

Crystal structures of the ubiquitin-conjugating enzyme E2-25K M172A mutant protein at pH 6.5 and pH 8.5 were determined to 1.9 and 2.2 Å resolution, respectively. Examination of the structures revealed domain–domain interactions between the UBC and UBA domains which have not previously been reported.
Keywords: huntingtin-interacting protein 2, E2-25K, ubiquitin-conjugating enzymes
Abstract
The ubiquitin-conjugating enzyme E2-25K has been identified as a huntingtin (the key protein in Huntington’s disease) interacting protein and has been shown to play a role in mediating the toxicity of Aβ, the principal protein involved in Alzheimer’s disease pathogenesis. E2-25K is a dual-domain protein with an ubiquitin-associated (UBA) domain as well as a conserved ubiquitin-conjugating (UBC) domain which catalyzes the formation of a covalent bond between the C-terminal glycine of an ubiquitin molecule and the ∊-amine of a lysine residue on the acceptor protein as part of the ubiquitin-proteasome pathway. The crystal structures of E2-25K M172A mutant protein at pH 6.5 and pH 8.5 were determined to 1.9 and 2.2 Å resolution, respectively. Examination of the structures revealed domain–domain interactions between the UBC and UBA domains which have not previously been reported.
1. Introduction
The ubiquitin-proteasome pathway is one of the means by which the cell degrades and recycles proteins. Tagging of a protein molecule with a polyubiquitin chain targets it for interaction with the proteasome, a multi-subunit cellular disposal system which proteolytically degrades the target protein and releases ubiquitin molecules to be recycled. Conjugation of a target protein with ubiquitin is a multi-step multi-enzyme process. The first step is the formation of a thioester bond between the C-terminal glycine residue of ubiquitin and the E1 ubiquitin-activating enzyme (Ciechanover, 1994 ▶). The ubiquitin is transferred to a cysteine residue of the E2 ubiquitin-conjugating enzyme and then covalently linked to a lysine residue of the target protein through the actions of the E2 and/or E3 (ubiquitin ligase) enzymes. Addition of ubiquitin molecules to the Lys48 residue of the conjugated ubiquitin builds a polyubiquitin chain that is recognized by the 19S subunit of the 26S proteasome (Deveraux et al., 1994 ▶). Deubiquitinating enzymes remove and break down the polyubiquitin chain, while the target protein is degraded by the proteasome. This system of protein degradation seems to be implicated in a number of protein-misfolding diseases. In particular, the presence of ubiquitin is noted in intranuclear inclusions in Huntington’s disease (Kalchman et al., 1996 ▶) as well as in fibrillary tangles in Alzheimer’s disease and other tauopathies (Song & Jung, 2004 ▶; Ciechanover & Brundin, 2003 ▶). This may indicate that a malfunction of the ubiquitin-proteasome pathway contributes to the accumulation of misfolded protein aggregates. E2-25K, a ubiquitin-conjugating enzyme, is upregulated in Alzheimer’s disease and its presence is required for the toxicity generated by Aβ fragments (Song et al., 2003 ▶). This same enzyme is also a binding partner for huntingtin, the key protein mutated in Huntington’s disease (HD; Kalchman et al., 1996 ▶). E2-25K is highly expressed in the brain and co-localizes with intranuclear neuronal inclusions in the brain tissue of patients with the polyglutamine disorders HD and spinocerebellar ataxia 3 (SCA3; de Pril et al., 2007 ▶).
E2-25K synthesizes Lys48-linked free polyubiquitin chains in vitro in the absence of an E3 ligase (Chen & Pickart, 1990 ▶). While it has not been definitively shown that this function is present in vivo, there is evidence of accumulation of Lys48-linked polyubiquitin chains in the cell (van Nocker & Vierstra, 1993 ▶). The function of these free polyubiquitin chains is not known, but it can be speculated that they play a role in the rapid turnover of certain protein substrates as well as potentially regulating the activity of the proteasome.
E2-25K represents a unique subgroup of class II E2 ubiquitin-conjugating enzymes which contain an ubiquitin-associated (UBA) domain C-terminal to the catalytic ubiquitin-conjugating (UBC) domain. The functional significance of the UBA domain in this protein is still undetermined. Initial studies suggested that the UBA domain was essential for the polyubiquitin chain-synthesizing activity (Haldeman et al., 1997 ▶). Removal of the UBA domain resulted in nonfunctional E2-25K; however, subsequent research indicated that the location of the truncation was of critical importance. Slightly longer constructs (still lacking the UBA domain) were able to catalyze polyubiquitin formation (Pichler et al., 2005 ▶), although the processivity was impaired relative to full-length E2-25K.
The amino-acid sequence of the UBA domain of E2-25K is well conserved among mammalian species, but diverges from the UBA-domain sequence found in nonmammalian homologs. This domain contains a highly conserved MGF loop (Met172, Gly173, Phe174) present in most UBA domains, which is an integral part of the ubiquitin-binding surface. Several key hydrophobic residues form the core of the domain. To better understand the functional role of the UBA domain, we prepared an M172A mutant protein for comparison with wild-type E2-25K. We have determined the crystal structures of E2-25K M172A mutant protein under two different conditions to 1.9 Å resolution (pH ∼6.5) and 2.2 Å resolution (pH ∼8.5). Comparison of these structures with that of the wild-type E2-25K revealed few structural differences overall, but demonstrated minor alterations in the domain–domain interface and flexibility in the position of a single catalytic site residue side chain.
2. Experimental procedures
2.1. Mutagenesis of E2-25K: molecular cloning
The pET30a expression vector encoding full-length wild-type E2-25K was a gift from Dr Seongman Kang of the Graduate School of Biotechnology, Korea University. The E2-25K M172A mutant was generated by site-directed mutagenesis following the manufacturer’s instructions (Stratagene). The following complementary primers were used to generate the E2-25K M172A mutant: forward, 5′-GAA AAC CTA TGT GCT GCG GGC TTT GAT AGG AAT GC-3′, and reverse, 5′- CTG CAT TCC TAT CAA AGC CCG CAG CAC ATA GGT TTT CTG-3′. The resulting protein contained an N-terminal histidine tag with sequence MHHHHHHSSGLVPRGSGMKETAAAKFERQHMDSPDLGTDDDDKAMADIGSEFD. Clones were sequence-verified (Functional Biosciences, Inc) and named E2-25K M172A.
2.2. Protein expression and purification
The E2-25K M172A mutant plasmid was transformed into BL21(DE3) Escherichia coli cells and grown at 310 K to an OD600 of ∼0.6 in Luria–Bertani medium containing 35 µg ml−1 kanamycin. Expression was induced by the addition of 1.0 mM IPTG and cells were grown for 3 h at 310 K. Cells were harvested by centrifugation at 6000g for 25 min and resuspended in lysis buffer (20 mM Tris–HCl pH 8.0, 500 mM NaCl, 5 mM imidazole) prior to cell lysis by ultrasonication. The supernatant solution was chromatographed on an Ni2+-chelating Sepharose (GE Healthcare) column and the E2-25K M172A mutant was eluted with 500 mM imidazole. Fractions containing the intact E2-25K M172A mutant, which included the tag from the pET30a expression vector, were pooled and dialyzed against 20 mM Tris–HCl pH 8.0, 50 mM NaCl, 1 mM 2-mercaptoethanol (BME). The protein was concentrated to 18 mg ml−1 using a Centricon ultrafiltration device (Millipore) and stored at 277 K until use in crystallization trials.
2.3. Crystallization
Initial crystallization trials were performed with commercially available reagents using the sitting-drop vapor-diffusion method in a 96-well Intelli-Plate (Art Robbins). Crystal Screen I (1–48) and Crystal Screen II (1–48) (Hampton Research, Laguna Hills, California, USA) were used as reservoir solutions (100 µl) in the initial trial. Protein:precipitant ratios of 1:1 (2 µl:2 µl) and 2:1 (2 µl:1 µl) were placed in sample wells. The plate was allowed to equilibrate undisturbed for one week at 294 K in a temperature-controlled incubator. Two sets of conditions [0.1 M sodium cacodylate trihydrate pH 6.5, 0.2 M calcium acetate hydrate, 18%(w/v) polyethylene glycol 8000 and 0.1 M Na HEPES pH 7.5, 0.2 M calcium chloride dihydrate, 28% PEG 400] produced poorly formed needle-like protein crystals. An optimization screen around both conditions was performed, targeting ionic strength and pH as screening parameters. Stock buffers with optimal buffering capacity at pH 4.6, 6.0 and 9.0 were mixed with the original reservoir solutions. Two optimal conditions were obtained and shown to produce well diffracting protein crystals. The final optimal condition for the low-pH condition was formulated by supplementing 50 µl reservoir solution consisting of 0.1 M sodium cacodylate trihydrate pH 6.5, 0.2 M calcium acetate hydrate, 18%(w/v) polyethylene glycol 8000 (Hampton Research) with 10 µl stock buffer containing 500 mM NaCl, 500 mM sodium acetate pH 4.6 (measured) and 10 mM EDTA. The final optimal conditions for the high-pH condition were formulated by supplementing 50 µl reservoir solution consisting of 0.1 M Na HEPES pH 7.5, 0.2 M calcium chloride dihydrate, 28% PEG 400 (Hampton Research) with 10 µl stock buffer containing 500 mM NaCl, 500 mM bicine pH 9.0 (measured) and 10 mM EDTA. Crystals were first observed after one week for the low-pH condition and after one month for the high-pH condition. The initial conditions that produced the low-pH crystals after optimization were similar to those reported for the wild-type crystals (PDB code 1yla).
2.4. Data collection and structure determination
Protein crystals suitable for X-ray analysis were soaked in a cryogenic solution consisting of the reservoir solution and 28%(v/v) glycerol prior to mounting in an appropriately sized nylon loop. Data collection was performed on SER-CAT beamline ID22 (Argonne National Laboratory, Chicago, Illinois, USA). The crystals diffracted to 1.9 Å (pH ∼6.5) and 2.2 Å (pH ∼8.5) resolution. A 125-frame data set was collected for the low-pH crystal form and a 75-frame data set was collected for the high-pH crystal form using 0.97 and 1.0 Å X-ray radiation, respectively, with 1° oscillation and 1 s per frame exposure time. X-ray diffraction was recorded with a MAR300 CCD (charge-coupled device) detector. The data were processed with the DENZO and SCALEPACK program packages from within HKL-2000 (Otwinowski & Minor, 1997 ▶). The data-collection statistics are recorded in Table 1 ▶. Both crystal forms indexed and scaled as I-centered tetragonal, with unit-cell parameters a = b = 134.5, c = 38.4 Å for the low-pH form and a = b = 134.8, c = 38.2 Å for the high-pH form. The asymmetric units contain one molecule and the crystals have a Matthews coefficient of 3.1 Å3 Da−1, corresponding to a solvent content of 60%.
Table 1. Data-collection and refinement statistics for E2-25K M172A mutant structures.
Data were collected on SER-CAT beamline ID22. Values in parentheses are for the highest resolution shell.
| Low-pH crystal (PDB code 3e46) | High-pH crystal (PDB code 3f92) | |
|---|---|---|
| Data collection | ||
| Wavelength (Å) | 1.0 | 0.97 |
| Space group | I4 | I4 |
| Molecules per ASU | 1 | 1 |
| Unit-cell parameters (Å, °) | a = b = 134.5, c = 38.4, α = β = γ = 90 | a = b = 134.8, c = 38.2, α = β = γ = 90 |
| Resolution (Å) | 50.0–1.86 (1.91–1.86) | 50.0–2.23 (2.31–2.23) |
| Rmerge† (%) | 8.1 (37.4) | 6.9 (37.7) |
| 〈I/σ(I)〉 | 19.65 (1.98) | 11.95 (2.39) |
| Completeness (%) | 93.4 (52.4) | 98.2 (88.8) |
| No. of reflections (No. unique) | 109109 (27414) | 48847 (16794) |
| Redundancy | 4.0 (1.4) | 2.9 (1.9) |
| No. of frames | 125 | 75 |
| Matthews coefficient (Å3 Da−1) | 3.1 | 3.1 |
| Refinement | ||
| Resolution (Å) | 32.38-1.86 | 42.64-2.23 |
| No. of reflections | 27404 | 15939 |
| Rwork/Rfree‡ (%) | 17.4/21.0 | 17.4/21.3 |
| No. of atoms/molecules | ||
| Protein | 1597 | 1599 |
| Non-water molecules | 1 | 5 |
| Water molecules | 207 | 89 |
| R.m.s.d. bonds (Å) | 0.018 | 0.024 |
| R.m.s.d. angles (°) | 1.5 | 1.85 |
| Ramachandran plot | ||
| Most favored region (%) | 99.0 | 99.0 |
| Additional allowed region (%) | 1.0 | 1.0 |
R
merge = 
 , where I
i(hkl) is the observed intensity of reflection i and 〈I(hkl)〉 is the average intensity of multiple observations.
, where I
i(hkl) is the observed intensity of reflection i and 〈I(hkl)〉 is the average intensity of multiple observations.
R
work and R
free = 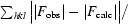
 , where R
free was calculated using 5% of the total reflections chosen at random and not used in the refinement.
, where R
free was calculated using 5% of the total reflections chosen at random and not used in the refinement.
The structure of each crystal form was solved by the molecular-replacement method using the program MOLREP (Vagin & Teplyakov, 1997 ▶) from the CCP4 program suite (Collaborative Computational Project, Number 4, 1994 ▶). Wild-type E2-25K (PDB code 1yla) was used as the search model for the low-pH crystal structure. Subsequently, the low-pH structure (PDB code 3e46) was used as the search model for the high-pH data set (PDB code 3f92). Refinement was carried out using the program REFMAC5 (Murshudov et al., 1997 ▶). The structure was visualized and modified with the program Coot (Emsley & Cowtan, 2004 ▶). Modifications were made to the protein molecule by manually fitting the calculated model against 2|F o| − |F c| and |F o| − |F c| electron-density maps using cutoffs of 1.0σ and 3.5σ, respectively, followed by subsequent iterative cycles of restrained refinement. An initial set of solvent atoms were added to each structure using ARP/wARP (Perrakis et al., 2001 ▶). Additional water molecules and ligands were added to the model manually after each round of fitting and refinement. Fig. 1 ▶ is a representation of the 2|F o| − |F c| electron-density map showing a close-up view of the Ca2+ ion-coordination geometry that represents a crystal contact between symmetry-related molecules that is present in both crystallization conditions. The low-pH structure contained 207 water molecules and one ligand (Ca2+) and refined with final R work and R free factors of 17.4% and 21.0%, respectively. The high-pH structure contained 89 water molecules, one Ca2+ ion, two PEG 400 molecules, one Tris molecule and one BME molecule. The high-pH structure refined with final R work and R free factors of 17.4% and 21.3%, respectively. The refinement statistics are summarized in Table 1 ▶. All figures were produced using the program PyMOL (DeLano, 2008 ▶).
Figure 1.

A close-up view of the Ca2+ ion-coordination geometry that represents a crystal contact between symmetry-related molecules present in both conditions with a 2|F o| − |F c| electron-density map contoured at 1.0σ for the low-pH crystal (PDB code 3e46) and bond lengths in Å.
2.4.1. Ligand binding and determination
The low-pH final structure contained one ligand (Ca2+) and the high-pH final structure contained one Ca2+ ion, two PEG 400 molecules, one Tris molecule and one BME molecule. The calcium ion in both crystal structures forms crystal contacts by coordinating Asp0 with Glu20 and Glu99 of symmetry-related molecules (Fig. 1 ▶). In order to identify the ligand type, the calcium ion was initially modeled as a water molecule. However, the temperature factor of the water was calculated to be 8.2 Å2, which was significantly lower than the temperature factors of either the surrounding water molecules or the surrounding protein atoms. Positive density was observed in the |F o| − |F c| electron-density map, suggesting that oxygen could not account for the observed density. The possibility of a sodium ion was also ruled out based upon the same criteria. The coordination shell surrounding the atom contains six O atoms with distances of less than 2.5 Å and angles characteristic of an octahedrally coordinated calcium ion. Furthermore, the crystallization of the protein was subsequently shown to be dependent upon the presence of calcium and the optimized condition contained a final concentration of 0.1 M calcium chloride. The two PEG 400 molecules, Tris and the BME in the high-pH final structure were identified based on best fit of the density using molecules known to be present in the crystallization solvent.
3. Results and discussion
3.1. Structure of the E2-25K mutant protein
The E2-25K M172A structure consists of a conserved 150-amino-acid N-terminal ubiquitin-conjugating (UBC) domain and a 50-amino-acid C-terminal ubiquitin-associated (UBA) domain (Fig. 2 ▶ a). Superposition of the M172A mutant structures and the wild-type enzyme structures resulted in an overall Cα r.m.s.d. of 0.34 Å. The UBC is comprised of an N-terminal α-helix followed by four antiparallel β-strands and then by an additional three α-helices. The C-terminal UBA domain is made up of a three-helix bundle.
Figure 2.

(a) Ribbon diagram of the E2-25K M172A mutant colored by domain. The catalytic UBC domain is shown in red and the C-terminal UBA domain is shown in blue. (b) Active-site superposition. The M172A mutant protein structures superimposed with wild-type E2-25K and catalytic domain structures: red, PDB code 3e46 (M172A at pH 6.5); green, PDB code 3f92 (M172A at pH 8.5); grey, PDB code 1yla (wild-type E2-25K at pH 6.5); blue, PDB code 2bep (wild-type E2-25K UBC domain at pH 8.5).
A Ramachandran plot calculated using MolProbity (Lovell et al., 2003 ▶) showed 99.0% (198/200) of the residues in most favored regions and the remaining residues in additionally allowed regions. The two residues that fall outside the most favored regions are Lys97 and Ala119. Ala119 is located at the end of helix 3, N-terminal to a flexible eight-amino-acid loop. Lys97 is located at the end of helix 2, C-terminal to the catalytic cysteine (Cys92). In UBC1, the E2-25K homolog in Saccharomyces cerevisiae, mutation of this lysine to arginine eliminated Lys48-linked polyubiquitin chain synthesis (Hodgins et al., 1996 ▶). Comparing both M172A structures with the wild-type structure reveals that most side chains in the active site are conformationally similar (Fig. 2 ▶ b). The exception is the side chain of Lys97, which exhibits multiple rotamer conformations as a function of the differing crystallization conditions.
The UBA domain is stabilized primarily by hydrophobic residues in the core of the domain. These residues include Leu169, Val179, Leu183, Val190 and Leu197. The relative positioning of the helices of the UBA domain of E2-25K is determined by ring stacking between the side chains of residues Tyr162 in helix 6 and Trp188 in the loop between helices 7 and 8. The UBC–UBA domain–domain interface consists of hydrophobic interactions between residues Met140, Leu147 and Val151 (helix 5) of the UBC domain and residues Ile166 (helix 6) and Ile180 (helix 7) of the UBA domain. Additionally, three hydrogen bonds exist between the side chains of Thr144 and Trp188, Trp148 and Ser184, and Tyr152 and Asn177 (Fig. 3 ▶).
Figure 3.

UBC/UBA-domain interactions between helix 5 (red) of the UBC domain and helix 6 and 7 (blue) of the UBA domain. Three hydrogen bonds between the two domains are shown as dashed lines with bond lengths in Å. The M172A mutation site is shown in green.
3.2. E2-25K M172A mutant UBA domain
Met172 is located in the loop between helices 6 and 7 of the UBA domain and forms part of the primary contact surface with ubiquitin (unpublished data). When compared with the wild-type structure, the M172A mutant has a slight rotation (∼10°) of the UBA domain relative to the UBC domain, with the axis of rotation along helix 7. However, this rotation did not impose structural changes on either the domain or the ubiquitin-binding surface. Furthermore, the M172A mutant structure did not significantly alter the primary contacts between the UBC and UBA domains, but did slightly decrease the length of the three interdomain hydrogen bonds (from 3.0, 3.0 and 2.6 Å in the wild type to 2.9, 2.9 and 2.5 Å in the M172A mutant).
4. Conclusions
Here, we have reported two crystal structures of the ubiquitin-conjugating enzyme E2-25K M172A. The structures have been solved to 1.9 and 2.2 Å resolution and subsequently compared with the wild-type structure (PDB code 1yla). We have shown that the E2-25K M172A mutant shows strong structural homology to the wild-type enzyme, demonstrating that the mutation does not disrupt the overall fold of the protein. We have discussed the basis for the interactions between the UBC and UBA domains and have shown that these interactions are not significantly altered by the mutation. These observations will be vital in further exploration of the effects of the mutation on the catalytic activity of the enzyme.
Supplementary Material
PDB reference: E2-25K, M172A mutant, 3e46, r3e46sf
PDB reference: 3f92, r3f92sf
Acknowledgments
Data were collected on Southeast Regional Collaborative Access Team (SER-CAT) 22-ID beamline at the Advanced Photon Source, Argonne National Laboratory. Use of the Advanced Photon Source was supported by the US Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. W-31-109-Eng-38. RCW and RCH were supported by National Science Foundation EPSCoR Graduate Research Fellowships. PDT and EJM were supported by the National Science Foundation EPSCoR Program (NSF # EPS-0447675). Additional support was provided by the Alpha Foundation.
References
- Chen, Z. & Pickart, C. M. (1990). J. Biol. Chem.265, 21835–21842. [PubMed]
- Ciechanover, A. (1994). Cell, 79, 13–21. [DOI] [PubMed]
- Ciechanover, A. & Brundin, P. (2003). Neuron, 40, 427–446. [DOI] [PubMed]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763.
- DeLano, W. L. (2008). The PyMOL Molecular Graphics System. http://www.pymol.org.
- Deveraux, Q., Ustrell, V., Pickart, C. & Rechsteiner, M. (1994). J. Biol. Chem.269, 7059–7061. [PubMed]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Haldeman, M. T., Xia, G., Kasperek, E. M. & Pickart, C. M. (1997). Biochemistry, 36, 10526–10537. [DOI] [PubMed]
- Hodgins, R., Gwozd, C., Arnason, T., Cummings, M. & Ellison, M. J. (1996). J. Biol. Chem.271, 28766–28771. [DOI] [PubMed]
- Kalchman, M. A., Graham, R. K., Xia, G., Koide, H. B., Hodgson, J. G., Graham, K. C., Goldberg, Y. P., Gietz, R. D., Pickart, C. M. & Hayden, M. R. (1996). J. Biol. Chem.271, 19385–19394. [DOI] [PubMed]
- Lovell, S. C., Davis, I. W., Arendall, W. B. III, de Bakker, P. I., Word, J. M., Prisant, M. G., Richardson, J. S. & Richardson, D. C. (2003). Proteins, 50, 437–450. [DOI] [PubMed]
- Murshudov, G. N., Vagin, A. A. & Dodson, E. J. (1997). Acta Cryst. D53, 240–255. [DOI] [PubMed]
- van Nocker, S. & Vierstra, R. D. (1993). J. Biol. Chem.268, 24766–24773. [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Perrakis, A., Harkiolaki, M., Wilson, K. S. & Lamzin, V. S. (2001). Acta Cryst. D57, 1445–1450. [DOI] [PubMed]
- Pichler, A., Knipscheer, P., Oberhofer, E., van Dijk, W. J., Korner, R., Olsen, J. V., Jentsch, S., Melchior, F. & Sixma, T. K. (2005). Nature Struct. Mol. Biol.12, 264–269. [DOI] [PubMed]
- Pril, R. de, Fischer, D. F., Roos, R. A. & van Leeuwen, F. W. (2007). Mol. Cell. Neurosci.34, 10–19. [DOI] [PubMed]
- Song, S. & Jung, Y. K. (2004). Trends Mol. Med.10, 565–570. [DOI] [PubMed]
- Song, S., Kim, S. Y., Hong, Y. M., Jo, D. G., Lee, J. Y., Shim, S. M., Chung, C. W., Seo, S. J., Yoo, Y. J., Koh, J. Y., Lee, M. C., Yates, A. J., Ichijo, H. & Jung, Y. K. (2003). Mol. Cell, 12, 553–563. [DOI] [PubMed]
- Vagin, A. & Teplyakov, A. (1997). J. Appl. Cryst.30, 1022–1025.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: E2-25K, M172A mutant, 3e46, r3e46sf
PDB reference: 3f92, r3f92sf
PDB reference: E2-25K, M172A mutant, 3e46, r3e46sf
PDB reference: 3f92, r3f92sf