

Abstract
Manganese superoxide dismutase (MnSOD or SOD2) is a key mitochondrial enzymatic antioxidant. Arguably the most striking phenotype associated with complete loss of SOD2 in flies and mice is shortened life span. To further explore the role of SOD2 in protecting animals from aging and age-associated pathology, we generated a unique collection of Drosophila mutants that progressively reduce SOD2 expression and function. Mitochondrial aconitase activity was substantially reduced in the Sod2 mutants, suggesting that SOD2 normally ensures the functional capacity of mitochondria. Flies with severe reductions in SOD2 expression exhibited accelerated senescence of olfactory behavior as well as precocious neurodegeneration and DNA strand breakage in neurons. Furthermore, life span was progressively shortened and age-dependent mortality was increased in conjunction with reduced SOD2 expression, while initial mortality and developmental viability were unaffected. Interestingly, life span and age-dependent mortality varied exponentially with SOD2 activity, indicating that there might normally be a surplus of this enzyme for protecting animals from premature death. Our data support a model in which disruption of the protective effects of SOD2 on mitochondria manifests as profound changes in behavioral and demographic aging as well as exacerbated age-related pathology in the nervous system.
Keywords: Drosophila, oxidative stress, SOD, aging, mitochondria
Introduction
A variety of genetic approaches have identified many genes that impact survival in worms, flies and mice. These studies provide valuable clues regarding the molecular maintenance of life span (Hekimi and Guarente, 2003; Murphy et al., 2003). Importantly, many of the genetic pathways identified by these studies are conserved throughout the animal kingdom (Tatar et al., 2003), suggesting that mechanisms of aging might be similarly conserved.
Age-related accumulation of oxidative damage is one of the mechanisms suggested by genetic and other studies to underlie aging (Harman 1956, 1972, 2006; Hekimi and Guarente, 2003; Longo and Finch, 2003). For example, oxidatively damaged macromolecules accumulate in mitochondria as a function of age (Sohal and Sohal, 1991; Sohal and Dubey, 1994; Sohal and Weindruch, 1996), augmentation of antioxidant defenses through genetic and dietary manipulations extend life span (review by Muller et al., 2007), and reduction in antioxidant defenses or exposure to exogenous oxidizing agents greatly shortens life span (Sohal et al, 2002). These studies and others are consistent with a model in which the rate of mitochondrial ROS production or damage to mitochondrial macromolecules determines the rate at which each species ages (Barja, 2002). Despite years of focused research, however, the connection between oxidative damage and aging remains somewhat tenuous (Sohal, 2002).
To further probe the relationship between oxidative stress and aging, we generated a series of Drosophila mutants with progressive reductions in mitochondrial superoxide dismutase (SOD2), the major SOD isoform in mitochondria. We analyzed indices of mitochondrial function, DNA damage, nervous system integrity, behavioral senescence, life span and mortality in these animals. We find that reduced SOD2 expression disrupts mitochondrial function, accelerates DNA strand breakage in the brain, causes precocious neurodegeneration, and accelerates senescence of olfactory behavior. Additionally, we find that reduced SOD2 expression increases age-specific mortality and that there is a non-linear relationship between SOD2 expression and this parameter of demographic aging. These mortality analyses suggest that normal levels of SOD2 allow aerobic organisms to survive within a wide range of oxidative stress. Together, our data indicate that SOD2 is an essential front line defense against oxidative damage, age-related pathology, functional senescence and mortality.
Experimental Methods
Western blots
Total protein was extracted with 1X sample buffer (0.5M TRIS.HCl pH 6.8, 3% glycerol; 0.4% SDS and 10mMDTT) from age-matched young flies. Equivalent amounts of protein samples were separated using 9% discontinuous SDS-PAGE and transferred to PVDF membranes. Transferred proteins were probed with rabbit anti-SOD2 primary antibody (Stressgen, Canada) at 1:5000 dilution and subsequently with HRP-conjugated anti-rabbit secondary antiserum (Calbiochem, USA) at 1:5000 dilution and detected with ECL using the manufacturer's protocol. Western blots were quantified by scanning the exposed films. The arbitrary luminescence units for SOD2 bands in each genotype were normalized and expressed as percentage values of KG06854R. Equal amounts of total protein in each lane were confirmed by staining the probed blot with Coomassie blue that reports all peptide fractions. The final values were compiled from three blots. The error bars represent the standard deviation in each genotype.
RNA isolation and real-time PCR
RNA was isolated from approximately 30 flies per sample using RNAeasy kit (Qiagen). Real-time PCR was performed in a two-step reaction using a cDNA preparation kit and Platinum SYBR green qPCR supermix from Invitrogen using random hexamer in the first step and gene specific primers in the second step. Sod2 mRNA was quantitated using 2−ΔΔCt method with RP49 as an internal control. The PCR efficiencies of Sod2 and RP49 were approximately equal and validation experiment was done spanning four log values. Graph of log input of cDNA versus ΔCt value for Sod2 and RP49 yielded straight lines with slopes < 0.1, thus validating the use of 2−ΔΔCtmethod (Giulietti et al., 2001).
Biochemical assays
Total SOD activity was measured in microtiter plates according to the manufacturer's instructions (Cayman Chemical Co., USA Superoxide Dismutases assay kit Cat#706002). Cu/Zn SOD (SOD1) activity was deduced by subtracting SOD2 activity from total SOD activity.
Aconitase activity was measured from whole fly extracts in reaction mixtures containing 0.6 mM MnCl2, 2 mM citric acid, and 50 mM Tris-HCl, pH 8.0. Aconitase activity was determined as the absorbance change at 240 nm which reflects the conversion of citrate to isocitrate. Fumarase activity was determined as the conversion of malate to fumarate measured at 240 nm (Rocker, 1950; Henson and Cleland, 1967).
Chromogenic detection of aconitase activity was performed according to Kirby et al., 2002.
Lifespan assays and mortality calculations
All lifespan studies were done in population cages with approximately 500 flies per cage. 2−3 day old flies were anesthetized in small batches, separated according to sex and counted. Males and females were allowed to recover for 24 hours and equal number of males and females were added to each population cage. Mortality cages were kept in insect chambers maintained at 24°C. Flies were cultured on regular fly media containing maize, yeast, agar and molasses. The numbers of dead flies were assessed daily. Age-specific mortality was calculated using the Gompertz's model of population aging. Ln values of instantaneous mortality (μx) were plotted against chronological time (days). All mortality calculations and maximum likelihood estimates were done using WinModest V1.02 (Pletcher, 1999) and plotted on GraphPad Prism V3.02, from GraphPad Software Incorporated.
TUNEL assays
TUNEL assays were performed using “In-Situ Cell Death Detection Kit, AP” (Cat#1684809910) Roche Diagnostics (USA), according to the manufacturer's instructions. Age-matched specimen heads were dissected, fixed in FAAG (4% formaldehyde, 5% acetic-acid, 1% glutaraldehyde dissolved in 80% ethanol), embedded in paraplast, and sectioned at 7μm. TUNEL detection was done using the optional alkaline phosphatase detection procedure (Roche catalogue no. 11684809910) and digitally imaged in bright field at 400X on a Zeiss Axioskop 2 plus microscope.
Olfactory Behavior
All flies for behavioral tests were reared and aged at 25°C, 60% relative humidity under a 12 hour light/dark cycle. Avoidance of 4-methylcyclohexanol (MCH, Sigma Chemical Co. St. Louis, MO, USA, dilution factor 1:100) was performed essentially as described (Stoltzfus et al., 2003). One- to four-day-old adults were briefly anesthetized with CO2, separated by sex, and then transferred in groups of 25 to fresh food vials. Flies at various ages were transferred to a T-maze. After one minute of rest, flies were allowed two minutes to choose between a maze arm containing an air stream with MCH and an opposing arm containing an air stream without an explicit odorant. After each two-minute choice test, flies were briefly anesthetized with CO2. Flies that moved into the two arms of the T-maze were counted and transferred together into a fresh food vial for aging until the next assessment. Avoidance indices were calculated as described (Stoltzfus et al., 2003) and then normalized to the performance of 3−5 day old w[CS] control flies (Cook-Wiens and Grotewiel, 2002) tested in parallel during each assessment. While males and females were tested separately, there were no effects of sex in any of the studies on olfactory behavior (three-way ANOVA, n.s.). Therefore, data from males and females were combined. Statistical analyses were performed with JMP (SAS, Cary, NC, USA).
Results
Flies with progressive reduction in SOD2 expression and activity
P-element insertion KG06854 resides in the 5’ untranslated region of the first exon in Sod2 (Fig. 1). Via imprecise excision of this P-element, we previously generated a strong loss of function allele, Sod2n283 (Fig.1), that has undetectable SOD2 activity and protein expression (Duttaroy et al., 2003). Additional molecular analyses revealed that the Sod2n283 chromosome carries a 167 bp deletion that removes portions of the first exon and intron of Sod2, including the putative ATG start codon (Fig. 1). As a control, a revertant allele, KG06854R, was derived from precise excision of KG06854 (Fig. 1). Thus, the Sod2n283 and KG06854R alleles are derivatives of the same KG06854 insertion line.
Figure 1.
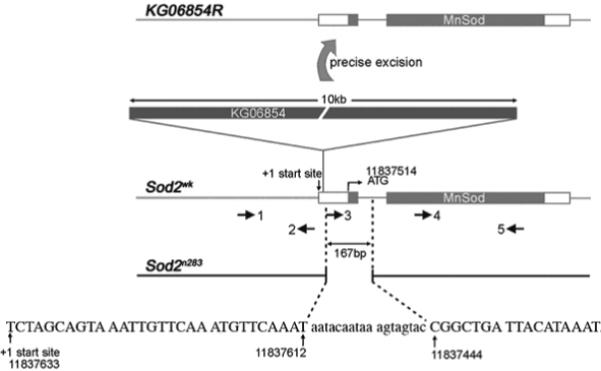
Molecular analysis of Sod2 alleles. Sod2n283 has a 167 bp deletion within exon 1 and part of intron 1. The span of the deletion was confirmed by sequencing a unique PCR band that arose in Sod2n283 when primer combination 1 and 5 were used. Sequence data revealed that 18 nt of P-element sequence (lower case letters) were left in Sod2n283. Sod2wk is the original P-insertion located in the 5’ untranslated region of Sod2. KG06854R is a precise excision derivative of KG06854 as revealed by DNA sequencing (not shown). Molecular coordinates are according to FlyBase release 3.0.
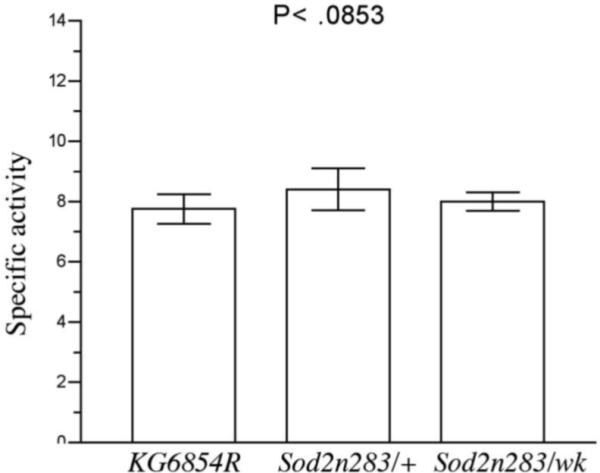
In addition to the null allele, we can now report that the single P-element insertion KG06854 into the Sod2 5’-UTR causes reduced expression of this gene, a characteristic shared by many P-insertions (Bellen et al., 2004) and it was designated as Sod2 weak (Sod2wk/wk). We performed a series of immunoblot, RNA and enzyme activity assays to determine the effects of the Sod2 alleles on SOD2 expression and function. Quantitation of Western blots revealed that with respect to the revertant, KG06854R, Sod2wk/wk homozygotes produced about 46% of SOD2 protein and heterozygous null Sod2n283/+ flies produce 44%, respectively (Fig. 2A). The Sod2 weak over null heterozygote (Sod2n283/wk) encodes only 22% of the SOD2 protein relative to the KG06854R (Fig. 2A). Reduced protein expression is a consequence of less Sod2 mRNA expression, which is evident from quantification of Sod2 mRNA in Sod2n283/+ heterozygotes, Sod2wk/wk homozogotes, and Sod2n283/wk (Fig. 2B). Total SOD activity assays indicated that Sod2n283/+ heterozygotes have 50% SOD2 activity compared to KG06854R controls while SOD2 activity was essentially undetectable in Sod2n283 homozygotes (Fig. 2C). Cu-ZnSOD activity was not changed in either of these genotypes (Fig 2C), suggesting that there is no compensatory upregulation of SOD1 in Sod2 mutants. These experiments demonstrate that Sod2n283 is a null allele and that Sod2wk is a partial loss of function allele at the Sod2 locus. Additionally, our analyses show that these alleles by themselves or in trans can be used to progressively reduce SOD2 expression and function.
Figure 2.

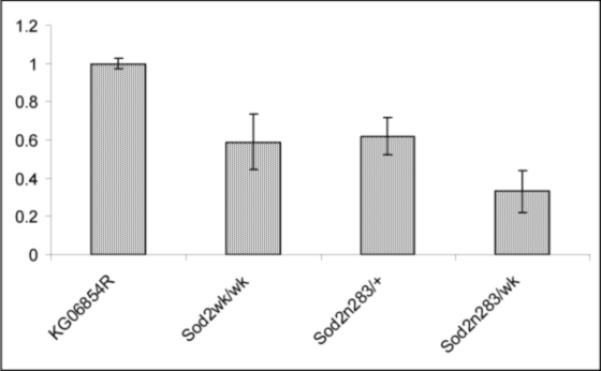

Levels of SOD2 protein, mRNA, and activities in various Sod2 alleles. (A) Histogram showing the expression of SOD2 protein in different alleles relative to the control KG06854R. Densitometric analysis revealed that Sod2wk/wk and Sod2n283/+ flies, respectively, encode about 46% and 44% of SOD2 protein relative to KG06854R. Sod2n283/wk double mutant expresses only 22% of SOD2 with respect to KG06854R. A transgenic line carrying four copies of Sod2 (obtained from Bill Orr, Southern Methodist University) encodes about 43% more SOD2 than KG06854R. Average of percent relative intensity values obtained from densitometric scans of three separate blots are shown in the table. Coomassie blue staining of the same blot established equal protein loading in each lane. Error bars indicate SD (n=3).
(B) Sod2 mRNA expression. Real-time PCR was used to measure mRNA expression of Sod2 relative to RP49 control. Compared to KG06854R flies Sod2wk/wk, Sod2n283/+ and Sod2n283/wk express 59%, 62% and 33% mRNA for Sod2, respectively.
(C) SOD2 activity. Sod2n283/+ heterozygotes have 50% of the SOD2 activity found in KG06854R controls whereas SOD2 activity is essentially undetectable in Sod2n283 homozygotes. The activity of SOD1 was unchanged in all three genotypes.
Reduced expression of SOD2 increases age-specific mortality
Complete loss of SOD2 function dramatically shortens life span in flies (Duttaroy et al., 2003). and mice (Li et al., 1995; Lebovitz et al., 1996). We performed survival studies to determine the impacts of progressive reduction of SOD2 function on life span and mortality. Revertant control flies had mean and maximum life spans of 50.4 and 86 days, respectively (Fig. 3A, Table 1). Mean and maximum life spans were reduced by 20−24% in Sod2n283/+ heterozygotes and Sod2wk/wk homozygotes (Fig. 3B and 3C, Table 1), flies that express ∼50% of normal levels of SOD2 activity (Fig. 2). Furthermore, reduction of SOD2 function by ∼75% in Sod2n283/wk transheterozygotes (Fig. 2) shortened mean and maximum life spans by 38% and 43%, respectively (Fig. 3D, Table 1). These data confirm that SOD2 activity is essential for normal life span.
Figure 3.

Life span and mortality in Sod2 mutants. Main plots are life span data. Insets are mortality data. In all graphs, solid lines are observed data and dotted lines are derived from the Gompertz's MLE parameters ‘a’ and ‘b’ using WinModest. In each genotype the predicted curve (dotted line) fits closely to the observed values (continuous line). (A) KG06854R controls. In B, C & D the grey continuous line in the mortality plots (insets) corresponds to the mortality trend of KG06854R and has been included for comparison of the difference in slope (initial mortality rate).
Table 1. Fitted Gompertz model and maximum likelihood estimates (MLE) values for intercept and slope.
Overall mortality data of individual genotypes as obtained by maximum likelihood estimates were best described with Gompertz parameters using WinModest v1.0.2 (Pletcher, 1999) (n= sample size, a= initial mortality rate, b= rate of demographic aging). Symbols *, †, indicates genetic background. Similar superscript in column for Gompertz parameters indicates no significant difference between populations. In column for Gompertz parameter ‘a’ significant difference is at p<0.01; in column for parameter ‘b’ significant difference is at p<0.001. A sensitivity analysis by varying the start parameters for ‘a’ and ‘b’ shows that the MLE parameters for each genotype is robust. The percent errors between the fitted and the actual mean life span for each genotype are small. The differences in mean lifespan were significantly different between KG06854R and Sod2n283/+ (P<0.0001), between KG06854R and Sod2wk/wk (P<0.0001), and between KG06854R and Sod2n283/wk (P<0.0001), but not between Sod2n283/+ and Sod2wk/wk (P=0.1958).
| Gompertz Parameters | Max LS (days) | Actual Mean LS (days) | Fitted Mean LS (days) | % Error between Actual & Fitted Mean LS | ||||
|---|---|---|---|---|---|---|---|---|
| a (intercept) | b (slope) | |||||||
| MLE value | Start Range | MLE value | Start Range | |||||
|
KG06854R * (n=679) |
0.00259 p | 0.0001 − 0.003 | 0.05115 p | 0.01 − 0.3 | 86 | 50.37850 | 50.51922 | 0.279 |
|
Sod2n283/+ * (n=1038) |
0.00288 p | 0.0001 − 0.004 | 0.06522 q | 0.01 − 0.4 | 69 | 41.49615 | 41.44730 | 0.118 |
|
Sod2wk/wk * (n=704) |
0.00250 p | 0.0001 − 0.002 | 0.07501 r | 0.01 − 0.2 | 65 | 39.63921 | 39.40065 | 0.602 |
|
Sod2wk/n283 * (n=687) |
0.00239 p | 0.0001 − 0.003 | 0.10728 s | 0.01 − 0.3 | 49 | 31.12664 | 30.97952 | 0.473 |
|
Sod2+/+;{Sod2+/+}† (n=227) |
0.00395 q | 0.0001 − 0.002 | 0.04072 u | 0.01 − 0.2 | 91 | 50.44934 | 50.06139 | 0.769 |
According to the Gompertz model of population aging, changes in life span can be caused by alteration of initial mortality, age-specific mortality rate, or both (Pletcher, 1999; Gompertz, 1825; Kowald, 2002). We first confirmed that all strains tested conformed to the Gompertz model without obvious errors (Fig. 3, Table 1). We then used maximum likelihood estimates (MLE) from WinModest software (Pletcher, 1999) to determine the initial mortality rate (parameter “a”) and the rate of demographic aging (change in mortality slope, parameter “b”) for each population. Reduced SOD2 expression had no effect on initial mortality (Table 1). In contrast, the rate of demographic aging was sensitive to SOD2 expression. All flies with reduced SOD2 levels had increased age-specific mortality (Fig. 3, insets). Furthermore, progressive reduction in SOD2 levels caused progressive increases in the mortality slopes (Table 1, Fig. 4A). We also performed a detailed mortality decomposition analysis that estimated the contribution of initial mortality and the change in mortality slope on life span in the Sod2 mutants (Pletcher, 1999). The increased age-specific mortality in Sod2 mutants was estimated to reduce mean life span by 7.5−21.5 days relative to revertant controls (Table 2). In contrast, any putative changes in initial mortality in Sod2 mutants was estimated to reduce life span by 1.6 days or actually increase life span by 2 days at most (Table 2). This analysis confirmed that loss of SOD2 affected life span primarily by increasing the slope of the mortality plot, but not through changes in the initial mortality rate (Table 2). Together, our life span and mortality analyses strongly support a role for SOD2 in determining the rate of demographic aging.
Figure 4.


Relationship between SOD2 expression and demographic parameters. Q= Sod2n283/wk, R= Sod2n283/+, S= Sod2wk/wk, T= KG06854R and U= Sod2+/+; P{Sod2+/+}. (A) A power law relationship was observed when the demographic rate of aging (parameter b from WinModest) was plotted versus percent SOD2 expressed. R2 = 0.9636. Inset shows projected rate of demographic aging as a function of percent SOD2 expression, indicating that a marginal decrease in the rate of demographic aging will occur with progressively increased levels of SOD2 expression. (B) Log mean lifespan versus Log SOD2 expression describes a straight line, indicating that the percent change in mean lifespan will eventually plateau after a certain level of SOD2 expression is achieved (inset).
Table 2. Mortality decomposition analysis.
The diagonal entries in bold, show the mean lifespan (in days ± standard error) for each genotype analyzed. The entries (in days) above the diagonal show the amount of the difference in longevity (explained in the methods) that is accounted for by variation in the slope (b) or the ‘rate of aging’ of the Gompertz model of aging. The entries (in days) below the diagonal show the amount of the difference in longevity that is accounted for by variation in the intercept (a) or the ‘initial mortality rate’ of the Gompertz model. An entry that contributes the most towards the observed effect in the variation has been italicized for comparison. Most observed variation between the populations analyzed has been due to differences in slope rather than the intercept.
| Variation due to effects of slope (b) (rate of aging) | |||||
|---|---|---|---|---|---|
| KG06854R | Sod2n283/+ | Sod2wk/wk | Sod2n283/wk | ||
| Variation due to effects of intercept (a) (Initial mortality rate) | KG06854R | 50.37 ±0.7 | −7.45 | −11.63 | −20.53 |
| Sod2n283/+ | −1.62 | 41.49 ±0.4 | −3.87 | −12.52 | |
| Sod2wk/wk | 0.52 | 1.82 | 39.63 ±0.5 | −8.88 | |
| Sod2n283/wk | 1.00 | 2.04 | 0.46 | 31.12 ±0.3 | |
What kind of relationship exists between SOD2 levels, demographic aging and life span? We found that SOD2 expression and the Gompertz rate of aging were related by an inverse power function (Y=1/Xn; r2 = 0.9636, Fig. 4A). This relationship suggests that the Gompertz rate of aging will be highly sensitive to SOD2 expression between 0 and ∼200% of normal level. Beyond that one can only make theoretical prediction that suggests greater than 2-fold overexpression of SOD2 will drive the Gompertz rate of aging toward zero (Figure 4A, inset). The effect of SOD2 on this aging parameter will become asymptotic at extremely high levels of expression similar to mean lifespan, which varied non-linearly (logarithmic) with SOD2 expression (Fig. 4B). This relationship predicts that the beneficial effect of SOD2 overexpression on this aging parameter plateaus at very high concentrations of SOD2. Our data corroborates well with an earlier report where life span studies were performed using the same transgenic line (Mockett et al., 1999).
SOD2 expression is essential for mitochondrial function
Aconitase is a citric acid cycle protein localized to the mitochondrial matrix. Aconitase activity is a useful index of mitochondrial function during normal aging and conditions with elevated oxidative stress (Fridovich and Gardner, 1991; Gardner et al., 1994; Yarian et al., 2006). We measured aconitase activity to determine whether reduced SOD2 function had consequences for the functional integrity of mitochondria. Since fumarase is a mitochondrial enzyme that is not sensitive to oxidative stress (Melov et al., 1999), we measured activity of this enzyme as a control. Aconitase activity was reduced by 75% in Sod2n283 homozygotes as well as in Sod2wk/Sod2n283 flies compared to revertant controls (Fig. 5A). Aconitase activity was found to be normal, in young Sod2n283/+ heterozygotes (Fig.5A). Total loss of mitochondrial aconitase activity was evident in Sod2n283 homozygotes by chromogenic detection of aconitase activity, which also indicated some reduction in aconitase activity in Sod2n283/wk (Fig, 5B). In contrast, fumarase activity was not altered in flies with reduced SOD2 expression (Fig.5C). The preferential inactivation of mitochondrial aconitase and not fumarase in Sod2wk/n283 indicates that reduced expression of SOD2 causes oxidative damage to aconitase and compromises mitochondrial function.
Figure 5.
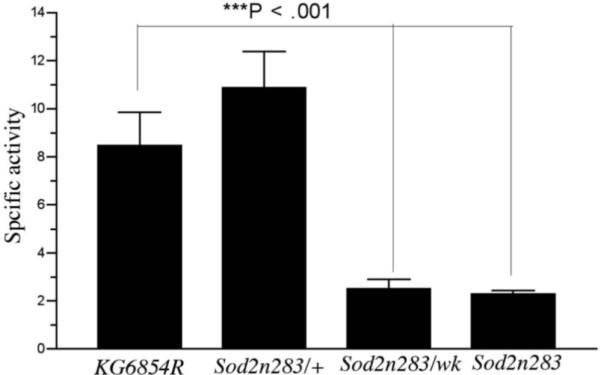
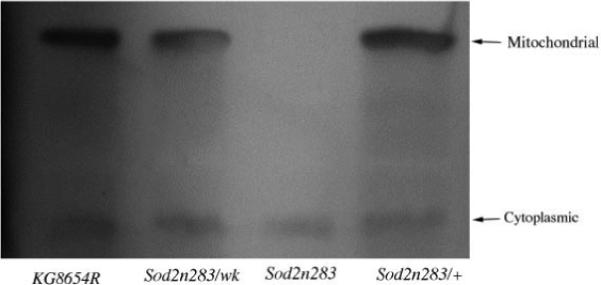
Aconitase and fumarase activity in Sod2 mutants. (A) Total aconitase activity was significantly less in Sod2n283 homozygotes and Sod2n283/wk transheterozygotes. Young Sod2n283/+ heterozygotes had normal aconitase activity. (B) Chromogenic detection of aconitase activity shows that mitochondrial aconitase consist of bulk of the celluar aconiatse. Complete absence of mitochondrial aconitase in Sod2n283 homozygotes suggests its breakdown by excessive ROS, (C) Fumarase activity remains unaltered in Sod2 mutants.
Reduced SOD2 expression exacerbates age-related cellular pathology and behavioral aging
Oxidative stress causes DNA strand breakage, a molecular change that can be monitored in situ with TUNEL assays (Kokoszka et al., 2001). We assessed TUNEL positive neurons in the fly brain to determine whether reduced SOD2 expression caused DNA strand breakage. TUNEL positive cells were essentially absent in 0−5 day-old revertant controls (Fig. 6A), whereas brains from Sod2n283 homozygote showed obvious TUNEL staining within the first 20 hours after eclosion (Fig. 6B). Interestingly, revertant controls (not shown) and Sod2n283/+ heterozygotes (Fig. 6C) did not have comparable numbers of TUNEL positive neurons until ∼9 weeks of age. These data suggest that reduced expression of SOD2 results in oxidative damage to DNA causing strand breakage.
Figure 6.

TUNEL staining and structural integrity of the brain in Sod2 mutants. (A-D) TUNEL staining. (A) TUNEL positive nuclei (in blue) in adult brain sections following the induction of DNA fragmentation with 2000U of DNase1. (B) Sod2n283 homozygote brains that are less than 20 hours old had many TUNEL positive nuclei due to either dying cells or DNA breaks induced by increased ROS production. (C) 0−5 day-old KG06854R brains had no TUNEL positive nuclei. (D) TUNEL positive nuclei appeared in Sod2n283/+ heterozygotes at ∼9 weeks of adulthood. (E-F) Hematoxylin-eosin stained sections of adult brains. (E) 29 day-old Sod2n283/wk transheterozygote with vigorous brain lesions. (F) 35 day-old KG06854R control with no detectable lesions.
Exposure to increased oxidative stress can cause loss of neurons (Melov et al., 1999). We assessed the structural integrity of the brain to determine whether reduced SOD2 expression is associated with neuronal loss. Brains from control flies do not exhibit signs of neuronal loss at 35 days of age (Fig. 6F) or greater ages (data not shown). Flies harboring Sod2n283 in trans to Sod2wk, however, had obvious vacuoles in the brain by as little as 29 days of age (Fig. 6E). The normal maintenance of neurons in the brain, therefore, requires SOD2 expression.
Olfactory abilities decline with age in a number of species (Grotewiel et al., 2005). We assessed odor avoidance as a function of age in flies to determine whether Sod2 influences age-related decline in olfactory behavior. Odor avoidance declined with age as expected in control flies (KG06854R) (Figure 7A and 7B). Odor avoidance across the first 5 weeks of life in Sod2wk/wk and KG06854R declined indistinguishably from that in controls (Figure 7A). In contrast, odor avoidance in flies with more severe reduction in Sod2 expression (Sod2n283/Sod2wk) declined more rapidly than in revertant controls (Figure 7B). These studies indicate that senescence of olfactory behavior in Drosophila is influenced by SOD2 and that substantial reductions in SOD2 expression are required before significant effects on age-related decline in olfactory behavior are observed.
Figure 7.
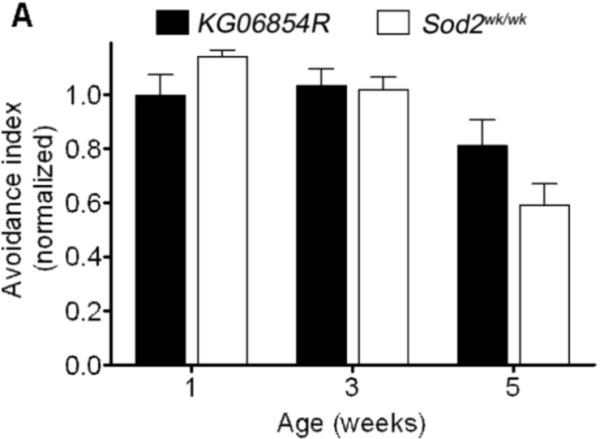
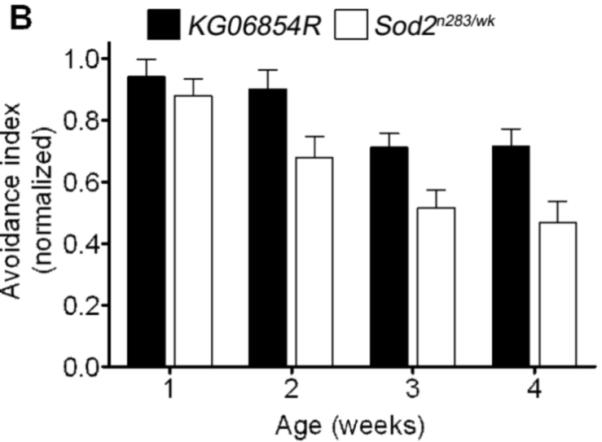
Senescence of olfactory behavior in Sod2 mutants. (A) Two-way ANOVA indicated that avoidance of MCH in KG06854R (black bars) and Sod2wk/wk flies (open bars) declined with age (p<0.0001, n = 13−16) but was not affected by genotype. (B) Avoidance of MCH in both KG06854R and Sod2n283/wk flies declined with age (two-way ANOVA, effect of age, p<0.0001, n = 28−32). MCH avoidance in the two genotypes was significantly different (p<0.0001). Data in A and B are compiled from 2 and 4 independent experiments, respectively.
Discussion
SOD2 is a key free radical scavenging enzyme within mitochondria (Landis and Tower, 2005). Severe reduction of SOD2 expression in mice and flies causes a number of phenotypes including short life span, neuronal loss, and mitochondrial dysfunction (Li et al., 1995; Lebovitz et al., 1996; Kirby et al., 2002; Duttaroy et al., 2003; Xia et al., 2006). Here, we used Drosophila to investigate the role of this enzyme in demographic mortality parameters, nervous system pathology and behavioral senescence. Drosophila Sod2 mutants have reduced mitochondrial aconitase activity, they exhibit increased apoptosis in the central nervous system along with early on-set neurodegeneration and accelerated senescence of olfactory behavior. Furthermore, life span is progressively shortened and age-specific mortality is increased in flies with reduced SOD2 activity. Thus, SOD2 might influence the on-set of age-related pathology, aging in general, or both.
Other studies have also investigated the consequences of reduced SOD2 function in model organisms. In contrast to previous studies, the data reported here were derived from a series of loss of function and hypomorphic mutations in Sod2 that allowed us to assess the consequences of progressively reducing SOD2 activity on mitochondrial function and several age-related phenotypes. Consistent with experiments in mice (Li et al., 1995; Williams et al., 1998; Lynn et al., 2005) and flies (Kirby et al., 2002), we find that the activity of mitochondrial aconitase, a citric acid cycle enzyme sensitive to oxidative damage (Das et al., 2001), is blunted in flies with reduced SOD2 function. Our data verify that SOD2 function is required to maintain mitochondrial aconitase activity and therefore mitochondrial function in animals. Interestingly, our results also show that cytoplasmic aconitase activity is not altered in Sod2 mutants, raising the possibility that the oxidative damage that occurs in these animals might be restricted largely to the mitochondria as suggested previously by studies in mice (Williams et al., 1998). In contrast to an earlier study in Drosophila where life span was measured under reduced SOD2 expression (<10% SOD2) using Sod2 RNAi (Kirby et al., 2002) this study uses bona fide Sod2 mutant alleles, which allowed us to test our hypothesis in mutant heterozygotes with 50% and 22% SOD2 activity remaining. Therefore our conclusions are based on a true gradient of SOD2 activity along with extensive demographic analysis.
The shortened life span in Drosophila Sod2 mutants reported here is consistent with previous studies in mice (Li et al., 1995; Lebovitz et al., 1996) and flies (Kirby et al., 2002; Duttaroy et al., 2003). By using a series of genotypes in which SOD2 activity was varied, however, we find that mean and maximum life span in flies is systematically shortened and their rate of aging is highly sensitive to progressive reductions in SOD2 activity. This finding highlights an intimate link between oxidative stress and life span determination. Moreover, these studies indicate that life span is closely correlated with the ability of animals to eradicate ROS at their primary cellular source, the mitochondria. One major caveat that remains with this conclusion is that Drosophila adults with ∼50% to up to 60% SOD2 activity can not achieve a normal life span yet a similar modest decrease in the amount of SOD2 enzyme did not impacted the life span of mice (Van Remmen et al., 2004). What is even more compelling is that like in Drosophila extensive tissue damages are also apparent in Sod2+/− mice yet their life span remains unaffected (Williams et al., 1998; Van Remmen 1999; Van Remmen 2001). While it is hard to reconcile this difference since catalase overexpression can extend the life span in mice (Schriner et al., 2005) but it is proposed recently that oxidative stress theory of aging can be better established in Drosophila than in mice (Muller et al., 2007). Considering the fact that certain strains of mice carries genetic modifier(s) like Nicotinamide Nucleotide Transhydrogenase which can positively influence the life span of Sod2−/− mice due to its mitochondrial antioxidant activity (Huang et al., 2006), it will be interesting to see if similar genetic modifier(s) might influence the life span of Sod2+/− mice as well. We now know that the life span of Sod2n283 remains uninfluenced in different genetic backgrounds (Duttaroy lab unpublished) nullifying the possibility of similar genomic modifiers in the Drosophila genome.
Reductions in life span can be due to alterations in initial mortality (a.k.a. baseline mortality), the rate of change in age-specific mortality (a.k.a. the mortality trajectory or demographic rate of aging), or both (Pletcher et al., 2000). Extensive demographic analyses revealed that while initial mortality was not altered in Sod2 mutants, the slope of the mortality plot was progressively increased as SOD2 activity was reduced. The increased slope in the mortality trajectory with unchanged initial mortality in Sod2 mutants seen in our studies, argues that SOD2 activity is principally required during adulthood to protect animals from progressive physiological defects associated with oxidative damage. Although no detail analysis is available at this time on how, when or if at all SOD2 activity is required during development, it is possible that there are other enzymes expressed that can compensate for the absence of SOD2 or that fewer reactive oxygen species are generated during this period (Kirby et al., 2002).
Consistent with previous reports (Mockett et al., 1999; Orr et al., 2003), we find that life span is not significantly extended in flies with overexpression of SOD2 throughout development and adulthood. In contrast, other studies show that conditional overexpression of SOD2 only during adulthood extends life span up to 33% (Sun et al., 2002; Sun et al., 2004). These apparently contradictory results can be reconciled by a model in which overexpression of SOD2 during development have negative consequences on adult life span while overexpression of SOD2 during adulthood has positive effects on adult life span. Our data showing increased initial mortality and decreased demographic rate of aging in flies constitutively overexpressing SOD2 support this model. Such a prediction seems to be gaining support from recent observations because over a certain amount SOD2 is toxic to the organism (Keaney et al 2004; Bayne et al., 2005; Ford et al., 2007). Furthermore, hyper production of SOD2 in mice can truly enhance the mitochondrial oxidative capacity (Silva et al., 2005) yet it failed to show any appreciable increase in mean life span although some increase in maximum life span was claimed (Hu et al., 2007). None of these studies however measured the Gompertz's rate of aging, so it is difficult to comment on how the mortality rate changes in these animals. Since excessive SOD might cause increased lipid peroxidation (Nelson et al., 1994; McCord and Edeas., 2005) or could disrupt signaling events requiring reactive oxygen species (Finkel, 2000; Griffiths, 2005; Gregg et al., 2004; Lo et al., 1996; Irani, 2000; Lehtinen et al., 2006) in young animals suggests that higher amounts of SOD may not be physiologically favorable therefore the existing amount of SOD allows the cell to function within a wide margin of ROS toxicity. Based on our observation we speculate that through evolution, aerobic organisms have struck a balance in their oxidative stress counter-measures and acquired the capacity to operate within a wide range (±40−50%) of mitochondrial SOD2 levels without a disastrous effect on lifespan.
It is intriguing that an association between SOD2 and neuropathology also exist in flies since this phenomenon was described in SOD2−/− mice as spongioform pathology of the brain (Melov et al., 2001, 2004). Further support for this association is incresaingly apparent because reduced SOD2 expression can modulate Alzheimer's disease like pathology in Amyloid precursor protein transgenic mice (Esposito et al., 2006) and in Drosophila brain enhanced oxidative stress resulting from less SOD2 expression facilitates tau-induced neurodegeneration (Dias-Santaga et al., 2007). Complete loss of SOD2 in mice causes a range of phenotypes related to nervous system function including lethargic locomotor behavior, seizures, ataxia, and head tremor (Li et al., 1995; Lebovitz et al., 1996; Melov et al., 1998; Lynn et al., 2005). These studies are largely qualitative and furthermore do not address whether any of these phenotypes progressively worsen with age. Our studies show that flies with 75% reductions in SOD2 activity exhibit accelerated loss of olfactory behavior as a consequence of age during the first 3−4 weeks of adulthood. These data confirm that SOD2 normally protects flies from age-dependent decline of olfactory system function. Interestingly, while flies with a 50% reduction in SOD2 activity have a greater than 20% reduction in mean life span, these animals do not have obvious changes in their olfactory behavior across age. Thus, life span and age-related decline of olfactory system function are differentially sensitive to loss of SOD2 activity.
In practice it can be difficult to determine whether manipulations that shorten life span or hasten age-related functional changes impact aging per se or, alternatively, engender a global pathological state unrelated to aging that nonetheless manifests as accelerated decline in age-related measures. Our studies with a series of Drosophila Sod2 mutants confirm the essential role of this gene in protecting flies from mitochondrial oxidative damage, neurodegeneration, age-related defects in behavior and early-onset mortality. These findings highlight the evolutionary conservation in SOD2 function from flies to mammals. Furthermore, these studies reinforce the utility of using the Drosophila model for investigating the role of this key enzyme in organismal biology.
Acknowledgement
The authors acknowledge Scott Pletcher for insightful comments on demographic analyses and Professor William Eckberg for critical reading of the manuscript and the technical assistance of Malcom Marphen. Some fly stocks were obtained from the Drosophila stock center in Bloomington, Indiana. Work supported by NIH grants AG024259 to M.S.G and U54NS039407 and AG025754-01 to A.D.
The content of this paper is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute on Neurological Disease and stroke or the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bayne VA, Mockett RJ, Orr WC, Sohal RS. Enhanced catabolism of mitochondrial superoxide/hydrogen peroxide and aging in transgenic Drosophila. Biochem J. 2005;391:277–284. doi: 10.1042/BJ20041872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barja G. Rate of generation of oxidative stress-related damage and animal longevity. Free Radic Biol Med. 2002;33:1167–1172. doi: 10.1016/s0891-5849(02)00910-3. [DOI] [PubMed] [Google Scholar]
- Bellen HJ, Levis RW, Liao G, He Y, Carlson JW, Tsang G, Evans-Holm M, Hiesinger PR, Schulze KL, Rubin GM, Hoskins RA, Spradling AC. The BDGP gene disruption project: single transposon insertions associated with 40% of Drosophila genes. Genetics. 2004;167:761–781. doi: 10.1534/genetics.104.026427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook-Wiens E, Grotewiel MS. Dissociation between functional senescence and oxidative stress resistance in Drosophila. Exp Gerontol. 2002;37:1347–57. doi: 10.1016/s0531-5565(02)00096-7. [DOI] [PubMed] [Google Scholar]
- Das N, Levine RL, Orr WC, Sohal RS. Selectivity of protein oxidative damage during aging in Drosophila melanogaster. Biochem J. 2001;360:209–16. doi: 10.1042/0264-6021:3600209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dias-Santagata D, Fulga TA, Duttaroy A, Feany MB. Oxidative stress mediates tau-induced neurodegeneration in Drosophila. J Clin Invest. 2007;117:236–245. doi: 10.1172/JCI28769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duttaroy A, Paul A, Kundu M, Belton A. A Sod2 null mutation confers severely reduced adult life span in Drosophila. Genetics. 2003;165:2295–2299. doi: 10.1093/genetics/165.4.2295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esposito L, Raber J, Kekonius L, Yan F, Yu GQ, Bien-Ly N, Puolivali J, Scearce-Levie K, Masliah E, Mucke L. Reduction in mitochondrial superoxide dismutase modulates Alzheimer's disease-like pathology and accelerates the onset of behavioral changes in human amyloid precursor protein transgenic mice. J Neurosci. 2006;26:5167–5179. doi: 10.1523/JNEUROSCI.0482-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkel T. Redox-dependent signal transduction. FEBS Lett. 2000;476:52–54. doi: 10.1016/s0014-5793(00)01669-0. [DOI] [PubMed] [Google Scholar]
- Fridovich I, Gardner PR. Superoxide sensitivity of the Escherichia coli aconitase. J Biol Chem. 1991;266:19328–193233. [PubMed] [Google Scholar]
- Ford D, Hoe N, Landis G, Tozer K, Luu A, Bhole D, Badrinath A, Tower J. Alteration of Drosophila life span using conditional, tissue-specific expression of transgenes triggered by doxycyline or RU486/Mifepristone. Experimental Gerontology. 2007;42:483–497. doi: 10.1016/j.exger.2007.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner PR, Nguyen DD, White CW. Aconitase is a sensitive and critical target of oxygen poisioning in cultured mammalian cells and in rat lungs. Proc Natl Acad Sci U S A. 1994;91:12248–12252. doi: 10.1073/pnas.91.25.12248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gompertz B. On the nature of the function expressive of the law of human mortality and on a new mode of determining life contingencies. Philos Trans R Soc Lond. 1825;115:513–585. doi: 10.1098/rstb.2014.0379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregg D, de Carvalho DD, Kovacic H. Integrins and coagulation: a role for ROS/redox signaling? Antioxid Redox Signal. 2004;6:757–64. doi: 10.1089/1523086041361604. [DOI] [PubMed] [Google Scholar]
- Griffiths HR. ROS as signalling molecules in T cells - evidence for abnormal redox signaling in the autoimmune disease, rheumatoid arthritis. Redox Rep. 2005;10:273–80. doi: 10.1179/135100005X83680. [DOI] [PubMed] [Google Scholar]
- Grotewiel MS, Martin I, Bhandari P, Cook-Wiens E. 2005Functional senescence in Drosophila melanogaster. Ageing Res Rev. 4:372–397. doi: 10.1016/j.arr.2005.04.001. [DOI] [PubMed] [Google Scholar]
- Giulietti A, Overburgh L, Valckx D, Decallonne B, Bouillon R, Mathieu C. An overview of Real-time quantitative PCR: Applications to quantify cytokine gene expression. Methods. 2001;25:386–401. doi: 10.1006/meth.2001.1261. [DOI] [PubMed] [Google Scholar]
- Harman D. Aging: A Theory Based on Free Radical and Radiation Chemistry. J. Gerontol. 1956;11:298–300. doi: 10.1093/geronj/11.3.298. [DOI] [PubMed] [Google Scholar]
- Harman D. The biologic clock: the mitochondria? J Am Geriatr Soc. 1972;20:145–147. doi: 10.1111/j.1532-5415.1972.tb00787.x. [DOI] [PubMed] [Google Scholar]
- Harman D. Free radical theory of aging: an update: increasing the functional life span. Ann N Y Acad Sci (United States) 2006;1067:10–21. doi: 10.1196/annals.1354.003. [DOI] [PubMed] [Google Scholar]
- Hekimi S, Guarente L. Genetics and the specificity of the aging process. Science. 2003;299:1351–1354. doi: 10.1126/science.1082358. [DOI] [PubMed] [Google Scholar]
- Henson CP, Cleland WW. Purification and kinetic studies of beef liver cytoplasmic aconitase. J Biol Chem. 1967;242:3833–3838. [PubMed] [Google Scholar]
- Hinerfeld D, Traini MD, Weinberger RP, Cochran B, Doctrow SR, Harry J, Melov S. Endogenous mitochondrial oxidative stress: neurodegeneration, proteomic analysis, specific respiratory chain defects, and efficacious antioxidant therapy in superoxide dismutase 2 null mice. J Neurochem. 2004;88:657–67. doi: 10.1046/j.1471-4159.2003.02195.x. [DOI] [PubMed] [Google Scholar]
- Hu D, Cao P, Edda T, Chu CT, Wu Gang-yi., Oury DT, Klann E. Hippocampul long term potentiation, memory and longevity in mice that overexpress mitochondrial superoxide dismutase. Neubiol. Learning and Memory. 2007;87:372–384. doi: 10.1016/j.nlm.2006.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang TT, Naeemuddin M, Elchuri S, Yamaguchi M, Kozy HM, Carlson EJ, Epstein CJ. Genetic modifiers of the phenotype of mice deficient in mitochondrial superoxide dismutase. Hum Mol Genet. 2006;15:1187–94. doi: 10.1093/hmg/ddl034. [DOI] [PubMed] [Google Scholar]
- Irani K. Oxidant signaling in vascular cell growth, death, and survival: a review of the roles of reactive oxygen species in smooth muscle and endothelial cell mitogenic and apoptotic signaling. Circ Res. 2000;87:179–183. doi: 10.1161/01.res.87.3.179. [DOI] [PubMed] [Google Scholar]
- Keaney M, Matthijssens F, Sharpe M, et al. Superoxide dismutase mimetics elevate superoxide dismutase activity in vivo but do not retard aging in the nematode Caenorhabditis elegans. Free Radic Biol Med. 2004;37:239–250. doi: 10.1016/j.freeradbiomed.2004.04.005. [DOI] [PubMed] [Google Scholar]
- Kirby K, Hu J, Hilliker AJ, Phillips JP. RNA interference-mediated silencing of Sod2 in Drosophila leads to early adult-onset mortality and elevated endogenous oxidative stress. Proc Natl Acad Sci U S A. 2002;99:16162–16167. doi: 10.1073/pnas.252342899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowald A. Lifespan does not measure ageing. Biogerontology. 2002;3:187–190. doi: 10.1023/a:1015659527013. [DOI] [PubMed] [Google Scholar]
- Landis GN, Tower J. Superoxide dismutase evolution and life span regulation. Mech Ageing Dev. 2005;126:365–379. doi: 10.1016/j.mad.2004.08.012. [DOI] [PubMed] [Google Scholar]
- Lebovitz RM, Zhang H, Vogel H, Cartwright J, Jr., Dionne L, Lu N, Huang S, Matzuk MM. Neurodegeneration, myocardial injury, and perinatal death in mitochondrial superoxide dismutase-deficient mice. Proc Natl Acad Sci U S A. 1996;93:9782–7. doi: 10.1073/pnas.93.18.9782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehtinen MK, Yuan Z, Boag PR, Yang Y, Villen J, Becker EB, DiBacco S, de la Iglesia N, Gygi S, Blackwell TK, Bonni A. A conserved MST-FOXO signaling pathway mediates oxidative-stress responses and extends life span. Cell. 2006;125:987–1001. doi: 10.1016/j.cell.2006.03.046. [DOI] [PubMed] [Google Scholar]
- Li Y, Huang TT, Carlson EJ, Melov S, Ursell PC, Olson JL, Noble LJ, Yoshimura MP, Berger C, Chan PH, et al. Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nat Genet. 1995;11:376–381. doi: 10.1038/ng1295-376. [DOI] [PubMed] [Google Scholar]
- Lo YY, Wong JM, Cruz TF. Reactive oxygen species mediate cytokine activation of c-Jun NH2-terminal kinases. J Biol Chem. 1996;271:15703–15707. doi: 10.1074/jbc.271.26.15703. [DOI] [PubMed] [Google Scholar]
- Lynn S, Huang EJ, Elchuri S, Naeemuddin M, Nishinaka Y, Yodoi J, Ferriero DM, Epstein CJ, Huang TT. Selective neuronal vulnerability and inadequate stress response in superoxide dismutase mutant mice. Free Radic Biol Med. 2005;38:817–28. doi: 10.1016/j.freeradbiomed.2004.12.020. [DOI] [PubMed] [Google Scholar]
- Longo VD, Finch CE. Evolutionary medicine: from dwarf model systems to healthy centenarians? Science. 2003;299:1342–1346. doi: 10.1126/science.1077991. [DOI] [PubMed] [Google Scholar]
- McCord JM, Edeas MA, SOD oxidative stress and human pathologies: a brief history and a future vision. Biomed Pharmacother. 2005;59:139–42. doi: 10.1016/j.biopha.2005.03.005. [DOI] [PubMed] [Google Scholar]
- Melov S, Coskun P, Patel M, Tuinstra R, Cottrell B, Jun AS, Zastawny TH, Dizdaroglu M, Goodman SI, Huang TT, Miziorko H, Epstein CJ, Wallace DC. Mitochondrial disease in superoxide dismutase 2 mutant mice. Proc Natl Acad Sci U S A. 1999;96:846–551. doi: 10.1073/pnas.96.3.846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melov S, Schneider JA, Day BJ, Hinerfeld D, Coskun P, Mirra SS, Crapo JD, Wallace DC. A novel neurological phenotype in mice lacking mitochondrial manganese superoxide dismutase. Nat Genet. 1998;18:159–63. doi: 10.1038/ng0298-159. [DOI] [PubMed] [Google Scholar]
- Melov S, Doctrow SR, Schneider JA, Haberson J, Patel M, Coskun PE, Huffman K, Wallace DC, Malfroy B. Lifespan extension and rescue of spongiform encephalopathy in superoxide dismutase 2 nullizygous mice treated with superoxide dismutase-catalase mimetics. J Neurosci. 2001;21:8348–53. doi: 10.1523/JNEUROSCI.21-21-08348.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mockett RJ, Orr WC, Rahmandar JJ, Benes JJ, Radyuk SN, Klichko VI, Sohal RS. Overexpression of Mn-containing superoxide dismutase in transgenic Drosophila melanogaster. Arch Biochem Biophys. 1999;371:260–9. doi: 10.1006/abbi.1999.1460. [DOI] [PubMed] [Google Scholar]
- Muller FL, Lustgarten MS, Jang Y, Richardson A, Van Remmen H. Trends in oxidative aging theories. Free Radic. Biol. Med. 2007;43:477–503. doi: 10.1016/j.freeradbiomed.2007.03.034. [DOI] [PubMed] [Google Scholar]
- Murphy CT, McCarroll SA, Bargmann CI, Fraser A, Kamath RS, Ahringer J, Li H, Kenyon C. Genes that act downstream of DAF-16 to influence the lifespan of Caenorhabditis elegans. Nature. 2003;424:277–283. doi: 10.1038/nature01789. [DOI] [PubMed] [Google Scholar]
- Nelson SK, Bose SK, McCord JM. The toxicity of high-dose superoxide dismutase suggests that superoxide can both initiate and terminate lipid peroxidation in the reperfused heart. Free Radic Biol Med. 1994;16:195–200. doi: 10.1016/0891-5849(94)90143-0. [DOI] [PubMed] [Google Scholar]
- Orr WC, Mockett RJ, Benes JJ, Sohal RS. Effects of Overexpression of Copper-Zinc and Manganese Superoxide Dismutases, Catalase, and Thioredoxin Reductase Genes on Longevity in Drosophila melanogaster. J, Biol. Chem. 2003;278:26418–26422. doi: 10.1074/jbc.M303095200. [DOI] [PubMed] [Google Scholar]
- Pletcher SD. Model fitting and hypothesis testing for age-specific mortality data. J. Evol. Biol. 1999;12:430–439. [Google Scholar]
- Pletcher SD, Khazaeli AA, Curtsinger JW. Why do life spans differ? Partitioning mean longevity differences in terms of age-specific mortality parameters. J Gerontol A Biol Sci Med Sci. 2000;55:B381–9. doi: 10.1093/gerona/55.8.b381. [DOI] [PubMed] [Google Scholar]
- Racker E. Spectrophotometric measurements of the enzymatic formation of fumaric and cis-aconitic acids. Biochim Biophys Acta. 1950;4:211–214. doi: 10.1016/0006-3002(50)90026-6. [DOI] [PubMed] [Google Scholar]
- Schriner SE, Linford NJ, Martin GM, Treuting P, Ogburn CE, Emond M, Coskun PE, Ladiges W, Wolf N, Van Remmen H, Wallace DC, Rabinovitch PS. Extension of murine life span by overexpression of catalase targeted to mitochondria. Science. 2005;24(5730):1909–1911. doi: 10.1126/science.1106653. 308. [DOI] [PubMed] [Google Scholar]
- Silva JP, Shabalina IG, Dufour E, Petrovic N, Backlund EC, Hultenby K, Wibom1 R, Nedergaard J, Cannon B, Larsso Nils-Go ran SOD2 overexpression: enhanced mitochondrial tolerance but absence of effect on UCP activity. The EMBO Journal. 2005;24:4061–4070. doi: 10.1038/sj.emboj.7600866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohal RS, Sohal BH. Hydrogen peroxide release by mitochondria increases during aging. Mech Ageing Dev. 1991;57:187–202. doi: 10.1016/0047-6374(91)90034-w. [DOI] [PubMed] [Google Scholar]
- Sohal RS, Dubey A. Mitochondrial oxidative damage, hydrogen peroxide release, and aging. Free Radic Biol Med. 1994;16:621–626. doi: 10.1016/0891-5849(94)90062-0. [DOI] [PubMed] [Google Scholar]
- Sohal RS, Weindruch R. Oxidative stress, caloric restriction, and aging. Science. 1996;273:59–63. doi: 10.1126/science.273.5271.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohal RS, Mockett RJ, Orr WC. Mechanisms of aging: an appraisal of the oxidative stress hypothesis. Free Radic Biol Med. 2002;33:575–586. doi: 10.1016/s0891-5849(02)00886-9. [DOI] [PubMed] [Google Scholar]
- Sohal RS. Oxidative stress hypothesis of aging. Free Radic Biol Med. 2002;33:573–4. doi: 10.1016/s0891-5849(02)00885-7. [DOI] [PubMed] [Google Scholar]
- Stoltzfus JR, Horton WJ, Grotewiel MS. Odor-guided behavior in Drosophila requires calreticulin. J Comp Physiol A Neuroethol Sens Neural Behav Physiol. 2003;189:471–83. doi: 10.1007/s00359-003-0425-z. [DOI] [PubMed] [Google Scholar]
- Sun J, Folk D, Bradley TJ, Tower J. Induced overexpression of mitochondrial Mnsuperoxide dismutase extends the life span of adult Drosophila melanogaster. Genetics. 2002;161:661–672. doi: 10.1093/genetics/161.2.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Jingtao., Molitor J, Tower J. Effects of simultaneous over-expression of Cu/ZnSOD and MnSOD on Drosophila melanogaster life span. Mech Aging and Dev. 2004;125:341–349. doi: 10.1016/j.mad.2004.01.009. 2004. [DOI] [PubMed] [Google Scholar]
- Tatar M, Bartke A, Antebi A. The endocrine regulation of aging by insulin-like signals. Science. 2003;299:1346–1351. doi: 10.1126/science.1081447. [DOI] [PubMed] [Google Scholar]
- Van Remmen H, Salvador C, Yang H, Huang TT, Epstein CJ, Richardson A. Characterization of the antioxidant status of the heterozygous manganese superoxide dismutase knockout mouse. Arch Biochem Biophys. 1999;363(1):91–97. doi: 10.1006/abbi.1998.1060. [DOI] [PubMed] [Google Scholar]
- Van Remmen H, Ikeno Y, Hamilton M, et al. Life-long reduction in MnSOD activity results in increased DNA damage and higher incidence of cancer but does not accelerate aging. Physiol. Genomics. 2004;16:29–37. doi: 10.1152/physiolgenomics.00122.2003. [DOI] [PubMed] [Google Scholar]
- Van Remmen H, Williams MD, Guo Z, Estlack L, Yang H, Carlson EJ, Epstein CJ, Huang TT, Richardson A. Knockout mice heterozygous for Sod2 show alterations in cardiac mitochondrial function and apoptosis. Am J Physiol Heart Circ Physiol. 2001;281:H1422–32. doi: 10.1152/ajpheart.2001.281.3.H1422. 2001. [DOI] [PubMed] [Google Scholar]
- Williams MD, Van Remmen H, Conrad CC, Huang TT, Epstein CJ, Richardson A. Increased oxidative damage is correlated to altered mitochondrial function in heterozygous manganese superocide dismutase knock out mice. J. Biol.Chem. 1998;273:28510–28415. doi: 10.1074/jbc.273.43.28510. [DOI] [PubMed] [Google Scholar]
- Xia XG, Zhou H, Samper E, Melov S, Xu Z. Pol II-expressed shRNA knocks down Sod2 gene expression and causes phenotypes of the gene knockout in mice. PLoS Genet. 2006:2–10. doi: 10.1371/journal.pgen.0020010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarian CS, Toroser D, Sohal RS. Aconitase is the main functional target of aging in the citric acid cycle of kidney mitochondria from mice. Mech Ageing Dev. 2006;127:79–84. doi: 10.1016/j.mad.2005.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]