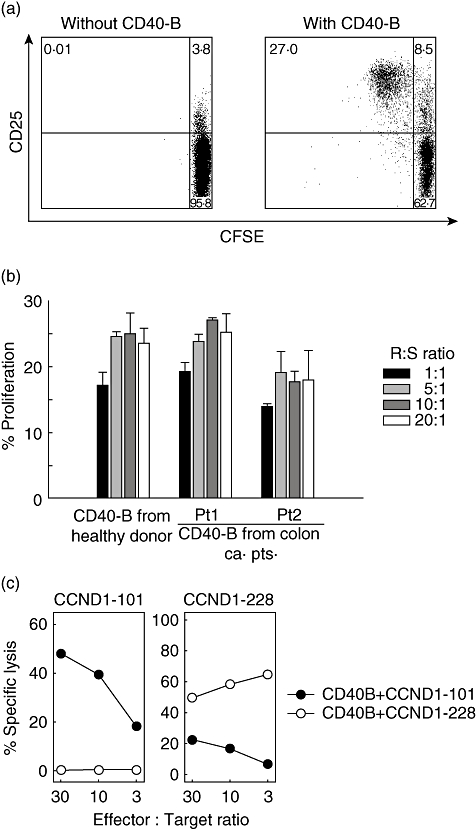
T cell stimulation by CD40-B cells from cancer patients. Allogenic CD4+ T cells were purified, stained with carboxyfluorescein succinimidyl ester (CFSE) and used as effector cells in a mixed lymphocyte reaction (MLR) assay. CD40-B cells from colon cancer patients were co-cultured for 4 days with CFSE-labelled CD4+ T cells at various effector: target ratios and T cell proliferation was assessed by flow cytometric analysis. (a) Representative dot plots of MLR. Unstimulated CD4+ T cells (negative control: left panel) and CD4+ T cells stimulated with CD40-B cells (right panel) are shown. (b) MLR proliferation by CD40-B cells from indicated donor (healthy or colon cancer patients) at various responder: stimulator ratios is shown. The percentage of cells with decreased CFSE staining intensity was reported as % proliferation. Error bars represent the mean ± standard error of the mean. (c) Generation of cyclin D1-specific cytotoxic T lymphocyte from cancer patients. Peripheral blood mononuclear cells from a colon cancer patient were stimulated with autologous CD40-B cells pulsed with indicated peptides. After four weekly stimulations, the lytic activity of the cytotoxic T lymphocyte line stimulated with CD40-B cells pulsed with CycD1_101–9 (left panel) or CycD1_228 (right panel) was tested by Europium release assay. Autologous CD40-B cells pulsed with CycD1_101–9 (filled circles) or CycD1_228 (open circles) were used as target cells at various effector: target ratios.
Next, we attempted to generate tumour antigen-specific T cells restricted to a self-human leucocyte antigen (HLA) molecule using CD40-B cells from colon cancer patients. Two HLA-A*0201-restricted synthetic nonamer peptides from cyclin D1 were used as model antigens. Cyclin D1 was expressed by the primary tumours of these patients and therefore represents a realistic scenario for cancer immunotherapy. PBMC from four colon cancer patients were stimulated with peptide-pulsed autologous CD40-B cells, and after four weekly stimulations the cytotoxicity of the bulk T cell cultures was examined by Eu release assay. T cells stimulated with CycD1_101 (LLGATCMFV) or CycD1_228 (RLTRFLSRV)-pulsed CD40-B cells could lyse relevant peptide-pulsed CD40-B cells, but not target cells pulsed with irrelevant peptide (Fig. 4c), indicating that CD40-B cells from cancer patients have the ability to induce tumour antigen-specific T cells.
Migration capacity of CD40-B cells from cancer patients
For the successful induction of immune responses, in vivo homing of APC (DC or CD40-B) to secondary lymphoid organs is crucial. As reported previously, CD40-B cells express chemokine receptors, CCR7 and CXCR4, which are critical for homing to secondary lymphoid tissues, and are therefore thought to stimulate T cells efficiently in the T cell area in lymphoid tissues. To address whether the migration capacity of CD40-B cells from cancer patients is impaired, we conducted a standard migration assay in the presence of chemokines on CCR7 and CXCR4. In the absence of chemokines, CD40-B cells showed very little spontaneous migration. CD40-B cells from cancer patients migrated efficiently towards CCL21/SLC and CXCL12/SDF-1, the cognate ligands for CCR7 and CXCR4 respectively (Fig. 5). No significant difference was observed between migration of CD40-B cells from colon cancer patients and healthy donors. Taken together, these data suggest that CD40-B cells from cancer patients have comparable migration capacity towards chemokines important for homing to secondary lymphoid organs with those from healthy donors.
Fig. 5.
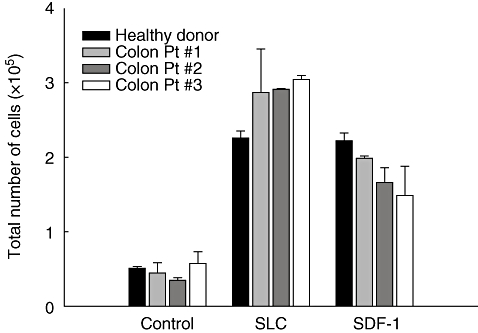
Migration of CD40-B cells from colon cancer patients. CD40-B cells from colon cancer patients or a control healthy donor were analysed for their migratory capacity towards the chemokines CCL21/SLC and CXCL12/SDF-1, which are ligands for CCR7 and CXCR4 respectively. Mean values and standard error of the mean of three independent experiments are shown.
Discussion
Cell-based vaccines are a promising cancer therapeutic. Induction of immunity in murine models and – in select cases – in humans certainly suggests significant potential to induce strong anti-tumour immune responses. However, so far clinical results remain disappointing [1]. An increasing knowledge of host–tumour interactions has shed light on tumour-protective mechanisms operative in the microenvironment [48–50], depending upon the stage of the disease. Several of these mechanisms have a direct impact on APC, e.g. rendering DC tolerogenic [4,51,52]. Therefore, the limited success of DC-based vaccines has been viewed recently in light of these tumour-derived factors.
However, generation of cellular vaccines remains technically challenging. Here, we show that production of significant quantities of a well-defined cellular agent that is free of contaminating bystander populations remains a rare exception. On the contrary, we feel that these basic requirements for any biological need to be met before we can be sure that the above inhibitory mechanisms in fact play a role. As suggested by Figdor and colleagues [45], it would be of extreme value to use sufficient doses of at least moderately pure, standard-phenotype DC using as few different-generation protocols as necessary.
Only with such a defined agent would it be possible to address the above biological questions in cancer patients. However, clinical settings involving cancer patients are a reality that is far from murine models and therefore these requirements are a significant challenge.
We and others have characterized CD40-activated B cells previously as cellular adjuvants [24,53–55]. In laboratory settings using peripheral blood from healthy donors, they could be expanded from small amounts (e.g. 10–20 cc) of peripheral blood over several months. When generated from healthy volunteers they reach greater 95% purity after 10–12 days and maintain their APC phenotype (CD80high CD83high CD86high MHC-class I/IIhigh) for several weeks, despite rapid expansion (doubling time 3–4 days). These cells prime naive T cells and induce expansion of tumour antigen-specifc CD4+ and CD8+ cells. Based on these findings, clinical trials are in preparation. However, they need to meet the same criteria as defined above in order to be of potential use in cancer patients and in order to enable the clinical researcher to learn from such an intervention.
Here, we show that activation and proliferation of B cells from colon cancer and mantle cell lymphoma patients is equivalent to that observed in healthy donors. Purity is always > 95% after 2 weeks and remains stable for several weeks. In this system cell numbers are 2–3 logs higher than those used in the DC trials analysed without even having to apherese the patient. APC function was assessed first by MLR and equivalent induction of T cell proliferation was identified. Furthermore, expansion of self-antigen-specific T cells was shown in HLA-A*0201+ patients. Furthermore, they express CCR7 and CD62L and demonstrate strong migration to CCL21 and CXCL12.
While CD40-B expansion was feasible in CD20-monoclonal antibody-naive patients, Rituximab treatment represents a challenge to the above approach. If B cells were expanded despite low initial counts, quality was comparable to healthy donors. However, it was not possible to overcome the effect of Rituximab in every patient. As B cell recovery takes about 1 year after completion of Rituximab infusion [56], it appears advisable to generate and cryopreserve CD40-B cells before the start of therapy or after B cell recovery. However, these considerations are relevant in only ∼0·5% of cancer patients.
Taken together, CD40-B cells can be generated in the majority of cancer patients meeting standard requirements for cellular adjuvants such as cell number and purity, as well as APC phenotype and function.
References
- 1.Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nat Med. 2004;10:909–15. doi: 10.1038/nm1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Luft T, Jefford M, Luetjens P, et al. Functionally distinct dendritic cell (DC) populations induced by physiologic stimuli: prostaglandin E2 regulates the migratory capacity of specific DC subsets. Blood. 2002;100:1362–72. doi: 10.1182/blood-2001-12-0360. [DOI] [PubMed] [Google Scholar]
- 3.Kuroda E, Sugiura T, Okada K, Zeki K, Yamashita U. Prostaglandin E2 up-regulates macrophage-derived chemokine production but suppresses IFN-inducible protein-10 production by APC. J Immunol. 2001;166:1650–8. doi: 10.4049/jimmunol.166.3.1650. [DOI] [PubMed] [Google Scholar]
- 4.von Bergwelt-Baildon M, Popov A, Saric T, et al. CD25 and indoleamine 2,3-dioxygenase are up-regulated by prostaglandin E2 and expressed by tumor-associated dendritic cells in vivo: additional mechanisms of T-cell inhibition. Blood. 2006;108:228–37. doi: 10.1182/blood-2005-08-3507. [DOI] [PubMed] [Google Scholar]
- 5.Kalinski P, Hilkens CM, Snijders A, Snijdewint FG, Kapsenberg ML. IL-12-deficient dendritic cells, generated in the presence of prostaglandin E2, promote type 2 cytokine production in maturing human naive T helper cells. J Immunol. 1997;159:28–35. [PubMed] [Google Scholar]
- 6.Sharma S, Stolina M, Lin Y, et al. IL-10 promotes lung cancer growth by suppressing both T cell and APC function. J Immunol. 1999;163:5020–8. [PubMed] [Google Scholar]
- 7.Steinbrink K, Graulich E, Kubsch S, Knop J, Enk A. CD4+ and CD8+ anergic T cells induced by interleukin-10-treated human dendritic cells display antigen-specific suppressor activity. Blood. 2002;99:2468–76. doi: 10.1182/blood.v99.7.2468. [DOI] [PubMed] [Google Scholar]
- 8.Sato K, Kawasaki H, Nagayama H, et al. TGF-{beta}1 reciprocally controls chemotaxis of human peripheral blood monocyte-derived dendritic cells via chemokine receptors. J Immunol. 2000;164:2285–95. doi: 10.4049/jimmunol.164.5.2285. [DOI] [PubMed] [Google Scholar]
- 9.Kobie J, Wu R, Kurt R, et al. Transforming growth factor {beta} inhibits the antigen-presenting functions and antitumor activity of dendritic cell vaccines. Cancer Res. 2003;63:1860–4. [PubMed] [Google Scholar]
- 10.Gabrilovich DI, Chen HL, Girgis KR, et al. Production of vascular endothelial growth factor by human tumors inhibits the functional maturation of dendritic cells. Nat Med. 1996;2:1096–103. doi: 10.1038/nm1096-1096. [DOI] [PubMed] [Google Scholar]
- 11.Dorrie J, Schaft N, Muller I, et al. Introduction of functional chimeric E/L-selectin by RNA electroporation to target dendritic cells from blood to lymph nodes. Cancer Immunol Immunother. 2008;57:467–77. doi: 10.1007/s00262-007-0385-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Serreze DV, Fleming SA, Chapman HD, Richard SD, Leiter EH, Tisch RM. B lymphocytes are critical antigen-presenting cells for the initiation of T cell-mediated autoimmune diabetes in nonobese diabetic mice. J Immunol. 1998;161:3912–18. [PubMed] [Google Scholar]
- 13.Kleindienst P, Brocker T. Concerted antigen presentation by dendritic cells and B cells is necessary for optimal CD4 T-cell immunity in vivo. Immunology. 2005;115:556–64. doi: 10.1111/j.1365-2567.2005.02196.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yan J, Harvey BP, Gee RJ, Shlomchik MJ, Mamula MJ. B cells drive early T cell autoimmunity in vivo prior to dendritic cell-mediated autoantigen presentation. J Immunol. 2006;177:4481–7. doi: 10.4049/jimmunol.177.7.4481. [DOI] [PubMed] [Google Scholar]
- 15.Fuchs EJ, Matzinger P. B cells turn off virgin but not memory T cells. Science. 1992;258:1156–9. doi: 10.1126/science.1439825. [DOI] [PubMed] [Google Scholar]
- 16.Gilbert KM, Weigle WO. Tolerogenicity of resting and activated B cells. J Exp Med. 1994;179:249–58. doi: 10.1084/jem.179.1.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Parekh VV, Prasad DV, Banerjee PP, Joshi BN, Kumar A, Mishra GC. B cells activated by lipopolysaccharide, but not by anti-Ig and anti-CD40 antibody, induce anergy in CD8+ T cells: role of TGF-beta 1. J Immunol. 2003;170:5897–911. doi: 10.4049/jimmunol.170.12.5897. [DOI] [PubMed] [Google Scholar]
- 18.Ritchie DS, Yang J, Hermans IF, Ronchese F. B-Lymphocytes activated by CD40 ligand induce an antigen-specific anti-tumour immune response by direct and indirect activation of CD8(+) T-cells. Scand J Immunol. 2004;60:543–51. doi: 10.1111/j.0300-9475.2004.01517.x. [DOI] [PubMed] [Google Scholar]
- 19.Coughlin CM, Vance BA, Grupp SA, Vonderheide RH. RNA-transfected CD40-activated B cells induce functional T-cell responses against viral and tumor antigen targets: implications for pediatric immunotherapy. Blood. 2004;103:2046–54. doi: 10.1182/blood-2003-07-2379. [DOI] [PubMed] [Google Scholar]
- 20.Fujiwara H, Melenhorst JJ, El Ouriaghli F, et al. In vitro induction of myeloid leukemia-specific CD4 and CD8 T cells by CD40 ligand-activated B cells gene modified to express primary granule proteins. Clin Cancer Res. 2005;11:4495–503. doi: 10.1158/1078-0432.CCR-04-2363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lapointe R, Bellemare-Pelletier A, Housseau F, Thibodeau J, Hwu P. CD40-stimulated B lymphocytes pulsed with tumor antigens are effective antigen-presenting cells that can generate specific T cells. Cancer Res. 2003;63:2836–43. [PubMed] [Google Scholar]
- 22.von Bergwelt-Baildon M, Shimabukuro-Vornhagen A, Popov A, et al. CD40-activated B cells express full lymph node homing triad and induce T-cell chemotaxis: potential as cellular adjuvants. Blood. 2006;107:2786–9. doi: 10.1182/blood-2004-01-0113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mason NJ, Coughlin CM, Overley B, et al. RNA-loaded CD40-activated B cells stimulate antigen-specific T-cell responses in dogs with spontaneous lymphoma. Gene Ther. 2008;15:955–65. doi: 10.1038/gt.2008.22. [DOI] [PubMed] [Google Scholar]
- 24.von Bergwelt-Baildon MS, Vonderheide RH, Maecker B, et al. Human primary and memory cytotoxic T lymphocyte responses are efficiently induced by means of CD40-activated B cells as antigen-presenting cells: potential for clinical application. Blood. 2002;99:3319–25. doi: 10.1182/blood.v99.9.3319. [DOI] [PubMed] [Google Scholar]
- 25.Barrou B, Benoit G, Ouldkaci M, et al. Vaccination of prostatectomized prostate cancer patients in biochemical relapse, with autologous dendritic cells pulsed with recombinant human PSA. Cancer Immunol Immunother. 2004;53:453–60. doi: 10.1007/s00262-003-0451-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bleumer I, Tiemessen DM, Oosterwijk-Wakka JC, et al. Preliminary analysis of patients with progressive renal cell carcinoma vaccinated with CA9-peptide-pulsed mature dendritic cells. J Immunother. 2007;30:116–22. doi: 10.1097/01.cji.0000211318.22902.ec. [DOI] [PubMed] [Google Scholar]
- 27.Burch PA, Breen JK, Buckner JC, et al. Priming tissue-specific cellular immunity in a phase I trial of autologous dendritic cells for prostate cancer. Clin Cancer Res. 2000;6:2175–82. [PubMed] [Google Scholar]
- 28.Fong L, Brockstedt D, Benike C, et al. Dendritic cell-based xenoantigen vaccination for prostate cancer immunotherapy. J Immunol. 2001;167:7150–6. doi: 10.4049/jimmunol.167.12.7150. [DOI] [PubMed] [Google Scholar]
- 29.Gitlitz BJ, Belldegrun AS, Zisman A, et al. A pilot trial of tumor lysate-loaded dendritic cells for the treatment of metastatic renal cell carcinoma. J Immunother. 2003;26:412–19. doi: 10.1097/00002371-200309000-00004. [DOI] [PubMed] [Google Scholar]
- 30.Heiser A, Coleman D, Dannull J, et al. Autologous dendritic cells transfected with prostate-specific antigen RNA stimulate CTL responses against metastatic prostate tumors. J Clin Invest. 2002;109:409–17. doi: 10.1172/JCI14364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Holtl L, Zelle-Rieser C, Gander H, et al. Immunotherapy of metastatic renal cell carcinoma with tumor lysate-pulsed autologous dendritic cells. Clin Cancer Res. 2002;8:3369–76. [PubMed] [Google Scholar]
- 32.Lee D. Autologous dendritic cells pulsed with prostatic acid phosphatase (APC8015) for patients with hormone-refractory prostate cancer with a Gleason score < or = 7. Clin Prostate Cancer. 2003;2:81–3. doi: 10.1016/s1540-0352(11)70024-5. [DOI] [PubMed] [Google Scholar]
- 33.Lodge PA, Jones LA, Bader RA, Murphy GP, Salgaller ML. Dendritic cell-based immunotherapy of prostate cancer: immune monitoring of a phase II clinical trial. Cancer Res. 2000;60:829–33. [PubMed] [Google Scholar]
- 34.Marten A, Flieger D, Renoth S, et al. Therapeutic vaccination against metastatic renal cell carcinoma by autologous dendritic cells: preclinical results and outcome of a first clinical phase I/II trial. Cancer Immunol Immunother. 2002;51:637–44. doi: 10.1007/s00262-002-0324-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mu LJ, Kyte JA, Kvalheim G, et al. Immunotherapy with allotumour mRNA-transfected dendritic cells in androgen-resistant prostate cancer patients. Br J Cancer. 2005;93:749–56. doi: 10.1038/sj.bjc.6602761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Murphy GP, Tjoa BA, Simmons SJ, Rogers MK, Kenny GM, Jarisch J. Higher-dose and less frequent dendritic cell infusions with PSMA peptides in hormone-refractory metastatic prostate cancer patients. Prostate. 2000;43:59–62. doi: 10.1002/(sici)1097-0045(20000401)43:1<59::aid-pros8>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 37.Oosterwijk-Wakka JC, Tiemessen DM, Bleumer I, et al. Vaccination of patients with metastatic renal cell carcinoma with autologous dendritic cells pulsed with autologous tumor antigens in combination with interleukin-2: a phase 1 study. J Immunother. 2002;25:500–8. doi: 10.1097/00002371-200211000-00006. [DOI] [PubMed] [Google Scholar]
- 38.Perambakam S, Hallmeyer S, Reddy S, et al. Induction of specific T cell immunity in patients with prostate cancer by vaccination with PSA146–154 peptide. Cancer Immunol Immunother. 2006;55:1033–42. doi: 10.1007/s00262-005-0090-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Small EJ, Fratesi P, Reese DM, et al. Immunotherapy of hormone-refractory prostate cancer with antigen-loaded dendritic cells. J Clin Oncol. 2000;18:3894–903. doi: 10.1200/JCO.2000.18.23.3894. [DOI] [PubMed] [Google Scholar]
- 40.Su Z, Dannull J, Heiser A, et al. Immunological and clinical responses in metastatic renal cancer patients vaccinated with tumor RNA-transfected dendritic cells. Cancer Res. 2003;63:2127–33. [PubMed] [Google Scholar]
- 41.Su Z, Dannull J, Yang BK, et al. Telomerase mRNA-transfected dendritic cells stimulate antigen-specific CD8+ and CD4+ T cell responses in patients with metastatic prostate cancer. J Immunol. 2005;174:3798–807. doi: 10.4049/jimmunol.174.6.3798. [DOI] [PubMed] [Google Scholar]
- 42.Thomas-Kaskel AK, Zeiser R, Jochim R, et al. Vaccination of advanced prostate cancer patients with PSCA and PSA peptide-loaded dendritic cells induces DTH responses that correlate with superior overall survival. Int J Cancer. 2006;119:2428–34. doi: 10.1002/ijc.22097. [DOI] [PubMed] [Google Scholar]
- 43.Waeckerle-Men Y, Uetz-von Allmen E, Fopp M, et al. Dendritic cell-based multi-epitope immunotherapy of hormone-refractory prostate carcinoma. Cancer Immunol Immunother. 2006;55:1524–33. doi: 10.1007/s00262-006-0157-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wierecky J, Muller MR, Wirths S, et al. Immunologic and clinical responses after vaccinations with peptide-pulsed dendritic cells in metastatic renal cancer patients. Cancer Res. 2006;66:5910–18. doi: 10.1158/0008-5472.CAN-05-3905. [DOI] [PubMed] [Google Scholar]
- 45.Figdor CG, de Vries IJ, Lesterhuis WJ, Melief CJ. Dendritic cell immunotherapy: mapping the way. Nat Med. 2004;10:475–80. doi: 10.1038/nm1039. [DOI] [PubMed] [Google Scholar]
- 46.Wiesner M, Zentz C, Mayr C, et al. Conditional immortalization of human B cells by CD40 ligation. PLoS ONE. 2008:3. doi: 10.1371/journal.pone.0001464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yamashita Y, Tsurumi T, Mori N, Kiyono T. Immortalization of Epstein–Barr virus-negative human B lymphocytes with minimal chromosomal instability. Path Int. 2006;56:659–67. doi: 10.1111/j.1440-1827.2006.02026.x. [DOI] [PubMed] [Google Scholar]
- 48.Dunn GP, Koebel CM, Schreiber RD. Interferons, immunity and cancer immunoediting. Nat Rev Immunol. 2006;6:836–48. doi: 10.1038/nri1961. [DOI] [PubMed] [Google Scholar]
- 49.Gajewski TF. Identifying and overcoming immune resistance mechanisms in the melanoma tumor microenvironment. Clin Cancer Res. 2006;12:2326s–30s. doi: 10.1158/1078-0432.CCR-05-2517. [DOI] [PubMed] [Google Scholar]
- 50.Mantovani A, Romero P, Palucka AK, Marincola FM. Tumour immunity: effector response to tumour and role of the microenvironment. Lancet. 2008;371:771–83. doi: 10.1016/S0140-6736(08)60241-X. [DOI] [PubMed] [Google Scholar]
- 51.Popov A, Schultze JL. IDO-expressing regulatory dendritic cells in cancer and chronic infection. J Mol Med. 2008;86:145–60. doi: 10.1007/s00109-007-0262-6. [DOI] [PubMed] [Google Scholar]
- 52.Rutella S, Danese S, Leone G. Tolerogenic dendritic cells: cytokine modulation comes of age. Blood. 2006;108:1435–40. doi: 10.1182/blood-2006-03-006403. [DOI] [PubMed] [Google Scholar]
- 53.Kondo E, Topp MS, Kiem HP, et al. Efficient generation of antigen-specific cytotoxic T cells using retrovirally transduced CD40-activated B cells. J Immunol. 2002;169:2164–71. doi: 10.4049/jimmunol.169.4.2164. [DOI] [PubMed] [Google Scholar]
- 54.Schultze JL, Michalak S, Seamon MJ, et al. CD40-activated human B cells: an alternative source of highly efficient antigen presenting cells to generate autologous antigen-specific T cells for adoptive immunotherapy. J Clin Invest. 1997;100:2757–65. doi: 10.1172/JCI119822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.von Bergwelt-Baildon MS, Schultze JL, Nadler LM. CD40-activated B-cells as antigen presenting cells: from immunobiology to immunotherapy. Modern Aspects Immunobiol. 2003;3:71–4. [Google Scholar]
- 56.McLaughlin P, Grillo-Lopez AJ, Link BK, et al. Rituximab chimeric anti-CD20 monoclonal antibody therapy for relapsed indolent lymphoma: half of patients respond to a four-dose treatment program. J Clin Oncol. 1998;16:2825–33. doi: 10.1200/JCO.1998.16.8.2825. [DOI] [PubMed] [Google Scholar]