

The crystallization and results of multiwavelength anomalous diffraction studies of a recombinant C3-inhibitory fragment of Efb from S. aureus are reported.
Keywords: Staphylococcus aureus, SCIN, complement component C3b
Abstract
Staphylococcus aureus secretes a number of small proteins that effectively attenuate the human innate immune response. Among these, the staphylococcal complement-inhibitor protein (SCIN) disrupts the function of the complement component 3 (C3) convertase that is initiated through either the classical or the alternative pathway and thereby prevents amplification of the complement response on the bacterial surface. Recent studies have shown that SCIN may affect the activities of the C3 convertase by binding in an equimolar fashion to C3b, which is itself an integral although non-enzymatic component of the convertase. In order to better understand the nature of the C3b–SCIN interaction, the hanging-drop vapor-diffusion technique was used to crystallize human C3b in the presence of a recombinant form of SCIN. These crystals diffracted synchrotron X-rays to approximately 6 Å Bragg spacing and grew in a primitive tetragonal space group (P41212 or P43212; unit-cell parameters a = b = 128.03, c = 468.59 Å). Cell-content analysis of these crystals was consistent with the presence of either two 1:1 complexes or a single 2:2 assembly in the asymmetric unit, both of which correspond to a solvent content of 51.9%. By making use of these crystals, solution of the C3b–SCIN structure should further our understanding of complement inhibition and immune evasion by this pathogen.
1. Introduction
Staphylococcus aureus is a highly disseminated human pathogen that causes a wide range of infections in humans (Lowy, 1998 ▶). Although it is armed with a suite of virulence-promoting factors, studies over the last several years have shown that this bacterium produces a number of proteins that attenuate or disrupt many of the body’s defense processes (Chavakis et al., 2007 ▶). This appears to be particularly true for systems of innate immunity; among these, the complement cascades are heavily targeted (Lambris et al., 2008 ▶; Geisbrecht, 2008 ▶). The staphylococcal complement inhibitor (SCIN; UniProt Accession No. Q931M7) is the prototypic member of a family of small (∼9.8 kDa) secreted proteins that disrupt complement-mediated immune responses (Rooijakkers et al., 2005 ▶; Jongerius et al., 2007 ▶). A 1.8 Å crystal structure of SCIN has recently been determined (Rooijakkers et al., 2007 ▶). Not only did this study help to shed light on important functional regions within the protein, it also revealed that the members of this family adopt a compact three-helix bundle fold that is evolutionarily related to staphylococcal protein A modules.
Native host regulators of complement activation typically limit complement activity by either (i) accelerating the decay of transiently stable C3 convertase assemblies and/or (ii) serving as a cofactor for complement factor I-mediated degradation of C3b (Morikis & Lambris, 2005 ▶). In contrast, the SCIN family of proteins instead exert their inhibitory effects by stabilizing the solid-phase C3 convertases, presumably in an inactive conformation. Interestingly, previous studies suggested that SCIN proteins could achieve this inhibitory stabilization even though these bacterial proteins did not appear to interact directly with any components of the C3 convertases (Rooijakkers et al., 2005 ▶). This observation raised questions regarding the nature of the interactions that culminate in SCIN-mediated inhibition of C3 convertases.
We recently discovered that SCIN binds to C3b with nanomolar affinity (Ricklin et al., 2009 ▶). This interaction apparently lies at the heart of SCIN function, because an inactive SCIN ortholog (known as SCIN-D or ORF-D; Rooijakkers et al., 2007 ▶) lacked detectable C3b-binding ability. To further characterize the C3b–SCIN interaction at the molecular level, we have initiated a series of experiments to determine the three-dimensional structure of the C3b–SCIN complex. In this report, we describe the crystallization of C3b in the presence of SCIN and present the preliminary results of diffraction studies on these crystals.
2. Materials and methods
2.1. Protein expression and purification
A designer gene fragment encoding residues 32–116 of SCIN (S. aureus strain Mu50) was PCR-amplified from purified bacterial genomic DNA using oligonucleotide primers that appended SalI and NotI sites at the 5′ and 3′ ends of the product, respectively. The resulting fragment was digested with the appropriate restriction endonucleases and subcloned into the vector pT7HMT (Geisbrecht et al., 2006 ▶) and the recombinant insert was sequenced in its entirety. This sequence-confirmed expression vector was transformed into Escherichia coli strain BL21 (DE3) for protein production.
Recombinant SCIN was overexpressed, initially purified by metal-ion affinity chromatography carried out in the presence of 8 M urea as a denaturant, refolded by rapid dilution and concentrated according to the general protocols set forth in a previous publication (Geisbrecht et al., 2006 ▶). Prior to crystallization screening, the vector-encoded amino-terminal affinity tag was removed from SCIN by proteolytic digestion with recombinant tobacco etch virus (TEV) protease (Geisbrecht et al., 2006 ▶) in a buffer compatible with metal-ion affinity-chromatography methods (20 mM Tris pH 8.0, 500 mM NaCl, 10 mM imidazole). Upon completion of this digestion, SCIN was purified further by metal-ion affinity chromatography; however, in this case the unbound fraction was retained. This fraction was buffer-exchanged into 20 mM acetate pH 5.0, applied onto a 6 ml Resource S column (GE Biosciences) and eluted with a linear gradient to 20 mM acetate pH 5.0, 1 M NaCl over 7.5 column volumes. The fractions which contained purified SCIN (as judged by SDS–PAGE) were pooled, dialyzed twice against 4 l double-deionized water and concentrated by ultrafiltration to 10 mg ml−1 (as determined by UV absorption, where ∊ = 8960 M −1 cm−1). The final preparation contained the residues Gly-Ser-Thr at the amino-terminus of the protein as an artifact of appending the SalI endonuclease recognition site at the 5′ end of the SCIN coding sequence.
Purified C3b was prepared by activated-thiol Sepharose (GE Biosciences) separation of a limited trypsin digestion of isolated human C3 according to previously established protocols (Lambris & Ross, 1982 ▶). Following purification, C3b was exchanged into phosphate-buffered saline, concentrated to 20 mg ml−1 (as determined by UV absorption, where ∊ = 168 390 M −1 cm−1), aliquoted and stored at 193 K until use.
2.2. Crystallization sample preparation and screening
Biochemical analyses indicate that SCIN binds C3b in an equimolar complex (Ricklin et al., 2009 ▶). As a result, solutions of each purified monomer were mixed to yield an equimolar complex of C3b–SCIN prior to buffer exchange by ultrafiltration into 10 mM HEPES–NaOH pH 7.4. Following buffer exchange, the sample was concentrated to 5 mg ml−1 complex (as judged by UV absorption; Fig. 1 ▶ a). A theoretical extinction coefficient equal to the sum of the contributions from each monomer present in the complex was used as a basis for sample quantitation (∊ = 177 350 M −1 cm−1).
Figure 1.

Sample preparation and crystallization. (a) Coomassie-stained SDS–PAGE analysis of the crystallization samples. Electrophoresis was performed under nonreducing conditions. Molecular-mass standards are shown in the left-hand lane, while the crystallization sample is shown in the right-hand lane. The positions of the bands corresponding to C3b and SCIN are indicated. (b) Coomassie-stained native PAGE analysis of C3b alone (lane 1), C3b–SCIN crystallization sample (lane 2) and C3b–C3-9 F(ab) (lane 3) as a positive control for binding. (c) Representative crystals derived from a sample of C3b–SCIN. The scale bar is approximately 50 µm.
Native PAGE was used to ensure that the C3b–SCIN sample described above was associated at the concentrations employed in crystallization screening (Fig. 1 ▶ b). 2 µl each of C3b alone and C3b–SCIN (both samples at 5 mg ml−1) were separated on a Novex 3–8% gradient Tris acetate gel. An equivalent sample of C3b bound to an equimolar quantity of the F(ab) from monoclonal anti-C3c antibody C3-9 was used as a control (Hack et al., 1988 ▶). C3-9 F(ab) was prepared and isolated as described previously (Nishida et al., 2006 ▶). Electrophoresis was carried out at 150 V for 120 min using an electrode buffer of 25 mM Tris and 192 mM glycine (pH 8.3).
Initial crystallization screening was performed using a hanging-drop vapor-diffusion sparse-matrix approach at 273 K. This identified approximately five potential crystallization conditions, although only a single condition yielded single crystals in a time-frame practical for routine sample reproduction.
2.3. X-ray data collection
An X-ray diffraction data set was collected at 93 K on beamline 22-ID of the Advanced Photon Source (Argonne National Laboratory). Prior to data collection, single crystals were briefly soaked in a fresh aliquot of the well buffer described above to rid the samples of fine amorphous precipitate. Individual samples were then flash-cooled by submersion in a dewar of liquid nitrogen. Diffraction data were collected with a 1° oscillation range. Because of the modest diffraction limits and large cell edge (Table 1 ▶) inherent to these crystals, the MAR300 CCD detector was maintained at a distance of 750 mm. The individual reflections were indexed, integrated and scaled using the HKL-2000 software package (Otwinowski, 1993 ▶; Otwinowski & Minor, 1997 ▶). Complete data-collection and processing statistics for these crystals are presented in Table 1 ▶.
Table 1. Data-collection and processing statistics for C3b–SCIN crystals.
Values in parentheses are for the highest resolution shell.
| Unit-cell parameters (Å) | a = b = 128.03, c = 468.59 |
| Wavelength (Å) | 0.9999 |
| Resolution limits (Å) | 500–6.0 |
| No. of reflections | 56928 |
| No. of unique reflections | 7294 |
| Completeness (%) | 69.5 (29.2) |
| 〈I/σ(I)〉 | 16.2 (1.7) |
| Rmerge† (%) | 9.9 (52.2) |
R
merge = 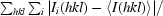
 , where I
i(hkl) is the ith measurement of reflection hkl and 〈I(hkl)〉 is a weighted mean of all measurements of hkl.
, where I
i(hkl) is the ith measurement of reflection hkl and 〈I(hkl)〉 is a weighted mean of all measurements of hkl.
3. Results and discussion
In vitro reconstitution was used to prepare a sample of the C3b–SCIN complex from the individual purified monomers, as shown in Figs. 1 ▶(a) and 1 ▶(b). This sample was used to carry out crystallization trials over the course of approximately eight weeks. A handful of potential crystallization conditions were identified near neutral pH values that contained ammonium sulfate (2.0–3.0 M) as the primary precipitant. However, these conditions required prolonged incubation times of between six and eight weeks and resulted in samples that were too small to manipulate successfully by hand. As a consequence, they were not pursued further.
An alternative condition that yielded small block-shaped single crystals within 3 d was also discovered. Optimization of the initial condition resulted in a final crystallization protocol in which 1 µl 5 mg ml−1 C3b–SCIN sample was mixed with 1 µl 0.1 M HEPES–NaOH pH 7.0, 30%(v/v) Jeffamine ED-2001–HCl pH 7.0 and equilibrated over 500 µl of the same buffer at 293 K. Block-shaped single crystals of appeared within 3 d and continued growing slowly over the course of 21 d to a final dimension of approximately 0.05 mm on the longest side (Fig. 1 ▶ c). Importantly, efforts to reproduce these samples in the absence of either component failed to yield similar crystals. Since both monomers have been crystallized separately from distinct conditions (Rooijakkers et al., 2007 ▶; Janssen et al., 2006 ▶), this strongly suggested that each protein component was present in the crystals reported here.
The size of the crystals shown in Fig. 1 ▶(c) permitted their manipulation, cryoprotection and subsequent diffraction analysis using synchrotron X-rays (Fig. 2 ▶). A data set consisting of 120° of oscillation diffraction images from a single flash-cooled crystal was collected and processed (Table 1 ▶). These data extended to modest resolution and there were few reflections present beyond approximately 6 Å Bragg spacing. In addition, these data were limited by completeness in the highest resolution shell. Attempts to address this issue were hampered by radiation-induced decay upon prolonged exposure to the X-ray source. In this respect, it is important to note that previous crystallographic studies of C3b, either free or bound with various ligands, also relied on crystal systems that diffracted to comparable resolution limits (Janssen et al., 2006 ▶; Wiesmann et al., 2006 ▶). This property is most likely to be a consequence of inherent flexibility between the dozen unique structural domains present in C3b, which itself is a rather large protein (∼176 kDa).
Figure 2.

Diffraction patterns resulting from the exposure of crystals to synchrotron radiation. The images shown in (a) and (b) are separated by a 45° rotation through the angle ϕ.
Despite these limitations, the crystals described here still revealed important features regarding this complex. To begin with, the C3b–SCIN crystals grew in a primitive tetragonal lattice. Subsequent analyses of systematic absences using the program SGPR4D (Fu et al., 2005 ▶) strongly suggested that the crystals belonged to space group P41212 or its enantiomorph P43212. Furthermore, crystals grown in this fashion exhibited a rather large unit cell. In particular, the longest cell edge, c, spanned nearly 470 Å. Depending upon the orientation of the crystallographic axes to the beam, this feature was readily visible in certain diffraction patterns [e.g. Fig. 2 ▶(b) versus Fig. 2 ▶(a)] even when the CCD detector was maintained at a distance of 750 mm.
Recent surface plasmon resonance and small-angle X-ray scattering studies have suggested that the C3b–SCIN complex exists as an equilibrium distribution between 1:1 and 2:2 arrangements (Ricklin et al., 2009 ▶). Understanding the nature of this equilibrium, as well as the physical basis of it structure, is important because the C3 convertases, which are the primary targets of SCIN and its analogs, occur to a large extent within the context of surfaces. This feature would be expected to favor the formation of the higher order 2:2 C3b–SCIN assembly (Grasberger et al., 1986 ▶). Cell-content analyses of the crystals reported here were consistent with the presence of either two 1:1 (186.3 kDa) complexes or a single 2:2 (372.6 kDa) complex in the asymmetric unit. These values corresponded to an empirical V M estimate of 2.6 Å3 Da−1 or a solvent content of 51.9%. Examination of self-rotation maps calculated from reflections between 10 and 12 Å Bragg spacing revealed the presence of only formal crystallographic symmetry axes (data not shown). Thus, it is not yet known whether the contents of the crystallographically unique unit reflect an asymmetric dimerization of the C3b–SCIN complex or whether the biological unit is perhaps divided between adjacent asymmetric units in the unit cell. Resolution of these issues will require solution of the C3b–SCIN structure by molecular or multiple isomorphous replacement methods. In turn, this will provide a basis for understanding the C3b–SCIN interaction and immune invasion by S. aureus more generally.
Acknowledgments
This work was supported by a grant from the National Institute of Allergy and Infectious Diseases (AI071028). Data were collected on Southeast Regional Collaborative Access Team (SER-CAT) 22-ID beamline at the Advanced Photon Source, Argonne National Laboratory. A list of supporting institutions may be found at http://www.ser-cat.org/members.html. Use of the Advanced Photon Source was supported by the US Department of Energy, Office of Science, Office of Basic Energy Sciences under contract No. W-31-109-Eng-38. We thank Drs Zhongmin Jin and Zheng-Qing Fu of SER-CAT for expert assistance with diffraction data collection.
References
- Chavakis, T., Preissner, K. T. & Herrmann, M. (2007). Trends Immunol.28, 408–418. [DOI] [PubMed]
- Fu, Z.-Q., Rose, J. & Wang, B.-C. (2005). Acta Cryst. D61, 951–959. [DOI] [PubMed]
- Geisbrecht, B. V. (2008). Adv. Exp. Med. Biol.632, 221–236. [PubMed]
- Geisbrecht, B. V., Bouyain, S. & Pop, M. (2006). Protein Expr. Purif.46, 23–32. [DOI] [PubMed]
- Grasberger, B., Minton, A. P., DeLisi, C. & Metzger, H. (1986). Proc. Natl Acad. Sci. USA, 83, 6258–6262. [DOI] [PMC free article] [PubMed]
- Hack, C. E., Paardekooper, J., Smeenk, R. J. T., Abbink, J., Eerenber, A. J. M. & Nuijens, J. H. (1988). J. Immunol.141, 1602–1609. [PubMed]
- Janssen, B. J. C., Christodoulidou, A., McCarthy, A., Lambris, J. D. & Gros, P. (2006). Nature (London), 444, 213–216. [DOI] [PubMed]
- Jongerius, I., Köhl, J., Pandey, M. K., Ruyken, M., van Kessel, K. P., van Strijp, J. A. & Rooijakkers, S. H. (2007). J. Exp. Med.204, 2461–2471. [DOI] [PMC free article] [PubMed]
- Lambris, J. D., Ricklin, D. & Geisbrecht, B. V. (2008). Nature Rev. Microbiol.6, 132–142. [DOI] [PMC free article] [PubMed]
- Lambris, J. D. & Ross, G. D. (1982). J. Immunol.128, 186–189. [PubMed]
- Lowy, F. D. (1998). N. Engl. J. Med.339, 520–532. [DOI] [PubMed]
- Morikis, D. & Lambris, J. D. (2005). Structural Biology of the Complement System. Boca Raton, USA: Taylor & Francis.
- Nishida, N., Walz, T. & Springer, T. A. (2006). Proc. Natl Acad. Sci. USA, 103, 19737–19742. [DOI] [PMC free article] [PubMed]
- Otwinowski, Z. (1993). Proceeding of the CCP4 Study Weekend: Data Collection and Processing, edited by L. Sawyer, N. Isaacs & S. Bailey, pp. 56–62. Warrington: Daresbury Laboratory.
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Ricklin, D., Tzekou, A., Garcia, B. L., Hammel, M., McWhorter, W. J., Sfyroera, G., Wu, Y.-Q., Geisbrecht, B. V. & Lambris, J. D. (2009). Submitted. [DOI] [PMC free article] [PubMed]
- Rooijakkers, S. H. M., Milder, F. J., Bardoel, B. W., Ruyken, M., van Strijp, J. A. G. & Gros, P. (2007). J. Immunol.179, 2989–2998. [DOI] [PubMed]
- Rooijakkers, S. H. M., Ruyken, M., Roos, A., Daha, M. R., Presanis, J. S., Sim, R. B., van Wamel, W. J. B., van Kessel, K. P. M. & van Strijp, J. A. G. (2005). Nature Immunol.6, 920–927. [DOI] [PubMed]
- Wiesmann, C., Katschke, K. J., Yin, J., Helmy, K. Y., Steffek, M., Fairbrother, W. J., McCallum, S. A., Embuscado, L., DeForge, L., Hass, P. E. & van Lookeren Campagne, M. (2006). Nature (London), 444, 217–220. [DOI] [PubMed]