

Abstract
Experimental autoimmune glomerulonephritis (EAG) can be induced in Wistar Kyoto (WKY) rats by immunization with the non-collagenous domain (NC1) of the alpha 3 chain of type IV collagen, α3(IV)NC1. In patients with Goodpasture's disease, the major B cell epitope is located at the N-terminus of α3(IV)NC1. In order to investigate whether B and T cell responses in EAG are directed towards immunodominant peptides within the same region of rat α3(IV)NC1, we immunized WKY rats with recombinant rat α3(IV)NC1 (positive control) and five 15-mer overlapping synthetic peptides from the N-terminus of rat α3(IV)NC1: pCol(17–31), pCol(24–38), pCol(31–45), pCol(38–52) and pCol(45–59). Positive control animals immunized with α3(IV)NC1 produced an antibody response directed towards α3(IV)NC1 and pCol(24–38). Splenic T cells from these animals proliferated in response to α3(IV)NC1 and pCol(24–38). No significant antibody or T cell responses were observed to the other peptides examined. Animals immunized with pCol(24–38) developed linear deposits of immunoglobulin G on the glomerular basement membrane, albuminuria and focal necrotizing glomerulonephritis with crescent formation by week 6 after immunization. Circulating antibodies from these animals recognized pCol(24–38) and α3(IV)NC1, and their T cells proliferated in response to pCol(24–38) and α3(IV)NC1. Animals immunized with the other peptides developed no significant immune response to α3(IV)NC1 and no disease. In conclusion, these results demonstrate that a 15-mer peptide from the N-terminus of α3(IV)NC1 [pCol(24–38)] is recognized by B and T cells from rats immunized with recombinant α3(IV)NC1, and that the same peptide is capable of inducing crescentic glomerulonephritis. Identification of this immunodominant peptide will be of value in designing new therapeutic strategies for inducing mucosal tolerance in EAG, which may be applicable to patients with glomerulonephritis.
Keywords: anti-GBM antibodies, B cell epitope, GBM, immunodominant peptides, T cell epitope
Introduction
Goodpasture's, or anti-glomerular basement membrane (GBM), disease is an autoimmune disorder characterized by rapidly progressive glomerulonephritis and lung haemorrhage [1,2]. The disease is caused by autoimmunity to a component of the GBM, which has been identified as the non-collagenous domain (NC1) of the α3 chain of type IV collagen, α3(IV)NC1 [3,4]. The immunodominant region for autoantibody binding in patients has been localized to the amino terminal of the α3(IV)NC1 molecule [5,6]. More recently, the major conformational epitopes have been mapped to residues 17–31 and 127–141 of the NC1 domain [7,8].
Experimental autoimmune glomerulonephritis (EAG), an animal model of Goodpasture's disease, can be induced in susceptible strains of rats and mice by immunization with GBM [9–11] or recombinant α3(IV)NC1 [12–14]. This results in the development of circulating and deposited anti-GBM antibodies, with focal necrotizing crescentic glomerulonephritis and lung haemorrhage. EAG shares many features with the human disease, in that the renal and lung pathology are very similar [15], and the anti-GBM antibodies show the same specificity for the main target antigen, α3(IV)NC1 [12–14].
There is now compelling evidence for the role of both humoral and cell-mediated immunity in the pathogenesis of EAG. The pathogenic role of anti-GBM antibodies has been demonstrated in a variety of passive transfer studies [11,16–18]. Transfer of disease has been demonstrated using antibodies pooled from the serum of nephritic mice [11], antibodies purified from the urine of nephritic rats [16], monoclonal antibodies (mAb) derived from rats with EAG [17] and antibodies eluted from the kidney of nephritic rats [18]. In the latter study, it was shown that deposited anti-GBM antibody has a higher functional affinity for GBM than circulating antibody.
The pathogenic role of T cells in EAG has also been demonstrated in several studies. T cells have been shown to be present in the glomeruli of animals with EAG [13,15], to proliferate in response to α3(IV)NC1 [14,19] and to transfer disease to naive recipients [11,20]. Glomerular T cells from rats with EAG show restricted T cell receptor CDR3 spectratypes, demonstrating that they are an oligoclonal antigen-driven population [21]. Anti-T cell immunotherapy has been shown to be effective in preventing or ameliorating disease [22–25]. Anti-CD4 mAb therapy is effective in the prevention of EAG [22], anti-CD8 mAb therapy is effective in both prevention and treatment of established disease [23], inhibition of T cell co-stimulation by blockade of either the CD28-B7 pathway [24] or the CD154-CD40 pathway [25] has been shown to reduce the severity of glomerulonephritis, and oral administration of GBM [26] or nasal administration of recombinant α3(IV)NC1 [27] has been shown to induce mucosal tolerance.
Further evidence supporting the role of T cell-mediated cellular immunity in the pathogenesis of EAG is documented in recent studies, including our own, demonstrating that synthetic peptides derived from α3(IV)NC1 can induce glomerulonephritis in Wistar Kyoto (WKY) rats [28–35]. Luo et al. showed that a 24-mer synthetic peptide, pCol(28–51), from the N terminus of α3(IV)NC1 was capable of inducing glomerulonephritis, although this was mild and inconsistent [29], while Wu et al. showed that a 13-mer peptide, pCol(28–40), containing a T cell epitope from α3(IV)NC1, induced severe crescentic glomerulonephritis [30]. In further characterization of this T cell epitope, it was shown that autoantibody deposition followed T cell-mediated damage to the kidney [31] and that only three residues within the peptide were critical for disease induction [32]. In addition, it has been reported that peptides containing the T cell epitope not only induced severe glomerulonephritis, but also triggered a diversified anti-GBM antibody response through B cell epitope spreading, suggesting that the autoantibody response to GBM antigens could be induced by a single nephritogenic T cell epitope [33–35].
In this study we have demonstrated that a 15-mer peptide from the N-terminus of α3(IV)NC1, pCol(24–38), is recognized by B and T cells from rats immunized with recombinant α3(IV)NC1, and that the same peptide is capable of inducing crescentic glomerulonephritis. Identification of this immunodominant peptide should be of value in designing new therapeutic strategies for mucosal tolerance in EAG, which may be applicable to patients with glomerulonephritis.
Materials and methods
Experimental animals
Male WKY rats, aged 8–10 weeks and weighing 120–150 g, were purchased from Charles River (Margate, Kent, UK). All animals were housed in standard conditions and had free access to normal laboratory diet and water. All experimental procedures were conducted in accordance with the UK Animals (Scientific Procedures) Act.
Production of recombinant rat α3(IV)NC1
Recombinant rat α3(IV)NC1 was produced from a stably transfected HEK293 cell line, as described previously [27]. Purification of recombinant rat α3(IV)NC1 from the supernatant was carried out by affinity chromatography using an anti-FLAG M2 affinity column (Sigma-Aldrich Company Ltd, Poole, UK), Recombinant rat α3(IV)NC1 was then characterized by Western blotting, using serum from an animal with EAG and control serum, as described previously [27].
Production of synthetic peptides
Five 15mer peptides, overlapping by eight amino acids (aa) and spanning the first 43 aa of rat α3(IV)NC1, were synthesized by the Advanced Biotechnology Centre, Charing Cross Campus, Imperial College London, UK. The aa sequence of the five peptides was as follows: peptide 1 pCol(17–31) – (TRMRGFIFTRHSQTT); peptide 2 pCol(24–38) – (FTRHSQTTANPSCPE); peptide 3 pCol(31–45) – (TANPSCPEGTQPLYS); peptide 4 pCol(38–52) – (EGTQPLYSGFSLLFV); and peptide 5 pCol(45–59) – (SGFSLLFVQGNEHAH).
Experimental protocol
Groups of WKY rats (n = 6) were given a single intramuscular (i.m.) injection of each of the synthetic peptides at a dose of 500 µg/rat in an equal volume of Freund's complete adjuvant (FCA, Sigma-Aldrich Company Ltd). In addition, groups of positive control rats (n = 6) were given a single i.m. injection of recombinant rat α3(IV)NC1 at a dose of 100 µg/rat in an equal volume of FCA [13], and groups of negative control rats (n = 6) were given FCA alone. Blood samples were taken by tail artery puncture under light anaesthesia (Isofluorane), and 24-h urine specimens were obtained at different time-points by placing animals in metabolic cages. All animals were killed at week 6 after immunization.
Assessment of antibody responses
Circulating antibody concentrations were measured in sera of experimental animals by a solid-phase enzyme-linked immunosorbent assay (ELISA), as described previously [10,13]. Briefly, recombinant rat α3(IV)NC1 or each of the synthetic peptides were coated onto ELISA plates (Life Technologies, Paisley, UK) at a concentration of 5 µg/ml by overnight incubation at 4°C. An optimum dilution of sera from animals immunized with recombinant rat α3(IV)NC1 or each of the synthetic peptides was then applied for 1 h at 37°C. Bound antibody was detected by alkaline phosphatase conjugated sheep anti-rat immunoglobulin (Ig) G (Sigma-Aldrich Company Ltd), and developed using the substrate p-nitrophenyl phosphate (NPP; Sigma-Aldrich Company Ltd). The absorbencies for each well were read at 405 nm using an Anthos Multiskan ELISA plate reader (Lab Tech International, Uckfield, UK), and the results calculated as mean optical density for each triplicate sample.
Assessment of T cell responses
T cell proliferative responses in the experimental animals were measured by standard tritiated thymidine incorporation assays, as described previously [26,27]. Briefly, spleens were dissociated into a single cell suspension, and plated out in round-bottomed 96-well plates (Invitrogen, Paisley, Scotland, UK) at a concentration of 5 × 105 cells/well. Cells were then cultured with α3(IV)NC1 or each of the synthetic peptides at a concentration of 10 µg/ml, in a humidified environment with 5% CO2 at 37°C for 72 h. Tritiated thymidine (Amersham Bioscience UK Limited, Little Chalfont, UK) was added at a concentration of 1 µCi/well at 16 h before harvesting, and thymidine incorporation was measured using an automated β counter (Amersham Bioscience UK Limited). Results were expressed as a stimulation index, which was calculated by dividing the counts per minute (cpm) in wells cultured with α3(IV)NC1 or peptides by the cpm in wells cultured with phosphate-buffered saline.
Assessment of disease after immunization with synthetic peptides
Deposition of IgG on the GBM
Deposits of IgG in the glomeruli were detected by direct immunofluorescence, as described previously [10,13,15]. Tissue was embedded in OCT embedding medium (Miles Inc, Elkhart, IN, USA) on cork disks, snap-frozen in isopentane (BDH Laboratory Supplies, Poole, Dorset, UK) pre-cooled in liquid nitrogen and stored at −70°C. Cryostat sections were cut 5 µm thick and were incubated with fluorescein isothiocyanate-labelled rabbit anti-rat IgG (Serotec Ltd, Oxford, UK). The degree of immunostaining was graded from 0 to 3+ by a ‘blinded’ observer.
Albuminuria
Urinary albumin concentrations were measured at different time-points in 24-h collections by rocket immunoelectrophoresis (Amersham Bioscience UK Limited), as described previously [10,13,15]. Briefly, urine samples from experimental animals were subjected to immunoelectrophoresis at 60 v in an electrophoresis tank containing barbitone buffer (BDH Laboratory Supplies), pH 9·5, for 6 h, using a 1% agarose gel (BDH Laboratory Supplies) containing rabbit anti-sera to rat albumin raised in our laboratory. Results were calculated using rat serum albumin standards (which were run at the same time) and expressed in mg per 24 h.
Glomerular abnormalities
Kidney tissue was fixed in 10% neutral buffered formalin, processed and embedded in paraffin wax for light microscopy by standard techniques (Histopathology Department, South Kensington Campus, Imperial College London, UK). Briefly, 3 µm sections were stained with haemotoxylin and eosin and periodic acid-Schiff. Fifty glomeruli per section were assessed and graded by a ‘blinded’ observer as: normal, mild (small areas of hypercellularity and/or focal necrosis), moderate (< 50% of the glomerulus affected by segmental necrosis and/or crescent formation) or severe (> 50% of the glomerulus affected by segmental necrosis and/or crescent formation), and expressed as a percentage of glomeruli examined [10,13,15].
Inhibition assay
The significance of binding of antibody from animals immunized with immunodominant peptide pCol(24–38) to recombinant rat α3(IV)NC1 was determined by inhibition studies. An optimum dilution of sera from animals immunized with pCol(24–38) was co-incubated in solution for 1 h at 37°C with pCol(24–38) or pCol (38–52) at concentrations of 1 µg/ml, 3 µg/ml, 10 µg/ml, 30 µg/ml, 100 µg/ml, 300 µg/ml or 1000 µg/ml. One hundred µl of each of the solutions was then transferred to ELISA plates coated with recombinant α3(IV)NC1 and incubated for 1 h at 37°C. Levels of bound antibodies were detected by alkaline phosphatase conjugated rabbit anti-goat IgG (Sigma-Aldrich Company Ltd), and developed and analysed as described above.
Statistical analysis
Differences between data were determined by the Mann–Whitney U-test. Analysis of variance was used to confirm differences between multiple data.
Results
Assessment of antibody responses
Sera from positive control animals immunized with α3(IV)NC1 showed a significant antibody response directed towards α3(IV)NC1 and pCol(24–38), but not to any of the other peptides. Animals immunized with pCol(24–38) generated an antibody response to the peptide and α3(IV)NC1, while those immunized with the other peptides did not make a significant antibody response to themselves or α3(IV)NC1. Results are shown in Fig. 1.
Fig. 1.
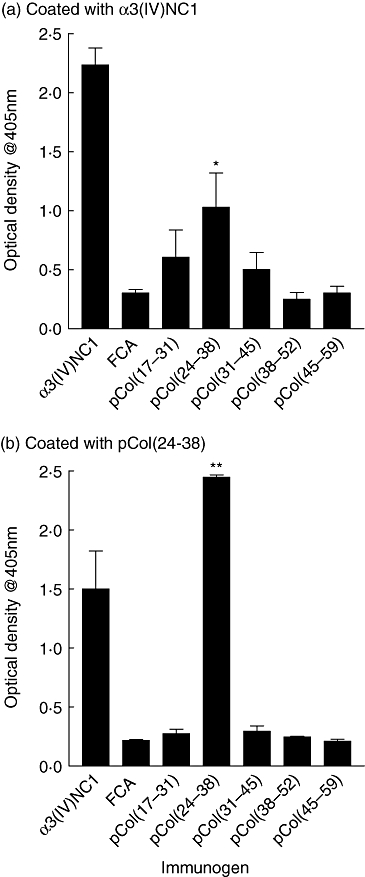
Circulating antibody levels from groups of Wistar Kyoto (WKY) rats (n = 6) immunized with alpha 3 chain of type IV collagen [α3(IV)NC1] or the five 15-mer overlapping peptides, directed towards: (a) α3(IV)NC1 and (b) pCol(24–38). Results shown represent the mean ± standard deviation of each group at week 6 after immunization. *P < 0·01; **P < 0·005, pCol(24–38) immunized versus Freund's complete adjuvant (FCA) alone. Each experiment was performed twice.
Assessment of T cell responses
Splenic T cells from positive control animals immunized with α3(IV)NC1, and cultured with either α3(IV)NC1 or pCol(24–38), showed a significant T cell proliferative response when compared with cells cultured with the other peptides. Splenic T cells from animals immunized with pCol(24–38), and cultured with either the peptide or α3(IV)NC1, also showed a significant T cell proliferative response. Splenic T cells from animals immunized with the other peptides did not show a significant T cell proliferative response when cultured with themselves or α3(IV)NC1. Results are shown in Fig. 2.
Fig. 2.
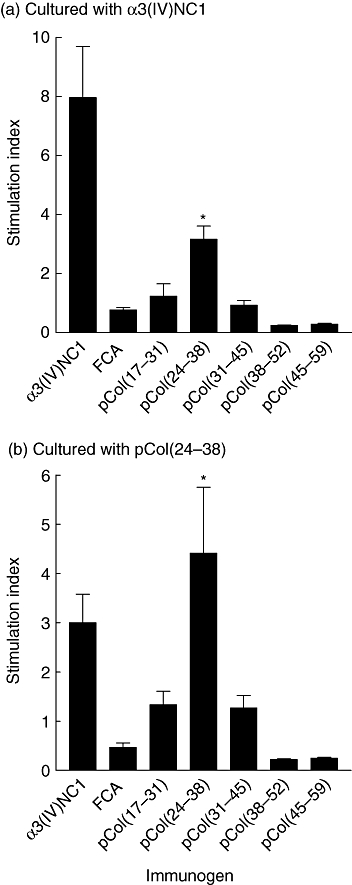
T cell proliferative responses from groups of Wistar Kyoto (WKY) rats (n = 6) immunized with alpha 3 chain of type IV collagen [α3(IV)NC1] or the five 15-mer overlapping peptides, cultured with: (a) α3(IV)NC1 and (b) pCol(24–38). Results shown represent the mean ± standard deviation of each group at week 6 after immunization. *P < 0·01, pCol(24–38) immunized versus Freund's complete adjuvant (FCA) alone. Each experiment was performed twice.
Assessment of disease after immunization with synthetic peptides
Deposits of IgG on the GBM
All positive control animals immmunized with α3(IV)NC1 developed strong linear deposits of IgG on the GBM by week 6 after immunization. All animals immunized with pCol(24–38) also developed strong linear deposits of IgG on the GBM by week 6 after immunization, while those immunized with the other peptides showed either no deposition of IgG on the GBM or weak, intermittent deposits of IgG. Negative control animals given FCA alone showed no deposition of IgG. Results are shown in Fig. 3a, and illustrated in Fig. 4.
Fig. 3.
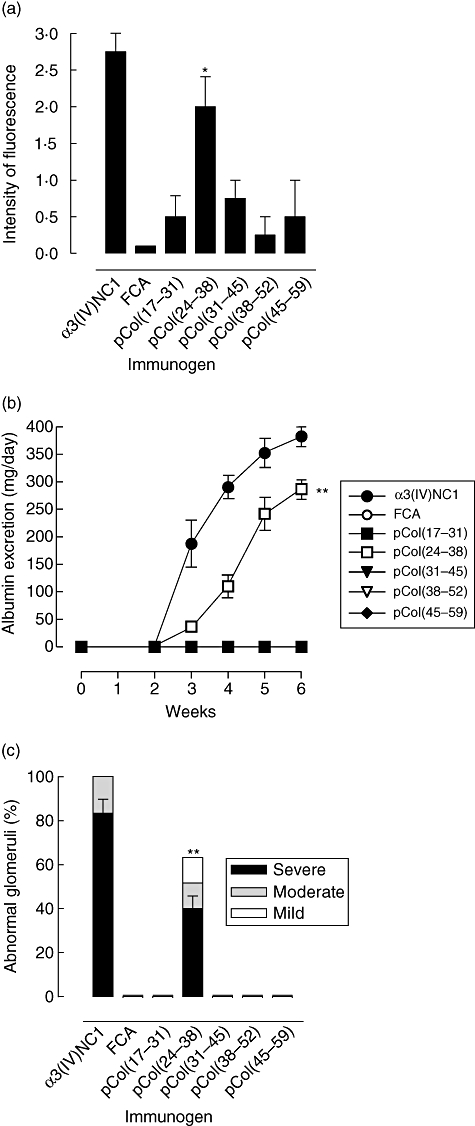
Induction of disease in groups of Wistar Kyoto (WKY) rats (n = 6) immunized with alpha 3 chain of type IV collagen [α3(IV)NC1] or the five 15-mer overlapping peptides, showing: (a) deposits of IgG on the glomerular basement membrane (GBM) at week 6 after immunization; (b) urinary albumin excretion at different time-points after immunization: α3(IV)NC1 (filled circles), Freund's complete adjuvant (FCA) (open circles), pCol(17–31) (filled squares), pCol(24–38) (open squares), pCol(31–45) (filled triangles), pCol(38–52) (open triangles), pCol(45–59) (filled diamonds) [note that albumin excretion was observed only in groups of animals immunized with α3(IV)NC1 or pCol(24–38); all other groups of animals remained at background levels throughout the experiment]; and (c) severity of glomerular abnormalities at week 6 after immunization. *P < 0·01; **P < 0·001, pCol(24–38) immunized versus FCA alone.
Fig. 4.
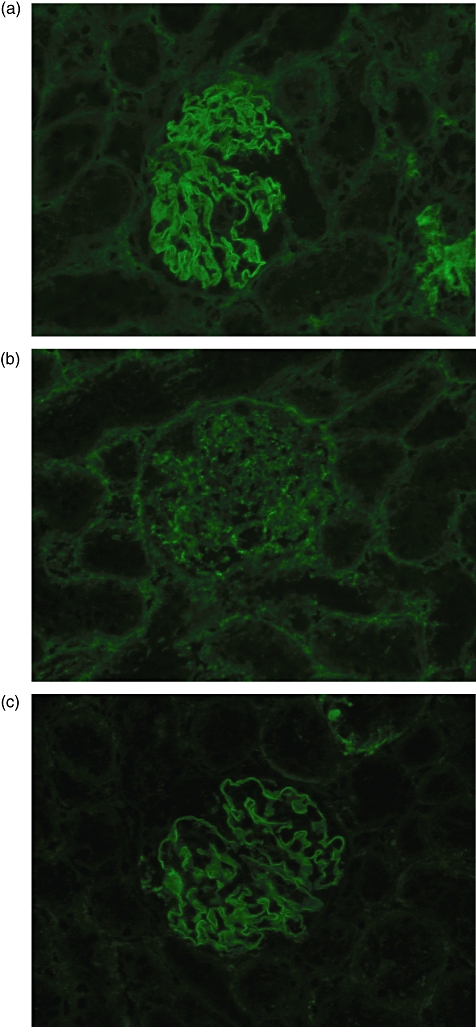
Direct immunofluorescence of kidney tissue at week 6 showing a representative example of: (a) strong linear deposits of immunoglobulin G (IgG) on the glomerular basement membrane (GBM) in a positive control Wistar Kyoto (WKY) rat immunized with alpha 3 chain of type IV collagen [α3(IV)NC1]; (b) no deposits of IgG on the GBM in a negative control WKY rat given Freund's complete adjuvant (FCA) alone; and (c) strong linear deposits of IgG on the GBM in a WKY rat immunized with peptide pCol(24–38). Magnification ×300.
Albuminuria
All positive control animals immmunized with α3(IV)NC1 showed detectable levels of albuminuria by week 3 after immunization, which increased to high levels by week 6. All animals immunized with pCol(24–38) also showed an increase in albuminuria by week 6 after immunization, while those immunized with the other peptides showed no increase in albuminuria. Negative control animals given FCA alone showed no increase in albuminuria. Results are shown in Fig. 3b.
Glomerular abnormalities
All positive control animals immmunized with α3(IV)NC1 developed focal proliferative glomerulonephritis affecting up to 100% of glomeruli, with segmental necrosis/crescent formation affecting up to 85% of glomeruli, by week 6 after immunization. All animals immunized with pCol(24–38) developed focal proliferative glomerulonephritis affecting up to 65% of glomeruli, with segmental necrosis/crescent formation affecting up to 45% of glomeruli. By contrast, those immunized with the other peptides showed normal glomerular architecture, similar to that seen in normal controls. Results are shown in Fig. 3c, and illustrated in Fig. 5.
Fig. 5.
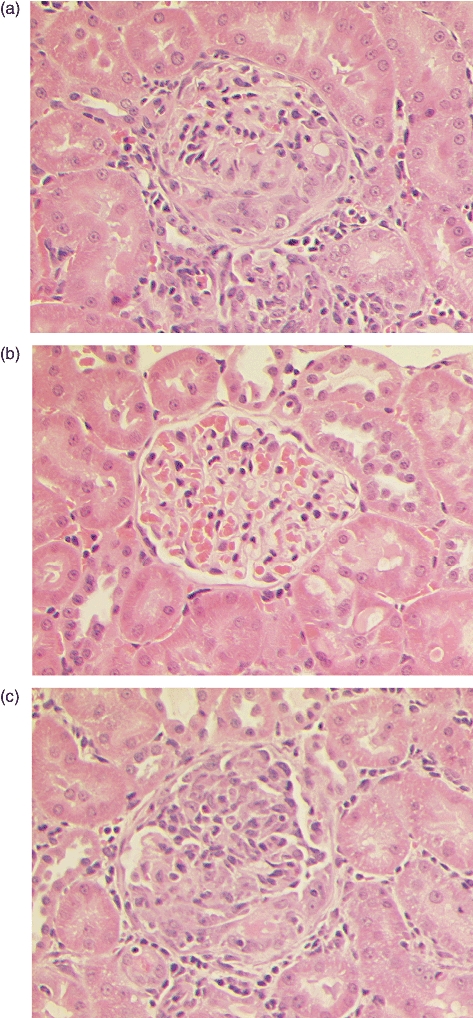
Light microscopy of kidney tissue at week 6 showing a representative example of: (a) marked segmental necrosis of the glomerular tuft with crescent formation in a positive control Wistar Kyoto (WKY) rat immunized with alpha 3 chain of type IV collagen [α3(IV)NC1]; (b) normal glomerular architecture in a negative control WKY rat given Freund's complete adjuvant (FCA) alone; and (c) severe crescentic glomerulonephritis in a WKY rat immunized with peptide pCol(24–38). Magnification ×300.
Inhibition assay
Inhibition studies showed that binding of antibodies from rats immunized with pCol(24–38) to recombinant α3(IV)NC1 could be reduced by co-incubation with pCol(24–38) from a concentration of 10 µg/ml, and inhibited totally at a concentration of 300 µg/ml. Co-inhibition with control peptide pCol(38–52) showed no inhibition of binding at any concentration. Results are shown in Fig. 6.
Fig. 6.
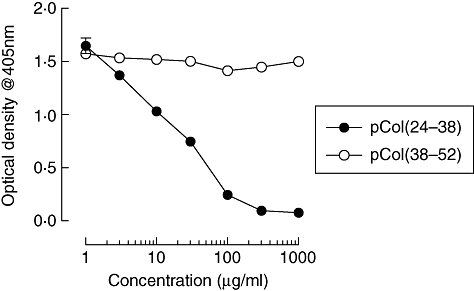
Inhibition studies showing binding of serum antibodies from animals immunized with pCol(24–38) to recombinant alpha 3 chain of type IV collagen [α3(IV)NC1], after co-incubation with different concentrations of pCol(24–38) (filled circles), or pCol(38–52) (open circles). Results shown represent the mean ± standard deviation at week 6 after immunization.
Discussion
In patients with Goodpasture's disease, the major B and T cell epitopes have been located to the N-terminus of α3(IV)NC1 [5–8,36]. In order to investigate whether B and T cell responses in EAG are directed towards immunodominant peptides within the same region of rat α3(IV)NC1, we immunized WKY rats with five 15-mer overlapping synthetic peptides spanning the region of the major human B cell epitope within the N-terminus of α3(IV)NC1. Positive control animals immunized with recombinant rat α3(IV)NC1 produced an antibody response directed towards α3(IV)NC1 and the second 15-mer peptide, pCol(24–38). Splenic T cells from these animals proliferated in response to α3(IV)NC1 and pCol(24–38). Interestingly, no significant antibody or T cell responses were observed to the other peptides examined. Moreover, animals immunized with pCol(24–38) developed circulating and deposited antibodies to the GBM, albuminuria and focal necrotizing glomerulonephritis with crescent formation by week 6 after immunization. Circulating antibodies from these animals recognized α3(IV)NC1 and pCol(24–38), while their T cells proliferated in response to α3(IV)NC1 and pCol(24–38). Animals immunized with the other peptides developed no significant immune response to α3(IV)NC1 and no disease. These results demonstrate for the first time that a 15-mer peptide, pCol(24–38), from the N-terminus of α3(IV)NC1 is recognized by both B and T cells from rats immunized with α3(IV)NC1, and that the same peptide is capable of inducing crescentic glomerulonephritis. It is intriguing that a single injection of synthetic peptide should be sufficient to induce such marked autoimmune disease. This finding emphasizes the importance of the pCol(24–38) sequence in the autoantigen, α3(IV)NC1. The WKY rat is known to be particularly susceptible to the induction of EAG when immunized with the autoantigen in Freund's complete adjuvant [18].
In parallel studies, a similar nephritogenic T cell epitope in EAG has been mapped by other groups. Luo et al. investigated the effect of immunizing WKY rats with overlapping peptides spanning the α3(IV)NC1 molecule, and showed that a 24-mer synthetic peptide from the N terminus of α3(IV)NC1, pCol(28–51), was capable of inducing a mild, inconsistent glomerulonephritis [29]. In this study most peptides elicited antibody responses exclusively to themselves, but not to native GBM, and T cells from GBM immunized rats proliferated in vitro to six of the synthetic peptides. In other studies, Wu et al. showed that a 13-mer peptide, pCol(28–40), containing the T cell epitope from α3(IV)NC1 was capable of inducing crescentic glomerulonephritis, without deposition of IgG on the GBM [30]. In subsequent studies the same group reported that animals immunized with pCol(28–40) did develop deposited antibodies by day 20 after immunization, suggesting that T cell-mediated glomerular injury may trigger de novo internal immunization with autoantigens released from damaged GBM, which could lead to activation of a group of GBM-specific B cells in the renal draining lymph node [31]. In our study, we looked for deposited antibodies only at a later time-point (day 42), therefore this explanation could be equally applicable.
In further characterization of the T cell epitope by Robertson et al., it was shown that only three residues in the T cell epitope were critical for disease induction [32]. Position 31 (threonine) was an anchor residue to the class II molecule, and positions 33 (asparagine) and 34 (proline) contributed to the specificity of the T cell epitope. Substitutions at those positions completely abrogated nephritogenicity of the peptide. In separate studies, Bolton et al. also demonstrated that asparagine at position 33 was critical for EAG induction. Interestingly, the aa sequence for human and rat α3(IV)NC1 is different at this position. Peptides that contained the human sequence for α3(IV)NC1, with isoleucine rather than asparagine at position 33, did not induce crescentic nephritis [33].
In the present study, both the second, pCol(24–38), and third, pCol(31–45), peptides contain the reported critical aa, threonine, asparagine and proline. However, only animals immunized with pCol(24–38) produced a significant immune response to α3(IV)NC1 and developed severe crescentic glomerulonephritis. Animals immunized with pCol(31–45) showed no evidence of disease and no significant immune response to α3(IV)NC1. Our results suggest that the position of these critical aa within the peptide, and the length of the peptide, may be important in the induction of EAG.
It has been reported that peptides containing the T cell epitope, pCol(28–40), not only induce severe glomerulonephritis, but also trigger an anti-GBM antibody response through B cell epitope spreading, suggesting that a diverse autoantibody response to GBM antigens can be induced by a single nephritogenic T cell epitope [34,35]. In our study, there is evidence that peptide pCol(24–38) itself contains a major B cell epitope, in that antibodies from animals immunized with α3(IV)NC1 bound to pCol(24–38), but showed no significant binding to the other peptides studied. Furthermore, circulating antibodies from animals immunized with pCol(24–38) bound to recombinant rat α3(IV)NC1, while animals immunized with the other peptides developed no humoral immune response to α3(IV)NC1. Animals immunized with pCol(24–38) also showed deposited IgG in a linear pattern on the GBM, confirming that the peptide induces antibodies specific for native α3(IV)NC1 in vivo. In addition, inhibition studies showed that binding of antibodies from rats immunized with pCol(24–38) to recombinant α3(IV)NC1 could be inhibited by co-incubation with pCol(24–38) but not pCol(38–52), demonstrating that sera from pCol(24–38) immunized animals do not contain anti-GBM antibodies that arose secondarily as a consequence of epitope spreading.
Further work to characterize the specific B and T cell responses to pCol(24–38) and to identify other potential B and T cell epitopes within α3(IV)NC1 are under way in our laboratory. Preliminary studies demonstrate that nasal administration of pCol(24–38) is effective in both the prevention and treatment of EAG (Reynolds et al., unpublished observation). This work should ultimately be of value in designing new therapeutic strategies in patients with glomerulonephritis.
Acknowledgments
This work was supported by grants from the Sir Jules Thorn Charitable Trust and the Hammersmith Hospital Charity Trustees. This work was presented in part at the 36th Annual Meeting of the American Society of Nephrology, San Diego, 17 November 2003.
References
- 1.Goodpasture EW. The significance of certain pulmonary lesions to the etiology of influenza. Am J Med Sci. 1919;158:863–70. [Google Scholar]
- 2.Wilson CB, Dixon FJ. Anti-glomerular basement membrane antibody-induced glomerulonephritis. Kidney Int. 1973;3:74–89. doi: 10.1038/ki.1973.14. [DOI] [PubMed] [Google Scholar]
- 3.Saus J, Wieslander J, Langeveld JPM, et al. Identification of the Goodpasture antigen as the α3 chain of collagen IV. J Biol Chem. 1988;263:13374–80. [PubMed] [Google Scholar]
- 4.Turner N, Mason PJ, Brown R, et al. Molecular cloning of the human Goodpasture antigen demonstrates it to be the α3 chain of type IV collagen. J Clin Invest. 1992;89:592–601. doi: 10.1172/JCI115625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ryan JJ, Mason PJ, Pusey CD, et al. Recombinant α-chains of type IV collagen demonstrate that the amino terminal of the Goodpasture antigen is critical for antibody binding. Clin Exp Immunol. 1998;113:17–27. doi: 10.1046/j.1365-2249.1998.00623.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hellmark T, Segelmark M, Ungar C, et al. Identification of the clinically relevant immunodominant region of collagen IV in Goodpasture's disease. Kidney Int. 1999;55:936–44. doi: 10.1046/j.1523-1755.1999.055003936.x. [DOI] [PubMed] [Google Scholar]
- 7.Netzer KO, Leinonen A, Boutaud A, et al. The Goodpasture autoantigen: mapping the major conformational epitope(s) of alpha3(IV) collagen to residues 17–31 and 127–141 of the NC1 domain. J Biol Chem. 1999;274:11267–274. doi: 10.1074/jbc.274.16.11267. [DOI] [PubMed] [Google Scholar]
- 8.David M, Borza DB, Leinonen A, et al. Hydrophobic amino acid residues are critical for the immunodominant epitope of the Goodpasture antigen. A molecular basis for the cryptic nature of the epitope. J Biol Chem. 2001;276:6370–77. doi: 10.1074/jbc.M008956200. [DOI] [PubMed] [Google Scholar]
- 9.Sado Y, Okigaki T, Takamiya H, et al. Experimental autoimmune glomerulonephritis with pulmonary haemorrhage in rats. The dose–effect relationship of the nephritogenic antigen from bovine glomerular basement membrane. J Clin Lab Immunol. 1994;15:199–204. [PubMed] [Google Scholar]
- 10.Reynolds J, Mavromatidis K, Cashman SJ, et al. Experimental autoimmune glomerulonephritis (EAG) induced by homologous and heterologous glomerular basement membrane in two sub-strains of Wistar Kyoto rat. Nephrol Dial Transpl. 1998;13:44–52. doi: 10.1093/ndt/13.1.44. [DOI] [PubMed] [Google Scholar]
- 11.Kalluri R, Danoff TM, Okada H, et al. Susceptibility to anti-glomerular basement membrane disease and Goodpasture's syndrome is linked to MHC class II genes and the emergence of T cell-mediated immunity in mice. J Clin Invest. 1997;100:2263–75. doi: 10.1172/JCI119764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sado Y, Boutaud AA, Kagawa M, et al. Induction of anti-GBM nephritis in rats by recombinant α3(IV) NC1 and α4(IV) NC1 of type IV collagen. Kidney Int. 1998;53:664–71. doi: 10.1046/j.1523-1755.1998.00795.x. [DOI] [PubMed] [Google Scholar]
- 13.Ryan JJ, Reynolds J, Norgan VA, et al. Expression and characterisation of recombinant rat α3(IV)NC1 and its use in the induction of experimental autoimmune glomerulonephritis. Nephrol Dial Transpl. 2001;16:253–61. doi: 10.1093/ndt/16.2.253. [DOI] [PubMed] [Google Scholar]
- 14.Hopfer H, Maron R, Butzmann U, et al. The importance of cell-mediated immunity in the course and severity of autoimmune anti-glomerular basement membrane disease in mice. FASEB J. 2003;17:860–68. doi: 10.1096/fj.02-0746com. [DOI] [PubMed] [Google Scholar]
- 15.Reynolds J, Moss J, Duda MA, et al. The evolution of crescentic nephritis and alveolar haemorrhage following induction of autoimmunity to glomerular basement membrane in an experimental model of Goodpasture's disease. J Pathol. 2003;200:118–29. doi: 10.1002/path.1336. [DOI] [PubMed] [Google Scholar]
- 16.Sado Y, Naito I, Okigaki T. Transfer of anti-glomerular basement membrane antibody-induced glomerulonephritis in inbred rats with isologous antibodies from urine of nephritic rats. J Pathol. 1989;158:325–32. doi: 10.1002/path.1711580410. [DOI] [PubMed] [Google Scholar]
- 17.Kohda T, Okada S, Hayashi A, et al. High nephritogenicity of monoclonal antibodies belonging to IgG2a and IgG2b subclasses in rat anti-GBM nephritis. Kidney Int. 2004;66:177–86. doi: 10.1111/j.1523-1755.2004.00719.x. [DOI] [PubMed] [Google Scholar]
- 18.Reynolds J, Albouainain A, Duda MA, et al. Strain susceptibility to active induction and passive transfer of experimental autoimmune glomerulonephritis in the rat. Nephrol Dial Transpl. 2006;21:3398–408. doi: 10.1093/ndt/gfl523. [DOI] [PubMed] [Google Scholar]
- 19.Wu J, Hicks J, Ou C, et al. Glomerulonephritis induced by recombinant collagen IV alpha 3 chain non-collagen domain 1 is not associated with glomerular basement membrane antibody: a potential T cell-mediated mechanism. J Immunol. 2001;167:2388–95. doi: 10.4049/jimmunol.167.4.2388. [DOI] [PubMed] [Google Scholar]
- 20.Wu J, Hicks J, Borillo J, et al. CD4+ T cells specific to a glomerular basement membrane antigen mediate glomerulonephritis. J Clin Invest. 2002;109:517–25. doi: 10.1172/JCI13876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Walters G, Habib A-M, Reynolds J, et al. Glomerular T cells are of restricted clonality and express multiple CDR3 motifs across different Vβ T cell receptor families in experimental autoimmune glomerulonephritis. Nephron Exp Nephrol. 2004;98:71–81. doi: 10.1159/000080682. [DOI] [PubMed] [Google Scholar]
- 22.Reynolds J, Pusey CD. In vivo treatment with a monoclonal antibody to T helper cells in experimental autoimmune glomerulonephritis. Clin Exp Immunol. 1994;95:122–27. doi: 10.1111/j.1365-2249.1994.tb06025.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reynolds J, Norgan VA, Bhambra U, et al. Anti-CD8 monoclonal antibody therapy is effective in the prevention and treatment of experimental autoimmune glomerulonephritis. J Am Soc Nephrol. 2002;13:359–69. doi: 10.1681/ASN.V132359. [DOI] [PubMed] [Google Scholar]
- 24.Reynolds J, Tam FWK, Chandraker A, et al. CD28-B7 blockade prevents the development of experimental autoimmune glomerulonephritis. J Clin Invest. 2000;105:643–51. doi: 10.1172/JCI6710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reynolds J, Khan SB, Allen AR, et al. Blockade of the CD154-CD40 T cell costimulatory pathway prevents the development of experimental autoimmune glomerulonephritis. Kidney Int. 2004;66:1444–52. doi: 10.1111/j.1523-1755.2004.00907.x. [DOI] [PubMed] [Google Scholar]
- 26.Reynolds J, Pusey CD. Oral administration of glomerular basement membrane prevents the development of experimental autoimmune glomerulonephritis. J Am Soc Nephrol. 2001;12:61–70. doi: 10.1681/ASN.V12161. [DOI] [PubMed] [Google Scholar]
- 27.Reynolds J, Prodromidi EI, Juggapah JK, et al. Nasal administration of recombinant rat α3(IV)NC1 prevents the development of experimental autoimmune glomerulonephritis. J Am Soc Nephrol. 2005;16:1350–55. doi: 10.1681/ASN.2004121026. [DOI] [PubMed] [Google Scholar]
- 28.Reynolds J, Haxby J, Juggapah JK, et al. Immunodominant B and T cell epitope in experimental autoimmune glomerulonephritis. J Am Soc Nephrol. 2003;14:633a. doi: 10.1111/j.1365-2249.2008.03833.x. Abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Luo AM, Fox JW, Chen L, et al. Synthetic peptides of Goodpasture's antigen in anti-glomerular basement membrane nephritis in rats. J Lab Clin Med. 2002;139:303–10. doi: 10.1067/mlc.2002.123623. [DOI] [PubMed] [Google Scholar]
- 30.Wu J, Borillo J, Glass WF, et al. T-cell epitope of α3 chain of type IV collagen induces severe glomerulonephritis. Kidney Int. 2003;64:1292–301. doi: 10.1046/j.1523-1755.2003.00227.x. [DOI] [PubMed] [Google Scholar]
- 31.Robertson J, Wu J, Ardens J, et al. Activation of glomerular basement membrane-specific B cells in the renal draining lymph node after T cell-mediated glomerular injury. J Am Soc Nephrol. 2005;16:3256–63. doi: 10.1681/ASN.2005040421. [DOI] [PubMed] [Google Scholar]
- 32.Robertson J, Wu J, Arends J, et al. Characterization of the T-cell epitope that causes anti-GBM glomerulonephritis. Kidney Int. 2005;68:1061–70. doi: 10.1111/j.1523-1755.2005.00498.x. [DOI] [PubMed] [Google Scholar]
- 33.Bolton WK, Chen L, Hellmark T, et al. Epitope spreading and autoimmune glomerulonephritis in rats induced by a T cell epitope of Goodpasture's antigen. J Am Soc Nephrol. 2005;16:2657–66. doi: 10.1681/ASN.2004100823. [DOI] [PubMed] [Google Scholar]
- 34.Chen L, Hellmark T, Pedchenko V, et al. A nephritogenic peptide induces intermolecular epitope spreading on collagen IV in experimental autoimmune glomerulonephritis. J Am Soc Nephrol. 2006;17:3076–81. doi: 10.1681/ASN.2006070688. [DOI] [PubMed] [Google Scholar]
- 35.Arends J, Wu J, Borillo J, et al. T cell epitope mimicry in anti-glomerular basement membrane disease. J Immunol. 2006;176:1252–58. doi: 10.4049/jimmunol.176.2.1252. [DOI] [PubMed] [Google Scholar]
- 36.Cairns LS, Phelps RG, Bowie L, et al. The fine specificity and cytokine profile of T-helper cells responsive to the alpha3 chain of type IV collagen in Goodpasture's disease. J Am Soc Nephrol. 2003;14:2801–12. doi: 10.1097/01.asn.0000091588.80007.0e. [DOI] [PubMed] [Google Scholar]