

The structure of M. tuberculosis N-acetylglucosamine-1-phosphate uridyltransferase (GlmU) was determined by the molecular-replacement method to 3.4 Å resolution in space group I432 and was refined to a final R work and R free of 0.285 and 0.321, respectively.
Keywords: Mycobacterium tuberculosis, bifunctional enzymes, acetyltransferases, uridyltransferases, GlmU
Abstract
GlmU is a bifunctional enzyme that catalyzes the final two steps in the biosynthesis of UDP-GlcNAc. Crystals of GlmU from Mycobacterium tuberculosis obtained using ammonium sulfate as a precipitant diffracted poorly (to 3.4 Å resolution) and displayed an unusually high solvent content (>80%) with sparse crystal packing that resulted in large solvent channels. With one molecule per asymmetric unit, the monomers from three neighbouring asymmetric units related by the crystal threefold formed a biological trimer. Although this is the first report of the structure of GlmU determined in a cubic crystal form, the trimeric arrangement here is similar to that observed for other GlmU structures determined in hexagonal (H3, H32, P6322) space groups.
1. Introduction
The Rv1018c (glmU) gene product of Mycobacterium tuberculosis is an N-acetylglucosamine-l-phosphate uridyltransferase (GlmU). GlmU, a bifunctional enzyme, catalyzes the final two steps (reactions 1 and 2 below) in the de novo biosynthesis of UDP-GlcNAc from acetyl-CoA, glucosamine-1-phosphate and UTP,
While the N-terminal domain of GlmU catalyzes uridyltransferase activity (reaction 2 above), acetyltransferase activity (reaction 1 above) at the C-terminal domain requires the formation of a biological trimer.
UDP-GlcNAc is an essential precursor for the biosynthesis of peptidoglycan and lipopolysaccharide, which are constituents of the bacterial cell wall (Barreteau et al., 2008 ▶; Zhang et al., 2008 ▶). With the emergence of mycobacterial multiple drug resistance during the treatment of tuberculosis, the biosynthetic pathway of UDP-GlcNAc might present an alternative target for new antibacterial agents. Recently, we reported modulation of the acetyltransferase activity of M. tuberculosis GlmU upon phosphorylation by the eukaryotic-like serine-threonine protein kinase B (PknB; Parikh et al., 2008 ▶).
The crystal structures of GlmU from Escherichia coli, Streptococcus pneumoniae, Haemophilus influenzae and M. tuberculosis have previously been reported in the hexagonal space groups H3, H32 and/or P6322 to resolutions better than 2.8 Å (Olsen & Roderick, 2001 ▶; Sulzenbacher et al., 2001 ▶; Mochalkin et al., 2008 ▶). Here, we report that GlmU also crystallizes in the cubic space group I432. However, it only diffracts to 3.4 Å resolution. This poor diffraction is correlated with a sparse crystal packing leading to the presence of large solvent channels in the crystal, unlike the hexagonal forms. The distinct crystal packing in these two forms seems to be a result of the involvement of different surfaces in crystal contacts.
2. Material and methods
2.1. Cloning, expression and purification
The gene encoding GlmU (Rv1018c, glmU) was cloned into a pQE2 (Qiagen) expression vector and the protein was purified as described in Parikh et al. (2008 ▶). Briefly, plasmid pQE2-GlmU was transformed in E. coli DH5α and the cells were grown in Luria broth (with 100 µg ml−1 ampicillin) and induced with 0.1 mM IPTG. Cells were lysed by sonication in phosphate buffer containing 5% glycerol, 1 mM β-mercaptoethanol and HIS-cocktail (Sigma). Clarified cell lysate was loaded onto a pre-equilibrated Ni–NTA column (His-Trap FF GE Healthcare). The protein was eluted using a linear gradient of imidazole to 500 mM in 50 mM Tris–HCl buffer pH 7.5 containing 150 mM NaCl. The protein was concentrated and subjected to size-exclusion chromatography using a 26/60 Superdex200 High Load (HL) gel-filtration column (GE Healthcare) equilibrated with 20 mM HEPES pH 7.5, 150 mM NaCl, 1 mM DTT. The elution profile revealed most GlmU to be present as a trimer.
2.2. Crystallization, data collection and processing
GlmU crystals in the ligand-free state were grown in VDX plates (Hampton Research) by the hanging-drop vapour-diffusion method at 277 K against 1 ml reservoir solution consisting of 25 mM MES pH 6.5, 1.8 M ammonium sulfate, 5% glycerol, 5 mM MgCl2. The drops contained 2 µl concentrated protein solution (∼15 mg ml−1 in 20 mM HEPES pH 7.5, 150 mM NaCl and 1 mM DTT) and 2 µl reservoir solution. Crystals appeared in 2–3 d and grew to a size suitable for diffraction experiments in 5–7 d. Crystals were cryoprotected in 2.0 M ammonium sulfate, 25 mM MES pH 6.5 containing 25% ethylene glycol and flash-frozen in liquid nitrogen.
X-ray diffraction data were collected from these crystals using an in-house Rigaku MicroMax007HF X-ray source with a copper rotating-anode generator equipped with Varimax optics, a MAR345dtb image-plate detector and an Oxford Cryosystem 700 series cryostream. A complete data set was collected to a resolution of 3.4 Å. The data were indexed, integrated and scaled using the XDS program package (Kabsch, 1993 ▶). The GlmU crystals belonged to space group I432 (No. 211) and contained one molecule per asymmetric unit with a solvent content of ∼82% (Matthews, 1968 ▶).
2.3. Structure determination and refinement
The structure of GlmU from cubic crystals was determined by molecular replacement using Phaser (CCP4i; Read, 2001 ▶) with M. tuberculosis GlmU (PDB code 3dj4; Parikh et al., 2008 ▶) as the search model. The top solution (LLG value 2694.4, Z score 46.6) showed a clear contrast with the next solution and was unambiguous for proceeding with phase refinement and model building. The initial model was built by several rounds of manual building in Coot (Emsley & Cowtan, 2004 ▶). The structure was refined with REFMAC5 (CCP4i) using the maximum-likelihood target function, employing rigid-body refinement followed by restrained refinement (Murshudov et al., 1997 ▶). Owing to the low resolution of the data, a weight term of 0.3 was employed to provide tight restraints during refinement. The model quality was assessed using PROCHECK (Laskowski et al., 1993 ▶). All figures were generated using CHIMERA (Pettersen et al., 2004 ▶). The final model of GlmU, consisting of 439 amino acids, was refined to a resolution of 3.41 Å with an R work of 28.5% and an R free of 32.1%.
3. Results and discussion
Preliminary crystals of GlmU from M. tuberculosis were obtained in conditions containing ammonium sulfate as precipitant at pH 6.5. These conditions were further optimized in order to produce crystals of suitable size for diffraction experiments. The crystals grew to approximately 300 µm in all dimensions (Fig. 1 ▶ a). However, they consistently yielded a poor and pathological diffraction pattern at room temperature as well as when cryoprotected at 100 K. An initial data set collected to 3.8 Å resolution revealed a very high solvent content in the crystals. Hence, they were dehydrated in an attempt to improve the diffraction quality. Dehydration was carried out by soaking crystals in increasing amounts of precipitant (ammonium sulfate), sodium malonate and glycerol. This resulted in improved diffraction and a full data set could be collected to 3.4 Å resolution at 100 K. The data were processed using the program package XDS (Kabsch, 1993 ▶).
Figure 1.

(a) Native crystal of GlmU obtained using ammonium sulfate as a precipitant. The crystals, which were grown in a hanging-drop setup, grew to dimensions of ∼0.3 × 0.3 × 0.3 mm. (b) The structure of GlmU. Each monomer contains an N-terminal domain (coloured pink) responsible for the uridyltransferase activity and a C-terminal domain with a left-handed β-helix fold (LβH; coloured gold) responsible for acetyltransferase activity. A hinge helix (coloured blue) connects the two domains and the C-terminal extensions are marked red. (c) GlmU forms a biological trimer. Two of the three monomers of the trimer are coloured dark grey and light blue, while the other is coloured as in (b). The N- and C-terminal parts of the trimer are defined as the ‘head’ and the ‘tail’.
The crystals belonged to the cubic space group I432, with unit-cell parameters a = b = c = 285.7 Å, and contained one molecule per asymmetric unit. The structure was determined by the molecular-replacement method using M. tuberculosis GlmU as the search model, as detailed in §2. GlmU consists of two domains: an N-terminal uridyltransferase domain with an α/β-like fold and a C-terminal acetyltransferase domain that forms a left-handed parallel β-helix structure (LβH) with the shape of an equilateral triangular prism consisting of ten turns. The two domains are connected by a long α-helix (Fig. 1 ▶ b). Although GlmU consists of 495 amino acids, a model could only be built for residues 1–472. The extended C-terminus present in M. tuberculosis GlmU was not well defined in the electron-density map. The N- and C-terminal domains are known to be responsible for the uridyltransferase and acetyltransferase activities, respectively (Parikh et al., 2008 ▶). The residues 149–150, 154–158, 164–177 and 200–206 that are part of the N-terminal active site are not well defined in the electron-density maps as the active site is devoid of bound ligands. Unlike the N-terminal active site, the formation of the C-terminal active site requires a trimeric arrangement (Fig. 1 ▶ c), as inferred from the structures of GlmU homologues (from E. coli and S. pneumoniae) bound to acetyl-CoA or CoA (Olsen & Roderick, 2001 ▶; Sulzenbacher et al., 2001 ▶). The biological trimer found in the current structure of GlmU is similar to that in the GlmU homologues, except that the active-site residues 398–403 are disordered.
The unusually high Matthews coefficient (V M = 9.17 Å3 Da−1) and high solvent content (>80%; Matthews, 1968 ▶) of the cubic crystals leads to the presence of large solvent channels. In contrast, most GlmU proteins crystallize in the H3/H32/P6322 form and do not show the presence of such large channels. This led us to compare the crystal packing in the cubic and hexagonal forms. GlmU exists as a biological trimer in solution. A trimeric arrangement is common to both crystal forms. It appears that different contacts between neighbouring trimers promote crystal formation, leading to either the cubic or hexagonal forms. To facilitate a comparison of crystal contacts, we define the N- and C-terminal parts of the trimer as the ‘head’ and the ‘tail’, respectively (Fig. 1 ▶ c). In the cubic crystals, monomers are arranged in a trimeric fashion along each of the body diagonals, i.e. the crystal threefolds (Fig. 2 ▶ a). Two such trimers pack against each other ‘tail to tail’ (indicated by a circle in Fig. 2 ▶ b) and their heads face the corner and the centre of the cube. Each diagonal therefore contains four such trimers. At the centre of the cube, where the four diagonals meet, several adjacent trimers meet head to head (Figs. 2 ▶ a and 2 ▶ b) which take part in crystal contacts (indicated by a square in Fig. 2 ▶ b). This arrangement of molecules in the crystal results in the unusually large solvent channels in the cubic form (Fig. 2 ▶ c). In the hexagonal form the trimer is along the c axis, which is the crystal threefold, but neighbouring trimers along this axis contact in a head-to-tail fashion (Fig. 3 ▶ a). In addition, along the a and b axis, head-to-head contacts stabilize the packing (Fig. 3 ▶ b). The region involved in head-to-head contacts in the cubic form, however, is distinct from those in the hexagonal form. The compact packing appears to be a result of the head-to-tail arrangement of trimers in the H3/H32 forms. Therefore, it appears that the tail-to-tail arrangement of trimers in the cubic form results in a sparse packing in the cubic crystal form of GlmU.
Figure 2.

Crystal packing in the cubic form. (a) A view along the body diagonal of the I432 unit cell with unit-cell parameter 285.7 Å depicts a sparse molecular packing. (b) A tail-to-tail crystal contact between trimers (blue circle) along the body diagonal and a head-to-head contact between the adjacent trimers (green square) at the centre of the cube stabilizes this arrangement. (c) The unique arrangement of the molecules in I432 crystals results in large solvent channels as viewed along the diagonal (left) and the plane (right) of the cube.
Figure 3.

Crystal packing in the hexagonal form, generated using the coordinates of M. tuberculosis GlmU determined in space group H3 (PDB code 3dk5; unit-cell parameters a = b = 79.6, c = 278.0 Å). (a) Crystal packing showing head-to-head packing of GlmU trimers along the c axis. These trimers contact each other in a head-to-tail fashion. (b) A head-to-head arrangement of trimers results in crystal contacts in the ab plane. (c) The arrangement of molecules in the hexagonal form results in a tight packing. Crystal packing in the ac and ab planes of the crystal is depicted.
Note added in proof: During the production of this manuscript another publication appeared on the structure of GlmU from Mycobacterium tuberculosis (Zhang et al., 2009 ▶).
Supplementary Material
PDB reference: GlmU, 3foq, r3foqsf
Table 1. X-ray data-collection and refinement statistics.
Values in parentheses are for the outer shell.
| Crystallization conditions | 25 mM MES pH 6.5, 1.8 M ammonium sulfate, 5% glycerol, 5 mM MgCl2 |
| Space group [No.] | I432 [No. 211] |
| Unit-cell parameter (Å) | a = b = c = 285.7 |
| Data-collection statistics | |
| Wavelength (Å) | 1.54179 |
| Resolution (Å) | 50.0–3.4 (3.50–3.41) |
| No. of observed reflections | 230992 |
| No. of unique reflections | 25629 |
| Completeness (%) | 94.6 (91.1) |
| I/σ(I), overall | 14.56 (4.53) |
| Rmeas† (%) | 19.9 (56.4) |
| Rmrgd-F‡ (%) | 10.4 (22.9) |
| Refinement statistics | |
| Resolution range (Å) | 48.97–3.41 |
| No. of reflections | 25629 |
| Rwork§ (%) | 28.5 |
| Rfree¶ (%) | 32.1 |
| R.m.s.d. bonds (Å) | 0.028 |
| R.m.s.d. angles (°) | 1.93 |
| Mean B value (Å2) | 49.3 |
| No. of protein atoms | 3186 |
| No. of ions | 4 |
| Ramachandran plot: main-chain torsion-angle statistics (%) | |
| Most favoured | 89.0 |
| Additionally allowed | 9.7 |
| Generously allowed | 0.8 |
| Disallowed | 0.6 |
R
meas = 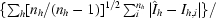
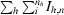 , where
, where 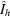 =
= 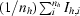 .
.
R
mrgd-F = 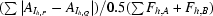 , where I
h,P and I
h,Q are a measure of the quality of the reduced amplitude.
, where I
h,P and I
h,Q are a measure of the quality of the reduced amplitude.
R
work = 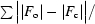
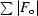 , where F
o and F
c are observed and calculated structure factors, respectively.
, where F
o and F
c are observed and calculated structure factors, respectively.
R free was calculated using 5% of data excluded from refinement.
Acknowledgments
This work was supported in part by funding provided by the Department of Biotechnology (DBT), India to VKN and BP and by grants from Wellcome Trust UK in the form of an International Research Fellowship to BP. We are also grateful to the Department of Science and Technology (DST), India for ‘DST-Fund for Improvement of S&T Infrastructure in Universities and Higher Educational Institutions’ to BSBE, IIT Kanpur. SKV is a Senior Research Fellow of the University Grants Commission, India. AP is a DBT postdoctoral fellow. We sincerely thank the members of the VKN and BP laboratories for their input during the course of the study. We thank Dr A. Sharma, ICGEB for the machine time for the collection of the initial data sets.
References
- Barreteau, H., Kovac, A., Boniface, A., Sova, M., Gobec, S. & Blanot, D. (2008). FEMS Microbiol. Rev.32, 168–207. [DOI] [PubMed]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Kabsch, W. (1993). J. Appl. Cryst.26, 795–800.
- Laskowski, R. A., MacArthur, M. W., Moss, D. S. & Thornton, J. M. (1993). J. Appl. Cryst.26, 283–291.
- Matthews, B. W. (1968). J. Mol. Biol.33, 491–497. [DOI] [PubMed]
- Mochalkin, I., Lightle, S., Narasimhan, L., Bornemeier, D., Melnick, M., Vanderroest, S. & McDowell, L. (2008). Protein Sci.17, 577–582. [DOI] [PMC free article] [PubMed]
- Murshudov, G. N., Vagin, A. A. & Dodson, E. J. (1997). Acta Cryst. D53, 240–255. [DOI] [PubMed]
- Olsen, L. R. & Roderick, S. L. (2001). Biochemistry, 40, 1913–1921. [DOI] [PubMed]
- Parikh, A., Verma, S. K., Khan, S., Prakash, B. & Nandicoori, V. K. (2008). J. Mol. Biol.386, 451–464. [DOI] [PubMed]
- Pettersen, E. F., Goddard, T. D., Huang, C. C., Couch, G. S., Greenblatt, D. M., Meng, E. C. & Ferrin, T. E. (2004). J. Comput. Chem.25, 1605–1612. [DOI] [PubMed]
- Read, R. J. (2001). Acta Cryst. D57, 1373–1382. [DOI] [PubMed]
- Sulzenbacher, G., Gal, L., Peneff, C., Fassy, F. & Bourne, Y. (2001). J. Biol. Chem.276, 11844–11851. [DOI] [PubMed]
- Zhang, Z., Bulloch, E. M. M., Bunker, R. D., Baker, E. N. & Squire, C. J. (2009). Acta Cryst. D65, 275–283. [DOI] [PMC free article] [PubMed]
- Zhang, W., Jones, V. C., Scherman, M. S., Mahapatra, S., Crick, D., Bhamidi, S., Xin, Y., McNeil, M. R. & Ma, Y. (2008). Int. J. Biochem. Cell Biol.40, 2560–2571. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: GlmU, 3foq, r3foqsf
PDB reference: GlmU, 3foq, r3foqsf