

Abstract
Haemolytic uraemic syndrome (HUS) is characterized by microangiopathic haemolytic anaemia, thrombocytopenia and renal failure because of thrombotic microangiopathy (TMA). It may be caused by infection with Shiga toxin-producing enteropathic bacteria (Stx-associated HUS) or with genetic defects in complement alternative pathway (CAP) regulation (atypical HUS). We hypothesized that defective complement regulation could increase host susceptibility to Stx-associated HUS. Hence, we studied the response of mice with heterozygous deficiency of the major CAP regulator, factor H, to purified Stx-2. Stx-2 was administered together with lipopolysaccharide to wild-type and Cfh+/− C57BL/6 animals. Forty-eight hours after administration of the first Stx-2 injection all animals developed significant uraemia. Renal histology demonstrated significant tubular apoptosis in the cortical and medullary areas which did not differ between wild-type or Cfh+/− Stx-2-treated mice. Uraemia and renal tubular apoptosis did not develop in wild-type or Cfh+/− animals treated with lipopolysaccharide alone. No light microscopic evidence of TMA or abnormal glomerular C3 staining was demonstrable in the Stx-2 treated animals. In summary, Stx-2 administration did not result in TMA in either Cfh+/− or wild-type C57BL/6 mice. Furthermore, haploinsufficiency of factor H did not alter the development of Stx-2-induced renal tubular injury.
Keywords: apoptosis, complement, thrombosis, toxin
Introduction
Thrombotic microangiopathy (TMA) is a vascular occlusive disease characterized by endothelial injury and formation of microthrombi composed of agglutinated platelets in the microvasculature [1]. Renal TMA is the renal lesion seen in haemolytic uraemic syndrome (HUS), a disorder characterized by thrombocytopenia, microangiopathic haemolytic anaemia (MAHA) and acute renal failure [2]. The most common form of HUS is due to infection with Shiga toxin (Stx)-producing bacteria such as Escherichia coli O157:H7 and some Shigella strains (Stx-associated HUS) [3]. Following infection with Stx-producing bacteria only a small percentage of individuals (lower than 10% in sporadic cases and lower that 30% in outbreaks) develop HUS, suggesting that other risk factors may be required for the infection to progress to HUS [4,5]. The cytotoxic effect of Stx is mediated by binding of the toxin to the globotriaosyl ceramide receptor (Gb3; CD77) [6]. Once bound to the Gb3 receptor, the toxin inhibits protein synthesis causing cell death [6,7]. The kidneys are the preferential extra-intestinal target for Stx because of the abundant expression of Gb3 receptors on human renal epithelial, endothelial and glomerular mesangial cells [7,8]. The action of Stx on renal endothelium can result in cell death and consequently the exposure of the basement membrane, factors that favour platelet activation in the subendothelium and platelet aggregation in the glomerular microcirculation [9,10]. Two major forms of Stx are produced by E. coli, Stx-1 and Stx-2. Stx-1 (Shiga toxin 1, also known as Shiga-like toxin 1 or SLT-1) differs from the Shiga toxin made by S. dysenteriae serotype I by only a single amino acid within the A subunit of the toxin. Stx-2 (also known as Shiga-like toxin 2 or SLT-2) is approximately 60% homologous with Stx-1.
Uncommonly, cases of HUS have no association with diarrhoea [termed diarrhoea-negative HUS or atypical HUS (aHUS)][11]. aHUS includes both sporadic and familial forms and is associated strongly with dysregulation of the alternative pathway (AP) of the complement system [12]. Complement dysregulation in aHUS is due most commonly to mutations in the genes encoding complement regulatory proteins such as factor H, factor I and membrane co-factor protein (MCP, CD46) and the complement AP activation protein, factor B [12]. Notably, these mutations are associated with incomplete penetrance, and it has now become clear that aHUS is a complex genetic trait requiring multiple complement genetic susceptibility factors [13] together with appropriate environmental triggers [14]. Supporting the latter is the observation that many patients with aHUS experienced episodes of infection prior to the onset of the syndrome [14].
Does the presence of genetic defects in complement regulation alter the phenotype of Stx-associated HUS? Recently, a mutation in the gene encoding MCP was reported in a child who developed fatal Stx-associated HUS [15]. While definitive studies assessing whether individuals who develop Stx-associated HUS are more likely to harbour complement gene mutations have not been performed, we hypothesized that the presence of a predisposing risk factor to HUS, namely a complement gene mutation, may contribute to the development of HUS following infection with Stx-producing bacteria.
Factor H is an abundant complement regulatory protein that inhibits AP activation [16] and heterozygous mutations in this protein are associated strongly with aHUS [12]. Mice with heterozygous deficiency of factor H (Cfh+/−) show mild impairment of plasma C3 regulation but do not develop spontaneous renal disease, including HUS [17]. Renal damage in mice infected with Stx-producing E. coli strains has been shown to be mediated by Stx-2 [18]. Furthermore, the toxic effects of Stx are enhanced by concomitant administration of lipopolysaccharide (LPS) [19]. Hence, purified Stx-2, sometimes in combination with LPS, has been used by several groups in an attempt to induce HUS in mice [20–27]. Therefore, to test our hypothesis, we chose to treat Cfh+/− mice with purified Stx-2 in the presence of LPS. Our protocol resulted in rapid clinical deterioration in both Cfh+/− and wild-type mice with the development of severe uraemia and renal tubular injury to an equivalent extent in both groups. Critically, in neither group could we detect MAHA or renal TMA. We conclude that administration of Stx-2 using our protocol resulted in renal tubular but not glomerular injury and that this lesion was not influenced by heterozygous deficiency of factor H.
Methods
Mice deficient in factor H
Heterozygous C57BL/6 factor H-deficient (Cfh+/−) mice were generated by crossing factor H-deficient mice [17], back-crossed onto the C57BL/6 genetic background for 10 generations, with C57BL/6 wild-type animals. Mice were age- and sex-matched and all experimental procedures were performed in accordance with institutional guidelines.
General reagents and antibodies
Purified Stx-2 derived from E. coli O157 and LPS derived from E. coli O111:B4 were obtained from Sigma-Aldrich (Poole, Dorset, UK).
Administration of purified Stx-2 in vivo
Wild-type and Cfh+/− C57BL/6 mice were injected intraperitoneally at day 0 with 50 ng (low dose, n = 6) or 200 ng (high dose, n = 6) of purified Stx-2. At day 1, mice received the same dose of Stx-2 and, in addition, an injection of 5 µg of LPS. A separate group of wild-type (n = 4) and Cfh+/− (n = 4) mice, that did not receive any Stx-2, were injected intraperitoneally with 5 µg of LPS on day 1 alone. All mice were killed at day 2.
Haematological parameters and assessment of renal function
The presence of red blood cell fragments was determined by light microscopic examination of peripheral blood smears prepared manually using blood collected in ethylenediamine tetraacetic acid. Haematocrit was determined by centrifuging peripheral blood that had been collected into heparinized capillary tubes (Merck, Hull, UK) and subsequently expressing the height of the centrifuged red cell mass as a percentage of the height of the total blood volume. Serum urea was measured by enzymatic bioanalysis using a commercial urea ammonia kit (R-Biopharm, Glasgow, UK) according to the manufacturer's instructions.
Histological studies
For light microscopy, kidneys were fixed in Bouin's solution embedded in paraffin and sections stained with periodic acid-Schiff reagent. Splenic, liver and lung tissue was fixed in formalin, embedded in paraffin and sections stained with haematoxylin and eosin. Glomerular histology was graded as follows: grade 0, normal; grade I, segmental hypercellularity in 10–25% of the glomeruli; grade II, hypercellularity involving > 50% of the glomerular tuft in 25–50% of glomeruli; grade III, hypercellularity involving > 50% of the glomerular tuft in 50–75% of glomeruli; and grade IV, glomerular hypercellularity in > 75% or crescents in > 25% of glomeruli. Histological analyses were performed in a blinded fashion, and 50 glomeruli per section were analysed for glomerular histology scoring. For assessment of tubular apoptosis several high-power field views of the tubulointerstitium were visualized and scored in a blinded fashion using the following cortical tubular apoptosis severity score: none = 0, mild = 1, moderate = 2, marked = 3.
Detection of renal cell apoptosis
Immunohistochemical detection of renal apoptosis was detected using an In Situ Cell Death Detection Kit (Roche Diagnostics, Burgess Hill, UK), according to the manufacturer's instructions. This utilizes the terminal deoxynucleotidyl transferase-mediated aUTP-biotin nick end labelling (TUNEL) method for detection of individual apoptotic cells.
Glomerular fibrinogen staining
Immunofluorescence analysis was performed on tissue cryosections. Sections were blocked for 30 min using 10% normal rabbit serum and then incubated for 1 h with a cross-reactive fluorescein isothiocyanate (FITC)-conjugated rabbit anti-human fibrinogen antibody (Dako, Cambridge, UK).
Renal complement C3 staining
Immunofluorescence analysis was performed on tissue cryosections. Sections were blocked for 30 min using 20% normal goat serum and then incubated for 1 h with FITC-conjugated goat antibody against mouse C3 (MP Biomedicals, Cambridge, UK). Quantitative immunofluorescence studies were performed as described previously [28] and results expressed as arbitrary fluorescence units.
Statistical analysis
Non-parametric data were given as the median with range of values in parentheses. The Mann–Whitney U-test was used for comparison of two groups while for analysis of three or more groups Bonferroni's multiple comparison test was used. Data were analysed using GraphPad Prism version 3·0 for Windows (GraphPad Software, La Jolla, CA, USA).
Results
Administration of purified Stx-2 in combination with LPS resulted in rapid clinical deterioration
Twenty-four hours following the first injection of Stx at either the low dose (50 ng) or high dose (200 ng) both wild-type and Cfh+/− animals showed no overt signs of illness. At this stage the animals received a second Stx injection together with an intraperitoneal injection of 5 µg LPS (E. coli O111:B4). Twenty-four hours after the second Stx dosing in combination with LPS the mice became moribund and were killed humanely and organs and serum collected for analysis. In contrast, animals that had received LPS alone remained well.
Stx-2 in combination with LPS induced a rapid rise in serum urea levels
We evaluated urea levels using plasma samples collected 48 h after the onset of the experiment at a time-point when mice that had received both Stx-2 and LPS were overtly unwell. A significant increase in plasma urea levels was evident in both the low-dose and high-dose Stx-2-treated mice compared with pretreatment values (Table 1). The degree of uraemia did not differ between the low-dose and high-dose Stx-2-treated groups. Median urea values did not increase significantly in either the wild-type or Cfh+/− animals treated with LPS alone. Furthermore, the degree of uraemia in all the experimental groups treated with Stx-2 and LPS was increased significantly when compared with the median urea values seen in either wild-type or Cfh+/− animals treated with LPS alone. Hence, Stx-2 treatment resulted specifically in marked uraemia to an equivalent extent in wild-type or Cfh+/− animals.
Table 1.
Plasma urea, haematocrit and blood film analysis 48 h after treatment with Shiga toxin (Stx-2) with or without lipopolysaccharide (LPS).
| LPS (5 µg) |
Stx (50 ng) LPS (5 µg) |
Stx (200 ng) LPS (5 µg) |
||||
|---|---|---|---|---|---|---|
| C57BL/6 (n = 4) | Cfh+/− (n = 4) | C57BL/6 (n = 6) | Cfh+/− (n = 6) | C57BL/6 (n = 6) | Cfh+/− (n = 6) | |
| Urea (mmol/l): pre | 9·6 (5·1–13·1) | 8·9 (6·8–10·3) | 12·5 (7·5–24·4) | 4·6 (2·2–5·7) | 9·8 (7–16·2) | 4·9 (4·1–7·5) |
| Urea (mmol/l): post | 13·3 (5–41·4) | 13·1 (10·2–39·7) | 40·4*† (38·6–41·3) | 41·3*† (36·6–42·1) | 41*† (39·6–42·4) | 41·6*† (40–41·9) |
| Haematocrit (%): pre | 35·5 (26–39) | 32·5 (29–35) | 34§ (28–40) | 35 (32–41) | 35 (32–39) | 34·5 (19–36) |
| Haematocrit (%): post | 29‡ (19–30) | 26‡ (16–29) | 27§ (20–30) | 17·5*§ (15–23) | 25*5 (18–29) | 18*§ (15–23) |
| Fragments | Negative | Negative | Negative | Negative | Negative | Negative |
Figures represent median with range of values in parentheses.
P < 0·05 versus respective pretreatment value, Mann–Whitney U-test;
P < 0·05 versus either wild-type or Cfh+/− LPS alone post-values, Bonferroni multiple comparison test.
Haematocrit performed in ‡ three of four,
four of six and
three of six animals at this time-point.
Stx-2 in combination with LPS did not induce red blood cell fragmentation
The presence of red blood cell fragments on the peripheral blood film, the hallmark of MAHA, is seen characteristically in patients with TMA. Hence, we examined peripheral blood smears in the Stx-2- and LPS-treated animals. Irrespective of genotype or Stx-2 dosing regimen, red cell fragmentation was not observed in any of the mice (Table 1). Administration of LPS alone also did not induce red cell fragmentation. However, there was a significant reduction in haematocrit readings in the peripheral blood of both wild-type and Cfh+/− mice injected with 200 ng Stx-2 and LPS (Table 1). In the mice treated with 50 ng Stx-2 and LPS there was a fall in the median haematocrit values, but this reached significance only in the Cfh+/− group. Importantly, the haematocrit value could not be assessed accurately in all Stx-2-treated mice because these animals were unwell at the time of killing. Similarly, while median haematocrit values fell in wild-type and Cfh+/− mice treated with LPS alone, in neither group did this reach statistical significance. However, the post-treatment haematocrit values among the Stx-2-treated mice did not differ significantly from the post-treatment haematocrit values of either wild-type or Cfh+/− mice treated with LPS alone. Hence, the reduction in haematocrit was not specific to Stx-treated animals.
Stx-2 in combination with LPS did not alter glomerular morphology
Light microscopic analysis of the kidneys showed no evidence of glomerular TMA in any of the mice injected with Stx-2 plus LPS, irrespective of the genetic background (Fig. 1). Immunostaining for glomerular fibrinogen was absent in all the experimental animals (Fig. 2). The only glomerular abnormality was the presence of small numbers of neutrophils in glomerular capillary lumens. In all experimental conditions median glomerular neutrophil numbers were < 1 per glomeruli and a similar influx of neutrophils was observed in both wild-type and Cfh+/− mice. Consistent with the lack of significant glomerular changes on light microscopy, glomerular staining for complement C3 did not show any abnormal deposition of complement C3 (data not shown).
Fig. 1.
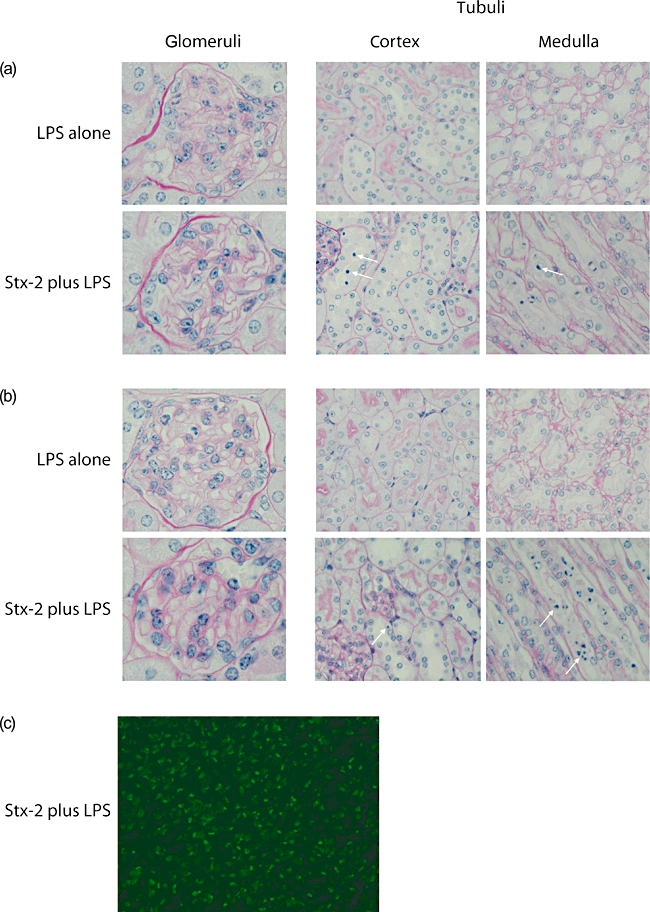
Renal histology in mice treated with Shiga toxin (Stx-2) and lipopolysaccharide (LPS). Light microscopic appearances of periodic acid-Schiff (PAS)-stained renal sections from (a) wild-type and (b) Cfh+/− animals treated with either LPS alone or 200 ng Stx-2 in combination with LPS. Representative glomerular, cortical and medullary tubular changes are shown. Arrows indicate examples of apoptotic tubular cells. Equivalent histological changes were seen in the tubules of mice treated with 50 ng Stx-2 in combination with LPS (data not shown). Original magnification was 40× for tubular and 100× for glomerular images. (c) Representative image of medullary terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end labelling (TUNEL) staining in wild-type mouse treated with Stx-2 and LPS. Equivalent staining was seen in Cfh+/− animals treated with Stx-2 and LPS.
Fig. 2.
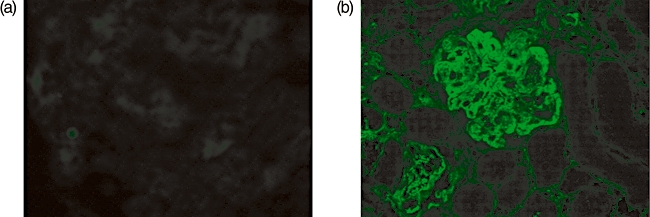
Glomerular fibrinogen staining in mice treated with Shiga toxin (Stx). (a) Glomerular staining in a wild-type mouse treated with 200 ng Stx-2 in combination with lipopolysaccharide (LPS). (b) Glomerular staining in a wild-type mouse subjected to serum nephrotoxic nephritis, an experimental model of immune complex-mediated glomerular disease characterized by glomerular inflammation and thrombosis. Marked staining is seen in the positive control (b), while staining is completely absent in the animal treated with Stx-2 and LPS.
Stx-2 in combination with LPS induced apoptosis in renal tubular cells
In contrast to minor glomerular changes, Stx-2 in combination with LPS induced apoptosis in renal tubular cells (Fig. 1) in both wild-type and Cfh+/− animals. Using a tubular apoptosis severity score (ranging from 0 = normal to 3 = marked) median cortical tubular apoptosis scores did not differ significantly between Cfh+/− (2, range 0–2, n = 6) and wild-type (2, range 0–3, n = 6) animals treated with low-dose Stx-2. Similarly, median cortical tubular apoptosis scores did not differ significantly between Cfh+/− (0·5, range 0–2, n = 6) and wild-type (2·5, range 0–3, n = 6) animals treated with high-dose Stx-2. Furthermore, the degree of tubular apoptosis was equivalent between the two Stx-2 doses. Features of tubular apoptosis included nuclear condensation with consequent sloughing of tubular cells into the lumen in some areas. Tubular apoptosis was evident in both cortical and medullary areas. While tubular apoptosis was readily evident by light microscopy, apoptosis was also confirmed using TUNEL staining (Fig. 1c). Similarly to the light microscopic assessment, the degree of tubular cell TUNEL staining did not differ between the two Stx-2 doses irrespective of factor H genotype. Importantly, no tubular changes were present in wild-type and Cfh+/− wild-type that had received LPS alone, indicating that this phenomenon was specific to administration of Stx-2. Immunostaining for complement C3 did not show any evidence of abnormal tubulointerstitial C3 deposition in any of the experimental animals (data not shown).
Stx-2 in combination with LPS induced marked splenic apoptosis
Stx-2 has the ability to bind to multiple tissues, including the lung and spleen [22]. Hence, we performed light microscopic analysis of lung, liver and spleen tissue in the experimental animals to determine if thrombi were present in non-renal microvasculature. No evidence of microvascular thrombosis was seen in lung, liver and spleen tissue sections in any of the experimental animals. However, marked splenic cell apoptosis was demonstrated in wild-type and Cfh+/− mice (Fig. 3) treated with both doses of Stx-2 in combination with LPS. Splenic apoptosis was not seen in wild-type and Cfh+/− mice treated with LPS alone (Fig. 2). Similarly to the observed renal tubular apoptosis, this phenomenon was therefore a specific effect of Stx-2.
Fig. 3.
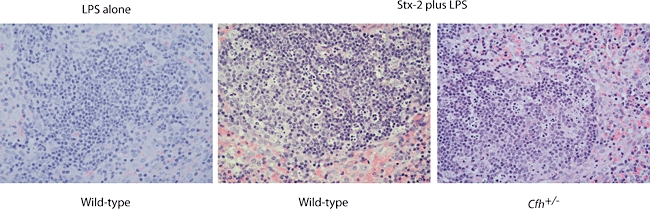
Splenic apoptosis in mice treated with Shiga toxin (Stx). Representative light microscopy images of haematoxylin and eosin-stained splenic tissue from mice treated with 50 ng Stx-2 in combination with lipopolysaccharide (LPS). Marked splenic cell apoptosis was demonstrated to an equivalent extent in both wild-type and Cfh+/− animals treated with either 50 ng or 200 ng Stx-2 in combination with LPS. Splenic apoptosis was not seen in wild-type or Cfh+/− mice treated with LPS alone. Original magnification ×40.
Discussion
The overwhelming association between aHUS and AP dysregulation [12], together with the recent report of a complement regulator mutation in an individual with lethal Stx-associated HUS [15], led us to hypothesize that complement dysregulation exacerbates Stx-induced renal pathology. To test this in an experimental model we chose to administer purified Stx-2 in combination with LPS to mice with haploinsufficiency of the major AP plasma regulator, factor H. Administration of purified Stx-2 in combination with LPS has been reported to result in glomerular TMA in mice in two recent studies [25,27]. However, using our protocol, the administration of purified Stx-2 to C57BL/6 mice resulted in acute tubular injury in the absence of any evidence of glomerular pathology.
Consistent with our observations, many studies in which mice have either been inoculated with Stx-producing E. coli strains [18,29,30] or injected with purified Stx with [20,21,25–27,31] or without LPS [22–24,32,33] have not developed evidence of glomerular thrombosis. We have summarized these studies in Table 2, where we list the experimental insult (e.g. Stx-producing bacterial infection, purified Stx) together with the reported renal phenotype [18,20–27,29–33]. The lack of typical glomerular pathological changes of TMA in these studies is striking. Thus, oral administration of E. coli O157:H7 strains to CD-1 mice resulted in death because of renal cortical tubular necrosis [30], a pathology that developed only in mice infected with E. coli O157:H7 strains that produced Stx-2 [18]. In these studies glomerular morphology remained normal by both light and electron microscopy [18,30]. Similarly, in a later study where C3H/HeN and C3H/HeJ mice were inoculated with E. coli O157:H7 strains, tubular cell necrosis developed but no glomerular thrombi were seen [29].
Table 2.
Summary of renal disease induced by Shiga toxin (Stx) in mice.
| Reference | Toxin or bacterial strain | LPS | Mouse strain | Renal pathology |
|---|---|---|---|---|
| [18,30] | E. coli O157:H7 orally | No | CD-1 | Bilateral renal cortical necrosis Glomeruli normal by light and electron microscopy |
| [31] | Stx-1, 0·01–2 µg intraperitoneally | E. coli O155:B8 | C3H/HeN | Mild to severe tubular injury with increasing Stx dose Tubular injury enhanced by administration of either LPS or TNF-α No glomerular abnormalities: no microvascular thrombosis |
| [23] | Stx-1 or Stx-2 1 µg–100 pg | None | CD-1 | Renal tubular damage greater in Stx-2 compared with Stx-1 treated mice |
| [29] | E. coli O157:H7 intragastrically | None | C3H/HeN (LPS-responder) C3H/HeJ (LPS non-responder) | Focal proliferation of mesangial cells and increased mesangial matrix. Tubular cell necrosis. No glomerular thrombi Renal histology did not differ between LPS-responder and LPS-non-responder mice |
| [20] | Purified Stx-2 250 pg intravenously | E. coli O111:B4 | BALB/c | Vascular congestion, diffuse interstitial inflammation, tubular cell necrosis |
| [21] | Purified Stx-2 600 pg intravenously | E. coli O111:B4 | BALB/c | Major lesions in cortical tubules No evidence of thrombi or renal microangiopathy |
| [33] | Purified Stx-1 500 pg intravenously | None | BALB/c | Moderate to severe widespread glomerular mesangial hypercellularity and crescent formation Treatment of mice with nitric oxide inhibitor resulted in PAS-positive amorphous material in the glomerular tufts consistent with thrombosis |
| [22] | Purified Stx-1 and Stx-2 1·5–4× LD50 | None | BALB/c | Tubular necrosis Glomerular morphology was normal |
| [32] | Purified Stx-1 50–100× LD50 intraperitoneally | None | BALB/c | Tubular dilatation and tubular cell apoptosis |
| [25] | Purified Stx-2 200 ng intraperitoneally | LPS (subtype not reported) | C3H/HeN | Focal proliferation of mesangial cells and infiltration of neutrophils Electron microscopy showed focal endothelial cell detachment, mild glomerular endothelial swelling and increased subendothelial space |
| [24] | Purified Stx-2 200–250 pg/g intravenously | None | ADAMTS13-deficient mice on (129/Sv × C57BL/6) or CASA/Rk genetic background | No disease in (129/Sv × C57BL/6) Adamts13−/− mice CASA/Rk Adamts13−/− mice developed red cell fragments on blood film, widespread vWF-rich, fibrin-poor thrombi in small vessels of multiple organs including kidney. Changes were consistent with thrombotic thrombocytopenic purpura |
| [27] | Purified Stx-2 225 ng/kg intraperitoneally | E. coli O55:B5 | C57Bl/6 | Glomerular red cell congestion and fibrin deposition Red cell and fibrin thrombi seen in glomerular arterioles Electron microscopy showed red cell congestion and electron-dense flocculent material and podocyte swelling |
| [26] | Purified Stx-2 4–12 ng intraperitoneally | E. coli O55:B5 | C57Bl/6 | Glomerular congestion with dilated capillary tufts filled with red cells and neutrophils |
aWF, von Willebrand factor; E. coli, Escherichia coli; LD50, lethal dose 50%; LPS, lipopolysaccharide; PAS, periodic acid-Schiff; TNF, tumour necrosis factor.
When purified Stx has been administered to mice, the pathological abnormalities within the kidney are predominantly that of tubular injury with little, if any, glomerular pathology (Table 2). Purified Stx-2 appears to result in greater renal tubular damage than purified Stx-1 [23]. This is perhaps not surprising, as the dose resulting in 50% death in treated animals lethal dose 50%) for Stx-2 was 400 times lower that of Stx-1, irrespective of whether the toxins were administered via the intravenous or intraperitoneal route [23]. Tubular lesions in mice given either Stx-1 or Stx-2 appear to affect predominantly renal cortical proximal tubules, with cortical distal tubules being relatively unaffected [23]. Lesions include epithelial cell necrosis and apoptosis [23]. While some studies have reported unremarkable glomerular pathology in mice given Stx-2 [22,23] or commented specifically that there was no evidence of renal TMA [21], others have reported focal proliferation of mesangial cells [25] and glomerular vascular congestion [20,26,27]. Furthermore, while ultrastructural examination of glomerular pathology in mice inoculated with Stx-2 producing E. coli strains was normal [18], some glomerular changes have been reported in mice given purified Stx-2 [25,27]. These have included evidence of endothelial damage, such as mild focal endothelial cell detachment and swelling [25], podocyte swelling [27] and red blood cell congestion and glomerular electron-dense flocculent material [27]. Podocyte injury can be seen in human HUS, and in vitro data using murine podocytes has shown that Stx-2 can induce podocyte endothelin-1 expression and actin remodelling [34].
Do the typical glomerular lesions seen in human TMA ever develop in Stx-treated mice? Only a few studies have reported glomerular lesions that reflect some of the typical changes seen in human renal TMA [24–27,33]. Importantly, some of these were provoked under special circumstances [24,33] and in none did the experimental animals develop the complete triad of thrombocytopenia, glomerular TMA and MAHA that comprise HUS. Thus, in one study in which purified Stx-1 was given to mice in the absence of LPS, glomerular capillary thrombosis and hypercellularity, in addition to tubular cell damage, was seen only in mice that had additionally received a nitric oxide synthase inhibitor [33]. In another, Stx-2 provoked small vessel von Willebrand factor (vWF)-rich fibrin-poor thrombi in multiple organs, including the kidney, in mice with complete deficiency of ADAMTS13 (a disintegrin-like and metalloproteinase with thrombospondin type 1 motif) on a permissive genetic background, a phenotype more consistent with thrombotic thrombocytopenic purpura rather than HUS [24]. Using LPS and purified Stx-2 at an identical dose to the high-dose group in our study, C3H/HeN mice developed uraemia, thrombocytopenia and mild glomerular endothelial cell injury evident using electron microscopy [25]. These mice did not develop MAHA, and surprisingly there was no report of tubular injury. Recently, it has been reported that administration of purified Stx-2 in combination with LPS to C57BL/6 mice results in renal injury that mimics human HUS [27]. Glomerular features included platelet clumping, red cell congestion and glomerular fibrin staining. While thrombocytopenia and anaemia also developed, red cell fragments were not reported [25]. Surprisingly, again there was no description of tubular injury.
The development of tubular injury in the absence of glomerular damage can be explained clearly by the localization of the Stx receptors in the mouse kidney. Using a monoclonal antibody directed against the trisaccharide moiety of the Gb3 receptor staining localized to the cytoplasm of tubular and collecting duct epithelial cells in the cortex and medulla of the mouse frozen kidney sections [23]. Similar staining patterns were noted when purified Stx toxins were used [23]. In a separate study, neither Stx-1 nor Stx-2 bound to murine glomeruli in vitro, while clear staining was seen for both toxins along murine renal tubules in vitro[22]. FITC-labelled Stx-1 also has been shown to localize to tubular structures but not glomeruli in vitro[35]. Furthermore, after in vivo injection of either Stx-1 or Stx-2 the toxins were localized to tubular cells but not glomerular structures [22], a finding confirmed using Stx-1 in another study [32]. Taken together, these studies indicate that murine renal tubules but not glomeruli express Gb3, the receptor for Stx, providing a sound physiological explanation for the development of tubular injury in the absence of glomerular damage in mice exposed to Stx. In our view, the subtle vascular and glomerular changes noted in some studies in mice (Table 2) could represent pathology that has developed secondary to tubular damage.
In addition to renal tubular cell injury we also demonstrated that florid splenic apoptosis developed in the mice exposed to Stx-2 and LPS, irrespective of factor H genotype or Stx-2 dose. A previous study has also reported the development of splenic apoptosis in mice treated with either purified Stx-1 or Stx-2 [22]. Studies of the biodistribution of radiolabelled Stx-1 and Stx-2 in mice have shown that both toxins target the lungs, nasal turbinates, spleen and kidney [22]. Stx-1 targeting to the spleen was twofold higher than that of Stx-2, while targeting to the lung was 10-fold higher for Stx-1 compared with Stx-2. Consistent with the preferential targeting of Stx-1 to the lung was the observation that Stx-1 induced lung haemorrhage and alveolitis in mice, while lung histology was unremarkable following Stx-2 administration [22]. Similarly, in our study Stx-2 and LPS did not result in any discernable lung abnormalities by light microscopy.
In summary, Stx-2 administration in combination with LPS did not result in TMA in either wild-type mice or mice with haploinsufficiency of factor H. The development of renal tubular damage and splenic apoptosis was equivalent between wild-type and Cfh+/− animals and no abnormal renal C3 staining was detected. The tropism of Stx-2-induced pathology mirrored the previously described Stx receptor tissue distribution in mice. We believe that the absence of the Stx receptor in murine glomeruli is the key factor that prevented the development of TMA not only in this study, but in the majority of the reported murine studies to date. Clearly, this fundamental limitation precluded our ability to test our original hypothesis in this experimental setting. While we cannot draw any conclusions on the effect of heterozygous factor H deficiency on Stx-2-induced TMA, our results indicated that heterozygous deficiency of complement factor H did not alter Stx-2-induced renal tubular damage or splenic apoptosis in mice.
Acknowledgments
We thank the staff of the Biological Services Unit at Imperial College, London, United Kingdom for the care of the animals involved in this study. D. P.-C. was funded by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) and a Marie Curie Fellowship (IMDEMI, 005632). M. C. P. is a Wellcome Trust Senior Fellow in Clinical Science (WT082291MA).
References
- 1.Nabhan C, Kwaan HC. Current concepts in the diagnosis and management of thrombotic thrombocytopenic purpura. Hematol Oncol Clin North Am. 2003;17:177–99. doi: 10.1016/s0889-8588(02)00085-0. [DOI] [PubMed] [Google Scholar]
- 2.Moake JL. Haemolytic-uraemic syndrome: basic science. Lancet. 1994;343:393–7. doi: 10.1016/s0140-6736(94)91227-0. [DOI] [PubMed] [Google Scholar]
- 3.Kulzer P, Wanner C. Thrombotic microangiopathy: a challenge with uncertain outcome. Nephrol Dial Transpl. 1998;13:2154–60. doi: 10.1093/ndt/13.8.2154. [DOI] [PubMed] [Google Scholar]
- 4.Le Saux N, Spika JS, Friesen B, et al. Ground beef consumption in noncommercial settings is a risk factor for sporadic Escherichia coli O157:H7 infection in Canada. J Infect Dis. 1993;167:500–2. doi: 10.1093/infdis/167.2.500. [DOI] [PubMed] [Google Scholar]
- 5.Carter AO, Borczyk AA, Carlson JA, et al. A severe outbreak of Escherichia coli O157:H7 – associated hemorrhagic colitis in a nursing home. N Engl J Med. 1987;317:1496–500. doi: 10.1056/NEJM198712103172403. [DOI] [PubMed] [Google Scholar]
- 6.Sandvig K, Dubinina E, Garred O, et al. Protein toxins: mode of action and cell entry. Biochem Soc Trans. 1992;20:724–7. doi: 10.1042/bst0200724. [DOI] [PubMed] [Google Scholar]
- 7.Lingwood CA. Verotoxin-binding in human renal sections. Nephron. 1994;66:21–8. doi: 10.1159/000187761. [DOI] [PubMed] [Google Scholar]
- 8.Ray PE, Liu XH. Pathogenesis of Shiga toxin-induced hemolytic uremic syndrome. Pediatr Nephrol. 2001;16:823–39. doi: 10.1007/s004670100660. [DOI] [PubMed] [Google Scholar]
- 9.Moake JL. Thrombotic thrombocytopenic purpura and the hemolytic uremic syndrome. Arch Pathol Lab Med. 2002;126:1430–3. doi: 10.5858/2002-126-1430-TTPATH. [DOI] [PubMed] [Google Scholar]
- 10.Karch H. The role of virulence factors in enterohemorrhagic Escherichia coli (EHEC) – associated hemolytic-uremic syndrome. Semin Thromb Hemost. 2001;27:207–13. doi: 10.1055/s-2001-15250. [DOI] [PubMed] [Google Scholar]
- 11.Ruggenenti P, Noris M, Remuzzi G. Thrombotic microangiopathy, hemolytic uremic syndrome, and thrombotic thrombocytopenic purpura. Kidney Int. 2001;60:831–46. doi: 10.1046/j.1523-1755.2001.060003831.x. [DOI] [PubMed] [Google Scholar]
- 12.Kavanagh D, Goodship TH. Update on evaluating complement in hemolytic uremic syndrome. Curr Opin Nephrol Hypertens. 2007;16:565–71. doi: 10.1097/MNH.0b013e3282f0872f. [DOI] [PubMed] [Google Scholar]
- 13.Esparza-Gordillo J, Jorge EG, Garrido CA, et al. Insights into hemolytic uremic syndrome: segregation of three independent predisposition factors in a large, multiple affected pedigree. Mol Immunol. 2006;43:1769–75. doi: 10.1016/j.molimm.2005.11.008. [DOI] [PubMed] [Google Scholar]
- 14.Caprioli J, Noris M, Brioschi S, et al. Genetics of HUS: the impact of MCP, CFH, and IF mutations on clinical presentation, response to treatment, and outcome. Blood. 2006;108:1267–79. doi: 10.1182/blood-2005-10-007252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fang CJ, Fremeaux-Bacchi V, Liszewski MK, et al. Membrane cofactor protein mutations in atypical hemolytic uremic syndrome (aHUS), fatal Stx-HUS, C3 glomerulonephritis, and the HELLP syndrome. Blood. 2008;111:624–32. doi: 10.1182/blood-2007-04-084533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pickering MC, Cook HT. Translational mini-review series on complement factor H: renal diseases associated with complement factor H: novel insights from humans and animals. Clin Exp Immunol. 2008;151:210–30. doi: 10.1111/j.1365-2249.2007.03574.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pickering MC, Cook HT, Warren J, et al. Uncontrolled C3 activation causes membranoproliferative glomerulonephritis in mice deficient in complement factor H. Nat Genet. 2002;31:424–8. doi: 10.1038/ng912. [DOI] [PubMed] [Google Scholar]
- 18.Wadolkowski EA, Sung LM, Burris JA, et al. Acute renal tubular necrosis and death of mice orally infected with Escherichia coli strains that produce Shiga-like toxin type II. Infect Immun. 1990;58:3959–65. doi: 10.1128/iai.58.12.3959-3965.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barrett TJ, Potter ME, Wachsmuth IK. Bacterial endotoxin both enhances and inhibits the toxicity of Shiga-like toxin II in rabbits and mice. Infect Immun. 1989;57:3434–7. doi: 10.1128/iai.57.11.3434-3437.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Palermo MS, Alves Rosa MF, Van Rooijen N, et al. Depletion of liver and splenic macrophages reduces the lethality of Shiga toxin-2 in a mouse model. Clin Exp Immunol. 1999;116:462–7. doi: 10.1046/j.1365-2249.1999.00925.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Palermo M, Alves-Rosa F, Rubel C, et al. Pretreatment of mice with lipopolysaccharide (LPS) or IL-1beta exerts dose-dependent opposite effects on Shiga toxin-2 lethality. Clin Exp Immunol. 2000;119:77–83. doi: 10.1046/j.1365-2249.2000.01103.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rutjes NW, Binnington BA, Smith CR, et al. Differential tissue targeting and pathogenesis of verotoxins 1 and 2 in the mouse animal model. Kidney Int. 2002;62:832–45. doi: 10.1046/j.1523-1755.2002.00502.x. [DOI] [PubMed] [Google Scholar]
- 23.Tesh VL, Burris JA, Owens JW, et al. Comparison of the relative toxicities of Shiga-like toxins type I and type II for mice. Infect Immun. 1993;61:3392–402. doi: 10.1128/iai.61.8.3392-3402.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Motto DG, Chauhan AK, Zhu G, et al. Shigatoxin triggers thrombotic thrombocytopenic purpura in genetically susceptible ADAMTS13-deficient mice. J Clin Invest. 2005;115:2752–61. doi: 10.1172/JCI26007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ikeda M, Ito S, Honda M. Hemolytic uremic syndrome induced by lipopolysaccharide and Shiga-like toxin. Pediatr Nephrol. 2004;19:485–9. doi: 10.1007/s00467-003-1395-7. [DOI] [PubMed] [Google Scholar]
- 26.Roche JK, Keepers TR, Gross LK, et al. CXCL1/KC and CXCL2/MIP-2 are critical effectors and potential targets for therapy of Escherichia coli O157:H7-associated renal inflammation. Am J Pathol. 2007;170:526–37. doi: 10.2353/ajpath.2007.060366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Keepers TR, Psotka MA, Gross LK, et al. A murine model of HUS: Shiga toxin with lipopolysaccharide mimics the renal damage and physiologic response of human disease. J Am Soc Nephrol. 2006;17:3404–14. doi: 10.1681/ASN.2006050419. [DOI] [PubMed] [Google Scholar]
- 28.Robson MG, Cook HT, Botto M, et al. Accelerated nephrotoxic nephritis is exacerbated in C1q-deficient mice. J Immunol. 2001;166:6820–8. doi: 10.4049/jimmunol.166.11.6820. [DOI] [PubMed] [Google Scholar]
- 29.Karpman D, Connell H, Svensson M, et al. The role of lipopolysaccharide and Shiga-like toxin in a mouse model of Escherichia coli O157:H7 infection. J Infect Dis. 1997;175:611–20. doi: 10.1093/infdis/175.3.611. [DOI] [PubMed] [Google Scholar]
- 30.Wadolkowski EA, Burris JA, O'Brien AD. Mouse model for colonization and disease caused by enterohemorrhagic Escherichia coli O157:H7. Infect Immun. 1990;58:2438–45. doi: 10.1128/iai.58.8.2438-2445.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Harel Y, Silva M, Giroir B, et al. A reporter transgene indicates renal-specific induction of tumor necrosis factor (TNF) by shiga-like toxin. Possible involvement of TNF in hemolytic uremic syndrome. J Clin Invest. 1993;92:2110–16. doi: 10.1172/JCI116811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wolski VM, Soltyk AM, Brunton JL. Tumour necrosis factor alpha is not an essential component of verotoxin 1-induced toxicity in mice. Microb Pathog. 2002;32:263–71. doi: 10.1006/mpat.2002.0501. [DOI] [PubMed] [Google Scholar]
- 33.Dran GI, Fernandez GC, Rubel CJ, et al. Protective role of nitric oxide in mice with Shiga toxin-induced hemolytic uremic syndrome. Kidney Int. 2002;62:1338–48. doi: 10.1111/j.1523-1755.2002.kid554.x. [DOI] [PubMed] [Google Scholar]
- 34.Morigi M, Buelli S, Zanchi C, et al. Shigatoxin-induced endothelin-1 expression in cultured podocytes autocrinally mediates actin remodeling. Am J Pathol. 2006;169:1965–75. doi: 10.2353/ajpath.2006.051331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu XH, Lingwood CA, Ray PE. Recruitment of renal tubular epithelial cells expressing verotoxin-1 (Stx1) receptors in HIV-1 transgenic mice with renal disease. Kidney Int. 1999;55:554–61. doi: 10.1046/j.1523-1755.1999.00278.x. [DOI] [PubMed] [Google Scholar]