

The expression, purification, refolding and crystallization of the outer membrane protein AlgE from P. aeruginosa is described. The crystals diffracted to 3.0 Å resolution.
Keywords: AlgE, alginate, Pseudomonas aeruginosa, refolding, outer membrane proteins, β-barrel, porins, cystic fibrosis, biofilms, exopolysaccharides
Abstract
AlgE is an outer membrane protein present in alginate-producing (mucoid) Pseudomonas aeruginosa. AlgE has been overexpressed in insoluble inclusion bodies, purified under denaturing conditions and refolded in a buffer containing decyl β-d-maltopyranoside. Purified refolded AlgE was detergent-exchanged into n-octyl tetraoxyethylene and diffraction-quality crystals were grown using the hanging-drop vapor-diffusion method. The crystals grew as small hexagons with unit-cell parameters a = 98.8, b = 156.8, c = 90.4 Å, α = β = γ = 90.0°. The crystals exhibited the symmetry of space group C222 and diffracted to a minimum d-spacing of 3.0 Å. On the basis of the Matthews coefficient (V M = 3.28 Å3 Da−1), one molecule is estimated to be present in the asymmetric unit.
1. Introduction
Pseudomonas aeruginosa is the predominant pathogen implicated in chronic pulmonary infections among cystic fibrosis (CF) patients (Ramsey, 1996 ▶). The difficulty in eradicating P. aeruginosa infections arises from the intrinsic resistance of the bacteria to many clinically administered antibiotics (Hancock, 1998 ▶). This multi-drug resistance occurs in part from the conversion of the bacteria from a nonmucoid to a mucoid phenotype that parallels the transition from acute to chronic lung infection in the patient. Mucoid P. aeruginosa are characterized by their ability to secrete copious amounts of the exopolysaccharide alginate, a β-1,4-linked polymer comprised of β-d-mannuronic acid and its C-5 epimer α-l-guluronic acid (Evans & Linker, 1973 ▶). The hydroxyl groups at the C-2 and C-3 positions of β-d-mannuronic acid can be selectively acetylated and have been shown to play a role in the avoidance of opsonic phagocytosis within the host (Pier et al., 2001 ▶).
Biosynthesis of alginate begins with the synthesis of the activated sugar-nucleotide precursor GDP-mannuronic acid from fructose-6-phosphate by the concerted actions of the AlgA, AlgC and AlgD enzymes (Remminghorst & Rehm, 2006b ▶). While the biosynthesis of GDP-mannuronate has been well characterized, much less is known about the assembly and secretion of the full-length alginate polymer. Ten proteins located on the algD operon have been implicated in the polymerization, modification and export of alginate. Alginate polymerization and transport across the inner membrane appears to require two inner membrane proteins, Alg8 and Alg44 (Remminghorst & Rehm, 2006a ▶,c ▶). Alg8 has multiple transmembrane-spanning domains that surround a large cytoplasmic domain with homology to family 2 glycosyl transferases (Oglesby et al., 2008 ▶). In contrast, Alg44 has a single transmembrane domain, a cytoplasmic N-terminal bis-(3′-5′)-cyclic GMP-binding PilZ domain and a C-terminal periplasmic domain that shows homology to MexA, the periplasmic membrane-fusion protein component of the multidrug transporter complex MexAB-OprM (Merighi et al., 2007 ▶; Oglesby et al., 2008 ▶). Once in the periplasm, the polymannuronate polymer is acetylated by an unknown mechanism through the combined activities of AlgI, AlgJ and AlgF (Franklin & Ohman, 2002 ▶) and selectively epimerized by the mannuronan C-5 epimerase AlgG (Franklin et al., 1994 ▶). The proteins AlgK, AlgX and AlgL have also been shown to be required for alginate biosynthesis. AlgL appears to have a dual role involving both the degradation of alginate within the periplasm and acting as a scaffold protein within the secretion complex (Jain & Ohman, 2005 ▶; Bakkevig et al., 2005 ▶), while the precise functions of AlgK and AlgX are currently unknown (Robles-Price et al., 2004 ▶; Jain & Ohman, 1998 ▶). Export of the mature polysaccharide is believed to occur through the outer membrane protein AlgE.
AlgE was first identified in protein-profiling experiments as a 54 kDa outer membrane protein that was highly expressed in mucoid strains but absent in nonmucoid strains of P. aeruginosa outer membranes (Grabert et al., 1990 ▶). Preliminary biochemical characterization of AlgE confirmed its localization to the outer membrane and demonstrated its ability to form ion channels in planar lipid bilayers (Rehm et al., 1994 ▶). Based on secondary-structure prediction, it has been suggested that AlgE forms an 18-stranded β-barrel with long extracellular loops and short periplasmic turns. In order to gain further insight into the mechanism by which alginate is transported across the outer membrane, we have undertaken structural studies of AlgE and describe here the overexpression, purification, refolding and crystallization of the protein.
2. Materials and methods
2.1. Cloning and expression
The nucleotide sequence for algE from P. aeruginosa PAO1 was obtained from the Pseudomonas Genome Database (Stover et al., 2000 ▶) and used to design gene-specific primers. The algE gene encoding the mature protein without its signal sequence, residues 33–462, was amplified from genomic DNA using the following primers: forward, 5′-CATATGGCCAACAGCGGCGAGGCGCC-3′ containing an NdeI site, and reverse, 5′-CTCGAGTCAGAAGCGCCAGATGAAGTC-3′ containing an XhoI site. The PCR product was cloned into the acceptor vector pCR-2.1 using the TOPO TA-cloning kit (Invitrogen) and sequenced to ensure that no errors were introduced during the amplification reaction. The acceptor vector containing mature algE was subsequently digested with NdeI and XhoI and subcloned into the pET28a expression vector (Novagen). The resulting expression plasmid (pET28a-AlgE) encodes the mature AlgE protein fused to an N-terminal His6 tag to facilitate purification.
Escherichia coli BL21 CodonPlus (DE3) cells (Stratagene) transformed with the AlgE expression vector were grown in 1 l Luria–Bertani (LB) broth containing 50 µg ml−1 kanamycin at 310 K until the OD600 of the cell culture reached 0.6, at which point protein expression was induced by the addition of isopropyl β-d-1-thiogalactopyranoside (IPTG) to a final concentration of 1.0 mM. The cells were incubated post-induction for an additional 4 h at 310 K before being harvested by centrifugation at 2392g for 20 min. The resulting cell pellet was resuspended in buffer A (20 mM Tris–HCl pH 8, 150 mM NaCl) and lysed by ten 30 s sonication pulses at 60% amplitude with 120 s cooling time between each pulse. The insoluble cell lysate was collected by centrifugation at 31 000g for 45 min and the soluble cell lysate was discarded. The insoluble cell lysate (containing inclusion bodies) was resuspended in buffer B (20 mM Tris–HCl pH 8, 150 mM NaCl, 8 M urea) and incubated overnight at room temperature (295 K). The remaining insoluble cellular debris was removed by centrifugation at 31 000g for 45 min and discarded. The urea-solubilized fraction containing AlgE was retained for purification.
2.2. Purification and refolding
The initial steps of protein purification were carried out under denaturing conditions. Firstly, AlgE solubilized in buffer B was diluted approximately tenfold in buffer C (20 mM Tris–HCl pH 8, 500 mM NaCl, 8 M urea, 10 mM imidazole) and incubated with 10 ml Ni–NTA agarose (Qiagen) equilibrated with buffer C for 30 min at room temperature (295 K). After packing the resin into a column, the sample was washed with 50 ml buffer C before elution with 20 ml buffer D (20 mM Tris–HCl pH 8, 500 mM NaCl, 8 M urea, 250 mM imidazole). The eluted urea-solubilized protein was estimated to be >95% pure as determined by SDS–PAGE (Fig. 1 ▶ a, lane 1). AlgE was subsequently refolded by rapid dilution into buffer E [20 mM Tris–HCl pH 8, 150 mM NaCl, 0.5%(w/v) decyl β-d-maltopyranoside (Anatrace)]. AlgE contains no cysteine residues, so the formation of disulfide bonds was not a concern. A typical refolding experiment involved the dilution of 1–2 ml of 5–10 mg ml−1 AlgE solubilized in buffer D by dropwise addition to 50 ml buffer E. The refolding of >20 mg AlgE during a single refolding experiment led to the formation of a white precipitate. AlgE diluted in buffer E was stirred overnight at 310 K and the extent of refolding was monitored by SDS–PAGE by taking advantage of the differences exhibited in the relative molecular weights of the folded and unfolded forms of the protein. Chemical (e.g. urea solubilization; Fig. 1 ▶ a, lane 1) or heat (i.e. boiling the sample prior to electrophoresis; Fig. 1 ▶ b, lane 2) denaturation of the protein resulted in a shift in molecular weight relative to the folded (untreated) form (Fig. 1 ▶ a, lanes 2 and 3; Fig. 1 ▶ b, lane 1). Heat modifiability is a well characterized property of many bacterial outer membrane β-barrel proteins (Conlan & Bayley, 2003 ▶, Kleinschmidt et al., 1999 ▶). As an additional verification that the protein was refolded, it was incubated overnight with trypsin [Sigma; 500:1(w:w)] at room temperature (295 K) in buffer E. Trypsin was observed to degrade the unfolded form of AlgE within 30 s of digestion (data not shown), while the folded species remained stable after 24 h (Fig. 1 ▶ a, lane 3). After refolding, greater than 75% of the AlgE was estimated by SDS–PAGE to be refolded and highly protease-resistant (Fig. 1 ▶ a, lane 2). As a final purification step, refolded AlgE was separated from soluble aggregates and other misfolded species by size-exclusion chromatography using a HiLoad 16/60 Superdex 200 prep-grade gel-filtration column (GE Healthcare). The final purity of refolded AlgE was judged to be >95% (Fig. 1 ▶ b). Before crystallization, the detergent was exchanged on a HiLoad 16/60 Superdex 200 prep-grade gel-filtration column equilibrated in buffer F [20 mM Tris–HCl pH 8, 150 mM NaCl, 0.45%(v/v) n-octyl tetraoxyethylene (Anatrace)]. Purified AlgE was concentrated to 8 mg ml−1 by Centricon ultrafiltration (30 kDa molecular-weight cutoff, Millipore).
Figure 1.

SDS–PAGE analysis of AlgE refolding and purification. Samples loaded on the gel were not boiled unless otherwise indicated. (a) Lane M, molecular-weight markers (kDa); lane 1, purified urea-solubilized AlgE after Ni-affinity chromatography; lane 2, AlgE after refolding in buffer containing decyl β-d-maltopyranoside; lane 3, refolded AlgE after digestion with 500:1(w:w) trypsin overnight at room temperature (295 K). (b) Lane M, molecular-weight markers (kDa); lane 1, refolded AlgE after size-exclusion chromatography; lane 2, as lane 1 except boiled for 5 min prior to SDS–PAGE analysis. Unfolded and folded forms of AlgE are designated U and F, respectively.
2.3. Crystallization
Refolded AlgE in buffer F was screened for crystallization conditions using commercially available crystal screens and the hanging-drop vapor-diffusion technique. Crystallization experiments were set up by mixing 1.0 µl AlgE solution with 1.0 µl reservoir solution and equilibrating the drop against 250 µl reservoir solution. Crystal trays were kept at room temperature (295 K) for the duration of the experiments. Hits were obtained from several conditions over a broad range of pH. All hits contained some form of citrate, either as a buffer or a salt. The best crystals were obtained from condition No. 24 [0.2 M diammonium hydrogen citrate, 10%(w/v) PEG 3350] of the Membrane Protein Screen I (Biogenova). Optimization of this condition [0.3 M diammonium hydrogen citrate, 12%(w/v) PEG 3350] yielded singular hexagonal crystals that grew to maximum dimensions (100 × 50 × 25 µm) after 7 d (Fig. 2 ▶).
Figure 2.

Crystals of refolded AlgE. The crystal dimensions are approximately 100 × 50 × 25 µm.
2.4. Data collection
Prior to data collection, the crystals were cryoprotected using a mixture of well solution and glycerol in a 3:1 ratio. 7.5 µl well solution was mixed with 2.5 µl 100% glycerol and the crystals were soaked in this solution for 10–15 s prior to vitrification in liquid nitrogen. Cryoprotected crystals were stored in liquid nitrogen until data collection. Diffraction data were collected on a Rigaku MicroMax-007 HF operated at 40 kV and 30 mA with Osmic multilayer optics. A total of 185 images of 1° Δϕ oscillation were collected on a MAR345 image-plate detector with a 250 mm crystal-to-detector distance and an exposure time of 120 s per image. The data were integrated, reduced and scaled using XDS (Kabsch, 1993 ▶). The data-collection statistics are summarized in Table 1 ▶.
Table 1. Data-collection statistics.
Values in parentheses are for the highest resolution shell.
| Wavelength (Å) | 1.54 |
| Temperature (K) | 100 |
| Space group | C222 |
| Unit-cell parameters (Å, °) | a = 89.8, b = 156.8, c = 90.4, α = β = γ = 90.0 |
| Resolution (Å) | 90.4–3.0 (3.1–3.0) |
| Total No. of reflections | 107206 |
| No. of unique reflections | 26753 |
| Redundancy | 7.4 (7.5) |
| Completeness (%) | 100 (100) |
| Average I/σ(I) | 13.5 (4.2) |
| Rmerge† (%) | 12.6 (40.8) |
R
merge = 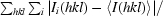
 , where Ii(hkl) and 〈I(hkl)〉 represent the diffraction-intensity values of the individual measurements and the corresponding mean values, respectively.
, where Ii(hkl) and 〈I(hkl)〉 represent the diffraction-intensity values of the individual measurements and the corresponding mean values, respectively.
3. Results
The outer membrane protein AlgE from P. aeruginosa has been expressed, refolded, purified and crystallized. Crystallization was facilitated by a refolding protocol that allowed the preparation of milligram quantities of this membrane protein. Successful refolding was verified both by heat modifiability and protease resistance (Fig. 1 ▶). Detergent exchange of refolded AlgE into the ethereal detergent n-octyl tetraoxyethylene (C8E4) allowed the growth of diffraction-quality crystals (Fig. 2 ▶). The crystals diffracted to 3.0 Å resolution and belonged to space group C222, with unit-cell parameters a = 98.8, b = 156.8, c = 90.4 Å, α = β = γ = 90.0°. The calculated solvent content based on the presence of a monomer in the asymmetric unit is 62.4% (V M = 3.28 Å3 Da−1; Matthews, 1968 ▶). We are currently in the process of determining the structure of this protein using selenomethionine incorporation and the multiple-wavelength anomalous diffraction technique (Hendrickson, 1991 ▶).
Acknowledgments
The authors thank the Advanced Protein Technology Centre at The Hospital for Sick Children for assistance with DNA sequencing. This work was supported by a research grant from the Canadian Institutes of Health Research (CIHR # MT13337) to PLH. PLH is the recipient of a Canada Research Chair. JCCW and AMN were funded in part by graduate scholarships from the Canadian Cystic Fibrosis Foundation, the Ontario Graduate Scholarship Program, the Ontario Student Opportunities Trust fund and The Hospital for Sick Children Foundation student scholarship program and by a CIHR postdoctoral fellowship, respectively. DEO was supported by Veterans Administration Medical Research Funds and Public Health Service Grant AI-19146 from the National Institute of Allergy and Infectious Disease.
References
- Bakkevig, K., Sletta, H., Gimmestad, M., Aune, R., Ertesvag, H., Degnes, K., Christensen, B. E., Ellingsen, T. E. & Valla, S. (2005). J. Bacteriol.187, 8375–8384. [DOI] [PMC free article] [PubMed]
- Conlan, S. & Bayley, H. (2003). Biochemistry, 42, 9453–9465. [DOI] [PubMed]
- Evans, L. R. & Linker, A. (1973). J. Bacteriol.116, 915–924. [DOI] [PMC free article] [PubMed]
- Franklin, M. J., Chitnis, C. E., Gacesa, P., Sonesson, A., White, D. C. & Ohman, D. E. (1994). J. Bacteriol.176, 1821–1830. [DOI] [PMC free article] [PubMed]
- Franklin, M. J. & Ohman, D. E. (2002). J. Bacteriol.184, 3000–3007. [DOI] [PMC free article] [PubMed]
- Grabert, E., Wingender, J. & Winkler, U. K. (1990). FEMS Microbiol. Lett.56, 83–87. [DOI] [PubMed]
- Hancock, R. E. (1998). Clin. Infect. Dis.27, Suppl. 1, S93–S99. [DOI] [PubMed]
- Hendrickson, W. A. (1991). Science, 254, 51–58. [DOI] [PubMed]
- Jain, S. & Ohman, D. E. (1998). J. Bacteriol.180, 634–641. [DOI] [PMC free article] [PubMed]
- Jain, S. & Ohman, D. E. (2005). Infect. Immun.73, 6429–6436. [DOI] [PMC free article] [PubMed]
- Kabsch, W. (1993). J. Appl. Cryst.26, 795–800.
- Kleinschmidt, J. H., Wiener, M. C. & Tamm, L. K. (1999). Protein Sci.8, 2065–2071. [DOI] [PMC free article] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol.33, 491–497. [DOI] [PubMed]
- Merighi, M., Lee, V. T., Hyodo, M., Hayakawa, Y. & Lory, S. (2007). Mol. Microbiol.65, 876–895. [DOI] [PubMed]
- Oglesby, L. L., Jain, S. & Ohman, D. E. (2008). Microbiology, 154, 1605–1615. [DOI] [PMC free article] [PubMed]
- Pier, G. B., Coleman, F., Grout, M., Franklin, M. & Ohman, D. E. (2001). Infect. Immun.69, 1895–1901. [DOI] [PMC free article] [PubMed]
- Ramsey, B. W. (1996). N. Engl. J. Med.335, 179–188. [DOI] [PubMed]
- Rehm, B. H., Boheim, G., Tommassen, J. & Winkler, U. K. (1994). J. Bacteriol.176, 5639–5647. [DOI] [PMC free article] [PubMed]
- Remminghorst, U. & Rehm, B. H. (2006a). Appl. Environ. Microbiol.72, 298–305. [DOI] [PMC free article] [PubMed]
- Remminghorst, U. & Rehm, B. H. (2006b). Biotechnol. Lett.28, 1701–1712. [DOI] [PubMed]
- Remminghorst, U. & Rehm, B. H. (2006c). FEBS Lett.580, 3883–3888. [DOI] [PubMed]
- Robles-Price, A., Wong, T. Y., Sletta, H., Valla, S. & Schiller, N. L. (2004). J. Bacteriol.186, 7369–7377. [DOI] [PMC free article] [PubMed]
- Stover, C. K. et al. (2000). Nature (London), 406, 959–964.