

Abstract
Mice deficient in the Flk-1 receptor tyrosine kinase are known to die in utero because of defective vascular and hematopoietic development. Here, we show that flk-1−/− embryonic stem cells are nevertheless able to differentiate into hematopoietic and endothelial cells in vitro, although they give rise to a greatly reduced number of blast colonies, a measure of hemangioblast potential. Furthermore, normal numbers of hematopoietic progenitors are found in 7.5-day postcoitum flk-1−/− embryos, even though 8.5-day postcoitum flk-1−/− embryos are known to be deficient in such cells. Our results suggest that hematopoietic/endothelial progenitors arise independently of Flk-1, but that their subsequent migration and expansion require a Flk-1-mediated signal.
The vascular endothelial growth factor (VEGF) receptor Flk-1 is known to play a key role in the regulation of embryonic vascular and hematopoietic development. In the mouse embryo, flk-1 expression can be detected in presumptive mesodermal yolk-sac blood-island progenitors as early as 7.0 days postcoitum (dpc) (1, 2). As has been described recently (3), mice deficient in Flk-1 do not develop blood vessels or yolk sac blood islands and die between 8.5 and 9.5 dpc. In chimeric aggregation studies with wild-type embryos, flk-1−/− embryonic stem (ES) cells fail to participate in vessel formation or to contribute to primitive or definitive hematopoiesis in vivo, suggesting that Flk-1 inactivation results in a cell autonomous endothelial and hematopoietic defect (4). Taken together, these studies suggest that Flk-1 may mark the common endothelial/hematopoietic precursor, the hemangioblast. The recently identified blast colony-forming cell (BL-CFC), an in vitro ES cell derivative, has confirmed the existence of such a cell. In response to VEGF, BL-CFCs form blast colonies containing Flk-1-expressing blast cells that give rise to primitive, definitive hematopoietic and endothelial cells in vitro (5, 6). Taken together, these findings strongly suggest that the VEGF/Flk-1 interaction is important for hemangioblast development.
Although all of these studies suggest that the Flk-1 receptor is important in embryonic endothelial and hematopoietic development, the precise nature of its role remains undefined. In particular, the contribution of Flk-1 to hemangioblast formation is unclear, as flk-1−/− and VEGF+/− mice show residual hematopoietic and endothelial activity (3, 7, 8) and Flk-1-deficient ES cells appear to retain a degree of hematopoietic potential in vitro (4). To define the developmental role of Flk-1 more systematically, we have characterized the hematopoietic and endothelial potential of flk-1−/− ES cells and embryos in greater detail. And in particular, as the BL-CFC assay should allow the developmental consequences of Flk-1 deficiency during the very early VEGF-dependent stage of hematopoiesis to be analyzed quantitatively (5), we have assessed the ability of flk-1−/− ES cells to give rise to blast colonies. We report that Flk-1-deficient BL-CFCs appear to develop normally within flk-1−/− embryoid bodies (EBs), but fail to respond to VEGF in forming blast colonies. Consequently, the number of blast colonies is greatly reduced. Nevertheless, Flk-1-deficient blast cells retain the potential to differentiate into hematopoietic and endothelial cells in culture. Consistent with this, flk-1−/− vs. flk-1+/+ or +/−EBs show similar levels of hematopoietic and endothelial cell differentiation potential. We also demonstrate that 7.5-dpc flk-1−/− embryos contain normal numbers of hematopoietic progenitors, even though 8.5-dpc flk-1−/− embryos are known to be profoundly deficient in such cells. Taken together, our results suggest that Flk-1 is required not for the formation of hematopoietic/endothelial progenitors but rather for the subsequent migration and expansion of such cells.
MATERIALS AND METHODS
Flk-1 Protein Analysis.
Day 7 EBs were pelleted and lysed in 1 ml of lysis buffer (1% Nonidet P-40/50 mM Tris⋅HCl, pH 7.5/150 mM NaCl/2 mM MgCl2/1 μg/ml aprotinin/1 μg/ml leupeptin/100 μg/ml phenylmethylsulfonyl fluoride) on ice for 30 min with occasional mixing. The lysate was then spun at 14,000 rpm for 30 min, the resultant supernatant was incubated with normal rabbit serum (25 μl/ml lysate) for 1 hr on ice, 200 μl protein A Sepharose CL-4B (Pharmacia) beads (100 μg/ml) were added, and the mixture was incubated at 4°C for another hour with rocking. The sample was then spun at 13,000 rpm for 2 min, the supernatant was removed and mixed with Flk-1 antiserum (1 μg/ml final concentration) and incubated on ice for 1 hr, followed by the addition of 100 μl protein A Sepharose CL-4B beads (100 μg/ml) and incubation on ice with rocking for 1 hr. Immune complex-containing beads were then collected by spinning at 4°C for 15 sec and were washed three times in cold lysis buffer. Beads were mixed with gel-loading buffer (2% SDS/100 mM DTT/60 mM Tris⋅HCl, pH 6.8), boiled for 5 min, size separated by SDS/PAGE, and transferred to nitrocellulose. Finally, Flk-1-specific bands were detected with Flk-1 antiserum followed by chemiluminescent detection.
Cell Culture.
ES cells were maintained on STO feeder cells in the presence of leukemia inhibitory factor (LIF). EBs, blasts, primitive erythroid (EryP), and myeloid colonies were generated as described previously (5). Briefly, blast colonies were generated by replating day 2.75 EBs in the presence of either VEGF (5 ng/ml), kit ligand (KL, 1% conditioned medium or 100 ng/ml purified) and D4T endothelial cell conditioned medium (25%) or VEGF (5 ng/ml) and D4T conditioned medium (25%). For the generation of endothelial cells, individual blast colonies were transferred to matrigel-coated (Collaborative Research) microtiter wells containing Iscove’s modified Dulbecco’s medium with 10% fetal calf serum (FCS) (HyClone), 10% horse serum (Biocell Laboratories), VEGF (5 ng/ml), insulin-like growth factor-1 (10 ng/ml), Epo (2 units/ml), basic fibroblast growth factor (10 ng/ml), interleukin-11 (50 ng/ml), KL (1% of the sup), endothelial cell growth supplement (100 ng/ml; Collaborative Research), l-glutamine (2 mM), and 4.5 × 10−4 M monothioglycerol (MTG) (Sigma). After 3 to 4 days of culture, nonadherent hematopoietic cells were removed by gentle pipetting, and the adherent cells were cultured for an additional 1–2 weeks in Iscove’s modified Dulbecco’s medium with 10% FCS (HyClone), 10% horse serum, VEGF (5 ng/ml), insulin-like growth factor-1 (10 ng/ml), basic fibroblast growth factor (10 ng/ml), endothelial cell growth supplement, 100 ng/ml; Collaborative Research), l-glutamine (2 mM), and 4.5 × 10−4 M MTG (Sigma) and then harvested by trypsinization for gene expression studies. EryP colonies were analyzed by replating day 4 EBs with plasma-derived serum (Antech; 10%), ascorbic acid (12.5 ng/ml), L-glutamine (2 mM), transferrin (300 ng/ml; Boehringer Mannheim), protein free hybridoma media II (GIBCO/BRL; 5%), and MTG (4.5 × 10−4 M) with erythropoietin (Amgen Biologicals, 2 units/ml). One ml of methylcellulose cultures containing 4 × 104 cells were put in 30 mm bacterial grade Petri dishes. EryP colonies were counted 4–6 days after replating. Other myeloid colonies were analyzed by replating day 9–10 EBs with plasma-derived serum (10%, Antech, Tyler, TX), ascorbic acid (12.5 ng/ml), l-glutamine (2 mM), transferrin (300 ng/ml; Boehringer Mannheim), protein-free hybridoma media II (GIBCO/BRL; 5%), and MTG (4.5 × 10−4 M) containing erythropoietin (2 units/ml), KL (1%), interleukin (IL)-3 (1%), IL-11 (25 ng/ml), granulocyte—macrophage colony-stimulating factor (CSF) (3 ng/ml), macrophage CSF (5 ng/ml), granulocyte CSF (30 ng/ml), IL-6 (5 ng/ml), LIF (1 ng/ml), and VEGF (5 ng/ml). Hematopoietic colonies were counted 7–10 days after the replating. IL-11, granulocyte—macrophage CSF, macrophage CSF, IL-6, LIF, and VEGF were purchased from R & D Systems. KL was obtained from medium conditioned by Chinese hamster ovary cells transfected with a KL expression vector (kindly provided by Genetics Institute) or purchased from R & D Systems. IL-3 was obtained from medium conditioned by X63 Ag8–653 myeloma cells transfected with a vector expressing IL-3 (9).
Gene Expression Analysis.
RNA isolation and reverse transcription—PCR (RT-PCR) were performed as described (10, 11). All RNA samples were treated with DNase I (amplification grade from GIBCO/BRL) before cDNA synthesis to eliminate any contaminating genomic DNA. Specific primers used are as follows (5). Primers for hprt, βmajor, and βH1 span the intron, such that ≈1,100 bp (hprt), 1,296 bp (βmajor), and 1,053 bp (βH1) genomic DNA fragment will be amplified. None of the samples showed genomic DNA amplification (not shown). The size of PCR products is indicated in parentheses.
hprt, sense, 5′-GCTGGTGAAAAGGACCTCT-3′; antisense, 5′-CACAGGACTAGAACACCTGC-3′ (249 bp). β-actin, sense, 5′-ATGAAGATCCTGACCGAGCG-3′; antisense 5′-TACTTGCGCTCAGGAGGA GC-3′ (443 bp). flk-1, sense, 5′-GCCCTGAGTCCTCAGGAC-3′; antisense, 5′-GGTCTCCACGCAGAACC-3′ (320 bp). lacZ (flk-1), sense, 5′-GCCCTGAGTCCTCAGGAC-3′; antisense, 5′-AAAGCGCCATTCGCCATTCA-3′ (320bp). α-globin, sense, 5′-CTCTCTGGGGAAGACAAAAGCAAC-3′; antisense, 5′-GGTGGCTAGCCAAGGTCACCAGCA-3′ (331 bp). flt-1, sense, 5′-TGTGGAGAAACTTGGTGACCT-3′; antisense, 5′-TGGAGAACAGCAGGACTGATGAT-3′ (504 bp). βH1, sense, 5′-AGTCCCCATGGAGTCAAAGA-3′; antisense, 5′-CTCAAGGAGACCTTTGCTCA-3′ (265 bp). βmajor, sense, 5′-CTGACAGATGCTCTCTTGGG-3′; antisense, 5′-CACAACCCCAGAAACAGACA-3′ (578 bp). tie-1, sense, 5′-GTCACTGCCCTCCTGACTGG-3′; antisense, 5′-CGATGTACTTGGATATAGGC-3′ (208 bp). PECAM-1, sense, 5′-GTCATGGCCGTCGAGTA-3′; antisense, 5′-CTCCTCGGCATCTTGCTGAA-3′ (260 bp). tie-2, sense, 5′-TTGAAGTGACGAATGAGAT-3′; antisense, 5′-ATTTAGAGCTGTCTGGCTGCTT-3′ (197 bp). VWF, sense, 5′-GTGGTGGGCATGATGGAGAGGTTA-3′, antisense, 5′-GCAAGGTCACAGAGGTAGCTGACT-3′ (484 bp).
Fluorescein di-β-d-galactopyranoside (FDG)/Fluorescence-Activated Cell Sorter Analysis.
EBs were trypsinized to produce a single-cell suspension. One × 106 cells were resuspended in PBS/10% FCS (HyClone)/10 mM Hepes, pH 7.2/300 μM chloroquine and incubated at 37°C for 30 min. Prewarmed 2 mM FDG (Molecular Probes) was added in equal volume to the cells and incubated for exactly 1 min. The FDG loaded into the cells (hypotonic state) and an isotonic shock was introduced with the addition of 1.8 ml of ice-cold staining medium [PBS/10% HyClone FBS/300 μM chloroquine/1 mM phenylethyl β-d-thiogalactopyranoside (PETG) and 1 μg/ml propidium iodide]. Cells were maintained at 4°C and in the dark until analysis. Endogenous β-galactosidase present in the lysosome was inhibited by pretreatment with chloroquine. In addition, a competitive inhibitor, PETG, was added to stop the reaction by outcompeting FDG from being cleaved by β-galactosidase.
Embryo Manipulation.
Embryos were removed at 7.5 dpc (noon on the day of the vaginal plug is 0.5 days) and were processed immediately for methylcellulose hematopoietic colony assay with factors described above. At the time of dissection, ectoplacental cone and trophoblast cells corresponding to each embryo were cultured in ES cell medium without LIF for 10–14 days to ensure loss of maternal cells. Embryos were then genotyped by Southern blot analysis of cultured cell DNA (not shown).
RESULTS
flk-1−/− EBs Give Rise to Reduced Numbers of Blast Colonies.
As previously described (4), flk-1−/− ES cells were derived by G418 selection from flk-1+/− cells produced by a gene-targeting event in which the lacZ gene was placed under the transcriptional control of flk-1 regulatory elements by insertion into the first exon of flk-1. β-galactosidase activity therefore acts as a histochemical marker of Flk-1 expression. To ensure that the formation of hematopoietic or endothelial cells in vitro could not be attributed to contamination of flk-1−/− cells with +/− or +/+ cells, we first verified that in vitro-differentiated flk-1−/− EBs did not produce Flk-1 and did not contain flk-1 transcripts (Fig. 1A).
Figure 1.

(A) flk-1−/− EBs do not express Flk-1 protein or flk-1 RNA. Lysates of EBs derived from flk-1−/− and +/− ES cells were analyzed by immunoprecipitation/immunoblotting by using polyclonal Flk-1 antiserum as described (19). The ≈200-kDa Flk-1-specific band is not seen in lysates from flk-1−/− EBs (Upper). A Flk-1-specific band was also not detected in lysates of [35S] methionine-labeled flk-1−/− EBs, while it was readily detected in flk-1+/−- derived samples (not shown). Total RNA was extracted from flk-1−/−, +/−, and +/+ EBs, cDNA was prepared, and PCR was performed with flk-1-specific probes as described (10, 11, and see Materials and Methods). Whereas flk-1-specific bands were detected in flk-1+/− and +/+ samples, they were not found in EBs derived from three independently derived flk-1−/− ES clones (Lower). Control PCRs performed with RNA in the absence of reverse transcription were negative in all cases. HPRT-specific RT-PCR serves as an RNA/cDNA loading control. −/−, flk-1−/−; +/−, flk-1+/−; +/+, flk-1+/+; numbers on left indicate position of molecular weight standards. (B) flk-1−/− ES cells give rise to greatly reduced numbers of blast colonies. The number of blast colonies from day 2.75 flk-1−/−, flk-1+/−, and +/+ EBs is shown. Similar results were obtained from three independent replatings. An average number from one such replating, obtained from triplicate plates, is shown. Error bars indicate standard deviations. Whereas the number of blast colonies from flk-1+/− EBs was somewhat lower than that from the flk-1+/+ EBs, the significance of the difference is not clear. R1, wild-type R1 ES cells; 8145, a flk-1+/− ES cell line; 47, a flk-1−/− ES cell line. (C) flk-1 and lacZ expression in flk-1+/+, +/−, and −/− blast colonies. Expression of flk-1, lacZ, and β-actin was determined by RT-PCR with 32P dCTP added to the PCR reaction. Bands were then visualized by autoradiography. −/−, flk-1−/−; +/−, flk-1+/−; H2O, PCR negative control. Day 6 EB cDNA, positive control.
Because the combined hematopoietic/endothelial defect observed in flk-1−/− embryos was consistent with impaired hemangioblast formation, we anticipated that the BL-CFC assay might allow the developmental consequences of Flk-1 deficiency during the very early VEGF-dependent stage of hematopoiesis and vasculogenesis to be analyzed quantitatively. We therefore assessed the ability of flk-1−/−, +/−, and +/+ ES cells to give rise to BL-CFCs, an in vitro measure of hemangioblast potential. When blast colony numbers were compared, far fewer blast colonies were obtained from flk-1−/− EB cells compared with flk-1+/− and +/+ cells (Fig. 1B). Moreover, the blast colonies produced from flk-1−/− ES cells were much smaller than those from +/− and +/+ cells (data not shown), suggesting that the proliferative capacity of blast cells is limited in the absence of Flk-1.
Because blast colonies are believed to arise from Flk-1-expressing progenitors in a VEGF-dependent manner, it was unclear how they could have developed in flk-1−/− EBs. Because it was possible that the blast colonies from flk-1−/− EBs could have originated from Flk-1-negative progenitors in response to some other factor(s) present in the blast colony replating medium, the cell origin of blast cells from flk-1−/− EBs was determined. We therefore analyzed individual flk-1+/+, +/−, and −/− blast colonies (28, 23, and 23, respectively) for flk-1 and β-galactosidase expression. By virtue of the lacZ gene-targeting event that underlies flk-1 inactivation, Flk-1-deficient cells that express β-galactosidase would normally express flk-1. As illustrated in Fig. 1C, all flk-1+/+ and flk-1+/− blast colonies expressed flk-1. In addition, all flk-1+/− blast colonies analyzed expressed lacZ. Whereas flk-1−/− blast colonies did not, as expected, express flk-1, they did however express lacZ. Thus, it is most likely that blast colonies from flk-1−/− EBs have arisen from the same Flk-1 (or β-galactosidase)-expressing precursors as have their +/− and +/+ counterparts.
Flk-1-Deficient Cells Proliferate Normally Within flk-1−/− EBs.
Whereas the reduced number of blast colonies found in flk-1−/− EBs was consistent with impaired BL-CFC formation in the absence of Flk-1, it was also possible that flk-1−/− BL-CFCs have developed normally, but then have failed to respond to VEGF to form blast colonies. The observation that flk-1−/−, +/−, and +/+ EBs were indistinguishable in size (data not shown) suggested that Flk-1-deficient cells were capable of extensive proliferation within flk-1−/− EBs in non-VEGF-dependent manners. To determine whether Flk-1-deficient cells developed normally within flk-1−/− EBs, we used β-galactosidase expression to identify Flk-1-deficient (but β-galactosidase+) cells within flk-1−/− EBs. β-galactosidase-expressing cells were quantified in days 2.75–9 flk-1−/− and +/− EBs by fluorescence-activated cell sorter analysis by using the fluorescein-conjugated β-galactosidase substrate FDG (12, 13). As shown in Table 1, flk-1−/− and +/− β-galactosidase+ cell numbers were similar in day 2.75 EBs and increased in parallel with ongoing EB differentiation. Thus, Flk-1-deficient precursors were able to proliferate normally within flk-1−/− EBs. We concluded therefore that the reduction of blast colony number and size was most likely because of the failure of Flk-1-deficient BL-CFCs to proliferate in response to VEGF when they are replated in methylcellulose cultures.
Table 1.
Kinetic analysis of β-galactosidase-expressing cells in flk-1+/+, +/−, and −/− EBs
| Exp | D2.75 | D4 | D6 | D9 | |
|---|---|---|---|---|---|
| 1 | flk-1+/− | 3.3 | 31.97 | 18.83 | |
| flk-1−/− | 4.6 | 47.26 | 18.75 | ||
| 2 | flk-1+/− | 3.9 | 18.9 | 23.9 | |
| flk-1−/− | 2.8 | 19.0 | 24.3 |
β-galactosidase-expressing cells were stained with FDG and quantified by FACS. The % of β-galactosidase-expressing cells present in day 2.75-9 EBs in two separate representative experiments is shown. The percentage of β-galactosidase-expressing cells in flk-1+/− EBs was similar to that of Flk-1-expressing cells as judged by Flk-1 monoclonal antibody staining (ref. 20, not shown).
The Developmental Potential of Flk-1-Deficient (lacZ-Expressing) Blast Cells in Vitro Is Not Impaired.
Because blast cells have been shown to give rise to both hematopoietic and endothelial cells (5), we next determined whether Flk-1- deficient blast cells retained this ability. Individual blast colonies were therefore transferred from methylcellulose to liquid culture containing both hematopoietic and endothelial cell growth factors (5). Sixty-nine of 156 (44%) and 29 of 72 (40%) of blast colonies from day 2.75 flk-1+/+ and flk-1+/− EBs, respectively, gave rise to both hematopoietic and endothelial cells. More importantly, 92 of 280 (33%) of flk-1−/− blast colonies, from day 2.75, also gave rise to both lineages, as assessed by the formation of hematopoietic cells with signs of hemoglobinization and adherent cells. The adherent cells that developed from flk-1−/− blast colonies were indistinguishable in morphology from those from +/+ or +/− blast cells and all expressed PECAM-1, tie-1, and tie-2, characteristic of endothelial cells (Fig. 2). Furthermore, while adherent cells derived from flk-1+/+ and +/− blast colonies expressed flk-1, all flk-1−/−-derived cells expressed lacZ. BL-CFCs found in flk-1−/− EBs therefore retain the potential for both endothelial and hematopoietic differentiation. Thus, Flk-1 expression is not an absolute requirement for the formation, or differentiation, of BL-CFCs/hemangioblasts.
Figure 2.

Expression of endothelial markers in blast colony-derived adherent cells. RNA obtained from adherent cells generated from flk-1+/+, +/−, and −/− blast colonies was analyzed by RT-PCR for the expression of flk-1, lacZ, and other endothelial transcripts. c-fms expression level was high in the first sample in the −/− series (lane 10 from the left, not shown). ∗, not done. H2O, PCR negative control. Day 6 EB RNA, positive control.
flk-1−/− ES Cells Are Capable of Hematopoietic and Endothelial Differentiation in Vitro.
Based on the observation that flk-1−/− ES cells were able to give rise to BL-CFCs that retained dual-lineage hemangioblast potential, we proceeded to determine the extent to which hematopoietic/endothelial differentiation potential was preserved. We therefore quantified hematopoietic progenitors found in day 4 and day 9 flk-1−/−, +/−, and +/+ EBs (14). As shown in Fig. 3A, flk-1−/− EBs contained a similar number of EryP progenitors compared with those of flk-1+/+ and flk-1+/− EBs. Furthermore, in contrast to the profound hematopoietic deficiency observed in 8.5 dpc flk-1−/− embryos (3), the ability of flk-1−/− ES cells to give rise to definitive erythroid and to macrophage/myeloid progenitors was indistinguishable from that of their heterozygous counterparts (Fig. 3B). Thus, the hematopoietic differentiation of flk-1−/− ES cells in vitro did not recapitulate the block to hematopoiesis previously observed in Flk-1-deficient embryos (3) and did not appear to manifest the cell autonomous hematopoietic defect seen in chimeric aggregation studies (4).
Figure 3.


(A) flk-1−/− ES cells give rise to EryP colonies in vitro. EryP progenitors were quantified from day 4 flk-1−/−, +/− and +/+ EBs using methylcellulose colony assay. R1, wild-type R1 ES cells; 8145, a flk-1+/− line; 47/48, two independent flk-1−/− ES cell lines. Results obtained from three independent replatings were similar. An average number from one such replating, obtained from triplicate plates, is shown. Error bars indicate standard deviations. The difference in the number of EryP colonies between 47 and 48 could reflect clonal variations. (B) flk-1−/− ES cells give rise to definitive erythroid and myeloid progenitors. Types of day 9 colonies produced: MAC, macrophage; ERYD, definitive erythroid; Ery/MAC, mixed erythroid/macrophage; GM, granulocyte/macrophage; Mast, mast cell; Mix, mixed myeloid. Results obtained from three independent replatings were similar. An average number from one such replating, obtained from triplicate plates, is shown. Error bars indicate standard deviations. In all cases, while clonal variation is observed among cell lines used, overall, the hematopoietic potential of flk-1−/− cells was indistinguishable from that of their +/− and +/+ counterparts. (C) Appearance of endothelial marker genes during differentiation of flk-1−/−, +/−, +/+ ES cells. RT-PCR was performed with total RNA prepared from timed EBs, but with primers specific to a series of endothelial marker genes. The appearance of endothelial markers in flk-1−/− day 3–day 7 EBs is indistinguishable from that of their +/− and +/+ counterparts. Marker gene expression was not detectable on day 0 (not shown). Transcripts detected are indicated on the right; d3–d7, days of differentiation; −/−, flk-1−/−; +/−, flk-1+/−; +/+, flk-1+/+.
In parallel with this study, we also examined the in vitro endothelial differentiation potential of flk-1−/− ES cells. Embryonic vascular development is characterized both in vivo and in vitro by the sequential activation of a series of “endothelial-specific” genes in a manner that is believed to reflect successive endothelial maturation steps (15). We therefore examined the expression of a series of such endothelial marker genes by RT-PCR at sequential time points of in vitro differentiation. As shown in Fig. 3C, with the exception of flk-1 transcripts, which were absent, the appearance of flt-1, tie-1, tek (tie-2), PECAM-1, and VWF transcripts during flk-1−/− ES cell differentiation was indistinguishable from that observed in flk-1+/− and +/+ EBs. Notably, flk-1−/− EBs contained tie-1 transcripts, which were absent in Flk-1-deficient embryos (3). Thus, as we had already observed in our hematopoietic assays, Flk-1-deficient endothelial differentiation proceeded further in vitro than in vivo, as well.
The in Vitro Hematopoietic Potential of flk-1−/− ES Cells Is Assay Dependent.
To determine whether the in vitro hematopoietic and endothelial potential of flk-1−/− ES cells might somehow be related to the specific culture conditions used, we next assessed their hematopoietic potential by using serum-free conditions (16). In this system, ES cells are differentiated in chemically defined medium (CDM) in the absence of serum, thereby avoiding the potentially confounding influence of factors found in serum. Hematopoietic differentiation can be triggered by the addition of bone morphogenetic protein-4 (BMP-4), which induces the formation of postero-ventral mesoderm, and the onset of hematopoiesis is scored by the detection of embryonic globin (βHI) mRNA by RT-PCR. As shown in Fig. 4, the addition of serum to flk-1+/− and −/− ES cells differentiating in CDM resulted in the induction of βHI expression. In addition, as expected, differentiation in CDM in the absence of BMP-4 did not result in βHI expression by either cell type. The addition of BMP-4, however, induced βHI expression in flk-1+/− cells but not in their −/− counterparts. Thus, under these conditions, Flk-1-deficient ES cells did appear to manifest a hematopoietic defect that could be identified in vitro.
Figure 4.
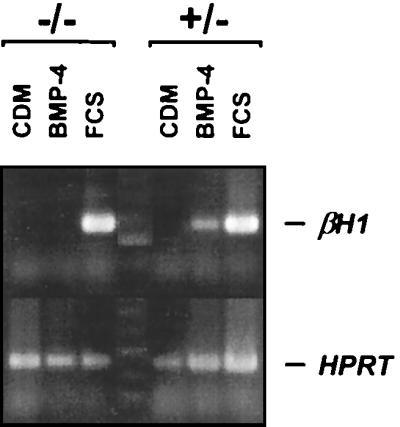
Flk-1-deficient ES cells do not initiate βHI globin expression under BMP-4-induced serum free differentiation conditions. flk-1−/− and +/− ES cells were differentiated in CDM in the presence or absence of added FCS or BMP-4, as described. Specifically, CDM contained Iscove’s modified Dulbecco’s medium plus Ham’s F-12 medium (both from GIBCO/BRL, Life Technologies, Grand Island, NY) at a 1:1 ratio, and supplemented with Glutamax-I (GIBCO/BRL)/BSA, 5 mg/ml (Boehringer Mannheim)/1X lipids (100 × chemically defined lipid concentrate; GIBCO/BRL)/human transferrin, 15 μg/ml (Boehringer Mannheim)/monothioglycerol, 450 μM (Sigma)/insulin, 7 μg/ml (Boehringer Mannheim)/LIF, 1 units/ml (GIBCO/BRL). Expression of βHI embryonic globin transcripts was detected by RT-PCR 5 days after differentiation. Whereas differentiating flk-1−/− and +/− ES cells both expressed βHI in the presence of added FCS, only flk-1+/− cells did so after the addition of BMP-4. Identical results were obtained in six separate experiments performed with two independent flk-1−/− ES cell lines (not shown). The absence of βHI expression in flk-1−/− CDM+ and BMP-4+ lanes was confirmed by Southern blotting with an internal βHI-specific oligonucleotide (not shown). −/−, flk-1−/−; +/−, flk-1+/−.
Hematopoietic Progenitors Are Present in 7.5-dpc flk-1−/− Embryos.
Previous studies indicated that 8.5-dpc flk-1−/− embryos contained virtually no hematopoietic progenitors (3). Studies of chimeric mice generated by the aggregation of flk-1−/− and +/+ ES cells revealed that Flk-1-deficient cells manifest a cell autonomous defect in hematopoiesis. Analysis of these chimeric embryos also suggested that this defect might relate to the failure of Flk-1-deficient cells to migrate appropriately from the posterior primitive streak to the extra-embryonic site of blood island formation and possibly also to intraembryonic sites of early hematopoiesis (4). If Flk−/+ cells truly failed to migrate to establish hematopoiesis and angiogenisis in the yolk sac, one would predict that it might be possible to rescue cells with hematopoietic potential from earlier embryos. We therefore isolated and genotyped 7.5-dpc embryos by Southern blot analysis of cultured ectoplacental cone and trophoblast cells (data not shown) and quantified erythroid, mixed, and myeloid hematopoietic progenitors as had previously been done at 8.5 dpc (3). As shown in Fig. 5, 7.5-dpc embryos contained hematopoietic progenitors of all types, in numbers only slightly lower than those of their +/− and +/+ counterparts. Thus, Flk-1-deficient embryos have the potential to initiate hematopoietic differentiation pathways.
Figure 5.

7.5-dpc flk-1−/− embryos contain hematopoietic progenitors. 7.5-dpc embryos (resulting from flk-1+/− × +/− matings) were treated with 0.25% collagenase/20% FCS, and cells were then plated in 1% methylcellulose in the presence of insulin (10 ng/ml), erythropoietin (2 units/ml), KL (100 ng/ml), IL-1 (650 units/ml), IL-3 (100 units/ml), granulocyte—macrophage CSF (50 units/ml) and macrophage CSF (100 units/ml) as described. Embryos were genotyped by Southern blot analysis of cultured ectoplacental cone and trophoblast cells (ref. 3; data not shown). Colonies were scored after 7 and 10 days of incubation. Mix, mixed colony; Ery, erythroid colony; M, myeloid/macrophage colony. Results from total eight embryos that are +/+ or +/− and four embryos that are −/− are shown.
DISCUSSION
In this report, we show that although flk-1−/− ES cells manifest a cell autonomous developmental defect in vivo, depending on the culture conditions used, they are able to give rise to primitive and definitive hematopoietic cells, endothelial cells, and to common hematopoietic/endothelial precursors in vitro. Furthermore, we demonstrate that flk-1−/− embryos do have the potential to produce hematopoietic (and endothelial) cells, although this ability is not normally realized in vivo. Cells capable of contributing to hematopoiesis do arise in flk-1−/− embryos, but they are detectable only during a brief developmental window before 8.5 dpc. Thereafter, such cells are no longer found. How can these disparate observations be reconciled? Our data are most consistent with the notion that common endothelial/hematopoietic precursors arise independently of a Flk-1 signal. In the absence of a functional Flk-1 receptor, however, such potential hematopoietic/endothelial cells are unable to complete this differentiation pathway: First, the observation that 7.5-dpc flk-1−/− embryos contain essentially normal numbers of hematopoietic progenitors, while 8.5-dpc yolk sacs contain practically none, is consistent with the idea that Flk-1-deficient precursors have failed to migrate appropriately to sites of future hematopoiesis and therefore have failed to receive the appropriate inductive or survival signals required for blood formation. This view is consistent with the reported accumulation of Flk-1-deficient cells in non-extraembryonic mesoderm, such as amnion, of flk-1−/−/+/+ chimeric embryos (4) and is further supported by the recent demonstration that VEGF functions as a chemoattractant for angioblast migration in Xenopus (17). Second, the reduced number and size of Flk-1-deficient colonies produced in the VEGF-dependent BL-CFC assay, contrasted with the essentially normal proliferation of Flk-1-deficient precursors during the formation of flk-1−/− EBs, suggests that the proliferation of Flk-1-deficient precursors may be impaired in vivo as well.
Because flk-1−/− ES cells could also differentiate and give rise to hematopoietic cells on OP9 stromal cells (not shown; ref. 18), it appears, at least in part, that serum may contain factor(s) that can deliver a VEGF/Flk-1 equivalent or bypassing signal in vitro, thereby promoting differentiation or proliferation or preventing cell death. This model requires that migration- and differentiation-blocked Flk-1-deficient cells are nevertheless mature enough to respond to such signals in vivo. The ability of 7.5-dpc flk-1−/− embryos to give rise to hematopoietic colonies with kinetics that are similar to those of +/+ or +/− embryos supports this idea. The nature of this serum effect remains to be defined. We believe, however, that it is likely not mediated by an alternative VEGF receptor, Flt1. Whereas flk-1−/− EBs and blast cells do express flt-1 transcripts (Fig. 3C and data not shown), as do flk-1−/− embryos (3), not only has Flt1 not been shown to mediate a proliferative signal, but the addition of VEGF and/or placenta growth factor (PlGF), a ligand for Flt-1, does not rescue Flk-1-deficient hematopoiesis under serum-free conditions (not shown). Furthermore, the addition of PlGF to blast colony replating media does not enhance blast colony formation (not shown).
Whereas total Flk-1 inactivation is known to result in lethal impairment of vascular and hematopoietic development (3), the role of specific Flk-1 receptor/substrate interactions in mediating hematopoietic/endothelial progenitor migration, proliferation, and differentiation remains obscure. The ability to isolate Flk-1-expressing progenitors and to generate ES cells and animals bearing more subtle Flk-1 mutations that interrupt individual receptor/substrate interactions, while leaving others intact, may provide insight into these questions.
Acknowledgments
We thank Wendy Horsfall, Fouad Shalaby, and Xiang-Fu Wu for technical assistance. We also thank Daved Fremont, Christopher Nelson, and Doreen Salli for critical comments on the manuscript. This work was supported by grants from the Medical Research Council of Canada and The Bayer/Canadian Red Cross Society Research and Development Fund (to A.C.S.) and from the National Institutes of Health (to K.C.).
ABBREVIATIONS
- VEGF
vascular endothelial growth factor
- dpc
days postcoitum
- ES
embryonic stem
- BL-CFC
blast colony-forming cell
- EB
embryoid body
- LIF
leukemia inhibitory factor
- EryP
primitive erythroid
- RT-PCR
reverse transcription–PCR
- FCS
fetal calf serum
- FDG
fluorescein di-β-d-galactopyranoside
- CDM
chemically defined medium
- BMP-4
bone morphogenetic protein-4
- KL
kit ligand
- IL
interleukin
- CSF
colony-stimulating factor
- MTG
monothioglycerol
References
- 1.Dumont D J, Fong G H, Puri M C, Gradwohl G, Alitalo K, Breitman M L. Dev Dyn. 1995;203:80–92. doi: 10.1002/aja.1002030109. [DOI] [PubMed] [Google Scholar]
- 2.Yamaguchi T P, Dumont D J, Conlon R A, Breitman M L, Rossant J. Development (Cambridge, UK) 1993;118:489–498. doi: 10.1242/dev.118.2.489. [DOI] [PubMed] [Google Scholar]
- 3.Shalaby F, Rossant J, Yamaguchi T P, Gertsenstein M, Wu X F, Breitman M L, Schuh A C. Nature (London) 1995;376:62–66. doi: 10.1038/376062a0. [DOI] [PubMed] [Google Scholar]
- 4.Shalaby F, Ho J, Stanford W L, Fischer K D, Schuh A C, Schwartz L, Bernstein A, Rossant J F. Cell. 1997;89:981–990. doi: 10.1016/s0092-8674(00)80283-4. [DOI] [PubMed] [Google Scholar]
- 5.Choi K, Kennedy M, Kazarov A, Papadimitriou J C, Keller G. Development (Cambridge, UK) 1998;125:725–732. doi: 10.1242/dev.125.4.725. [DOI] [PubMed] [Google Scholar]
- 6.Kennedy M, Firpo M, Choi K, Wall C, Robertson S, Kabrun N, Keller G. Nature (London) 1997;386:488–493. doi: 10.1038/386488a0. [DOI] [PubMed] [Google Scholar]
- 7.Carmeliet P, Ferreira V, Breier G, Pollefeyt S, Kieckens L, Gertsenstein M, Fahrig M, Vandenhoeck A, Harpal K, Eberhardt C, et al. Nature (London) 1996;380:435–439. doi: 10.1038/380435a0. [DOI] [PubMed] [Google Scholar]
- 8.Ferrara N, Carver-Moore K, Chen H, Dowd M, Lu L, O’Shea K S, Powell-Braxton L, Hillan K J, Moore M W N. Nature (London) 1996;380:439–442. doi: 10.1038/380439a0. [DOI] [PubMed] [Google Scholar]
- 9.Karasuyama H, Melchers F. Eur J Immunol. 1988;18:97–104. doi: 10.1002/eji.1830180115. [DOI] [PubMed] [Google Scholar]
- 10.Chomczynski P, Sacchi N. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 11.Chelly J, Kaplan J C, Maire P, Gautron S, Kahn A. Nature (London) 1988;333:858–860. doi: 10.1038/333858a0. [DOI] [PubMed] [Google Scholar]
- 12.Nolan G P, Fiering S, Nicolas J F, Herzenberg L A. Proc Natl Acad Sci USA. 1988;85:2603–2607. doi: 10.1073/pnas.85.8.2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reddy S, Rayburn H, von Melchner H, Ruley H E. Proc Natl Acad Sci USA. 1992;89:6721–6725. doi: 10.1073/pnas.89.15.6721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Keller G, Kennedy M, Papayannopoulou T, Wiles M. Mol Cell Biol. 1993;13:473–486. doi: 10.1128/mcb.13.1.473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vittet D, Prandini M H, Berthier R, Schweitzer A, Martin-Sisteron H, Uzan G, Dejana E D. Blood. 1996;88:3424–3431. [PubMed] [Google Scholar]
- 16.Johansson B M, Wiles M V. Mol Cell Biol. 1995;15:141–151. doi: 10.1128/mcb.15.1.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cleaver O, Krieg P A. Development (Cambridge, UK) 1998;125:3905–3914. doi: 10.1242/dev.125.19.3905. [DOI] [PubMed] [Google Scholar]
- 18.Nakano T, Kodama H, Honjo T. Science. 1994;265:1098–1101. doi: 10.1126/science.8066449. [DOI] [PubMed] [Google Scholar]
- 19.Harlow E, Lane D. Immunoprecipitation in Antibodies: A Laboratory Manual. Plainview, NY: Cold Spring Harbor Lab. Press; 1988. pp. 423–470. [Google Scholar]
- 20.Kabrun N, Buhring H-J, Choi K, Ullrich A, Risau W, Keller G. Development (Cambridge, UK) 1997;124:2039–2048. doi: 10.1242/dev.124.10.2039. [DOI] [PubMed] [Google Scholar]