

A joint X-ray and neutron crystallographic study has been initiated to determine the specific water network and the protonation states of the hydrophilic residues that coordinate it in human carbonic anhydrase II.
Keywords: carbonic anhydrase II, neutron diffraction
Abstract
Carbonic anhydrases catalyze the interconversion of CO2 to HCO3 −, with a subsequent proton-transfer (PT) step. PT proceeds via a proposed hydrogen-bonded water network in the active-site cavity that is stabilized by several hydrophilic residues. A joint X-ray and neutron crystallographic study has been initiated to determine the specific water network and the protonation states of the hydrophilic residues that coordinate it in human carbonic anhydrase II. Time-of-flight neutron crystallographic data have been collected from a large (∼1.2 mm3) hydrogen/deuterium-exchanged crystal to 2.4 Å resolution and X-ray crystallographic data have been collected from a similar but smaller crystal to 1.5 Å resolution. Obtaining good-quality neutron data will contribute to the understanding of the catalytic mechanisms that utilize water networks for PT in protein environments.
1. Introduction
The positions of H atoms in macromolecules are never guaranteed using X-ray crystallographic techniques, even when atomic resolution (≤1.0 Å) can be obtained. If atomic resolution can be achieved, the interpretation of electron-density maps can still be ambiguous, especially when assigning the positions of H atoms of water molecules with high (>20 Å2) thermal parameters (Gutberlet et al., 2001 ▶). However, neutron crystallographic data at medium (∼2.0 Å) resolution can complement medium- to high-resolution (∼2.0–1.0 Å) X-ray crystallographic data and allow the placement and analysis of key H atoms (Katz et al., 2006 ▶; Bennett et al., 2006 ▶; Fisher, Anderson et al., 2007 ▶; Blakeley, Ruiz et al., 2008 ▶; Coates et al., 2008 ▶; Blum et al., 2009 ▶).
H has a neutron scattering length (−3.7 × 10−15 m or −3.7 fm) that is similar in magnitude but opposite in sign to those of other atoms found in proteins (O, 5.8 fm; N, 9.4 fm; C, 6.6 fm; S, 2.8 fm), while deuterium (D) has a positive scattering length (6.7 fm) and a significantly smaller incoherent neutron-scattering cross-section (∼2.0 barns versus 80 barns for H). Consequently, the signal-to-noise ratio can be markedly improved in neutron scattering when D atoms can be substituted for H atoms. In contrast to neutrons, the diffraction of X-rays depends on the number of electrons; as H and D are relatively electron-poor compared with the heavier atoms found in proteins, they diffract very weakly and appear practically invisible. All these factors together make it much easier to locate D or H in resulting nuclear density maps (Shu et al., 2000 ▶) and neutron crystallography can readily provide information on the protonation states of amino-acid residues, ligands and the nature of bonds involving hydrogen (Blakeley, Langan et al., 2008 ▶). Neutron crystallography can also be used to identify H atoms that are exchanged with D and the extent of this replacement, thus providing a tool for identifying isotopically labeled structural features, for studying solvent accessibility and macromolecular dynamics and for identifying minimal protein-folding domains (Bennett et al., 2008 ▶).
Neutron crystallography is also a powerful tool for studying the hydration of macromolecules, especially when it is combined with X-ray crystallography in joint (XN) refinement procedures (Blum et al., 2009 ▶). In electron-density maps, a water molecule is usually represented as a spherical density peak corresponding to the position of the O atom. However, in neutron scattering density maps, owing to the strong scattering contribution from D, the density associated with D2O may no longer be spherical but rather extended. It can often be difficult to interpret these extended neutron scattering density peaks. However, we have found that using both X-ray and neutron data together can greatly help in this interpretation by allowing placement of the O atom and the subsequent interpretation of the extended neutron scattering density peak as either one or two D atoms. For this kind of analysis it is ideal to have neutron data to 2.0 Å resolution or better. Both the ability to accurately place and orient D2O and to view the resulting hydrogen-bonded patterns they participate in can give enormous advantages in understanding how enzymes work.
Carbonic anhydrases (CAs) are ubiquitous enzymes that are found in all phyla of life and are intricately involved in many physiological processes (Tufts et al., 2003 ▶). Of all the isoforms, human carbonic anhydrase II (HCA II) is the most studied and probably the best understood. CAs catalyze the reversible hydration/dehydration of CO2/HCO3 −. The reaction occurs in two distinct steps: the conversion of CO2 to HCO3 − and a subsequent proton-transfer (PT) step to regenerate the active site (see equations below; E = enzyme, B = H+ acceptor/donor, which can be buffer, His64 or bulk solvent; Christianson & Fierke, 1996 ▶; Silverman & Lindskog, 1988 ▶).
The first step is fairly well understood and a crystal structure of the enzyme–substrate complex was recently determined showing the substrate-binding pocket for the first time (Domsic et al., 2008 ▶; PDB code 3d92). The second step, which is also the rate-limiting step of catalysis, is less well defined but is believed to proceed with the transfer of a H+ from the Zn-bound water to a proton-shuttling residue, His64, via a hydrogen-bonded network of water molecules that span the 8 Å distance to the bulk solvent (Lindskog & Silverman, 2000 ▶; Cui & Karplus, 2003 ▶; Fisher et al., 2005 ▶; Fisher, Maupin et al., 2007 ▶). His64 sits on the edge of the cone-shaped active site and is ∼8 Å away from the ZnOH−/H2O. In the crystal structures of HCA II determined at various pH values it has been observed that this proton shuttle can occupy two distinct conformations. The two conformations are the so-called ‘in’ and ‘out’ positions and it has been suggested that flexibility is a requirement for efficient PT from the solvent network to the bulk solvent (Nair & Christianson, 1991 ▶; Fisher et al., 2005 ▶; Maupin & Voth, 2007 ▶; Silverman & McKenna, 2007 ▶). There are several important residues (Tyr7, Asn62, Asn67, Thr199 and Thr200) that line the active site that participate in hydrogen bonds with these water molecules and it is thought that they are important for ordering the network (Fisher, Tu et al., 2007 ▶).
Despite the abundance of kinetic and structural information on HCA II, the PT events in the catalytic cycle are not well understood or characterized. Even the determination of high-resolution X-ray crystal structures has failed to reveal the hydrogen-bonded details of the water network and the protonation states of the residues involved in the PT process (Duda et al., 2003 ▶; Fisher, Maupin et al., 2007 ▶). This enzyme provides a model system for the structure–function analyses of the role of hydrogen-bonded chains in rapid intramolecular PT over considerable distances in protein environments. HCA II and other CAs from diverse sources, overexpressed in bacterial systems, are also being widely developed for biomimetic carbon sequestration. CAs can be cheaply produced, are easily immobilized and work at a very high rate over a broad pH range, but their optimization for industrial application in carbon sequestration will require a detailed understanding of their catalytic cycle and the rate-limiting PT. Combining neutron crystallography with X-ray crystallography provides an ideal technique for observing the hydrogen-bonding networks and understanding their relation to PT in these and similar systems. To this end, H/D-exchanged crystals of HCA II have been prepared and X-ray and neutron crystallographic data have been collected at room temperature (RT) for joint XN crystallographic refinement of the water structure. Here, we report the preliminary data and feasibility of such a study.
2. Crystallization of the HCA II enzyme
HCA II was prepared as described previously (Fisher, Maupin et al., 2007 ▶). Briefly, HCA II was expressed in BL21 DE* pLysS Escherichia coli. The cells were grown in LB medium and expression was induced with 1 mM IPTG; 1 mM ZnSO4 was added at the time of expression. The cell lysate was loaded onto pAMBS (Sigma) resin and after several wash steps (0.2 M sodium sulfate, 100 mM Tris pH 9.0 and 7.0), the HCA II was eluted with 0.4 M sodium azide. The eluted protein was then buffer-exchanged into 50 mM Tris pH 8.0 using a desalting column from Pharmacia. Purified protein was concentrated to 15 mg ml−1 using a Millipore Amicon Ultra centrifugal filter unit with a molecular-weight cutoff of 10 kDa. Crystallization of the enzyme was carried out according to the sitting-drop method employing the Sandwich Box from Hampton Research. 400 µl crystallization drops were set up in a nine-well siliconized glass plate by mixing 200 µl well solution (containing 1.3 M sodium citrate, 0.1 M Tris pH 9.0) with 200 µl protein sample. No crystallization occurred after a week, so the concentration of sodium citrate in the well solution was increased to ∼1.6 M by the direct addition of sodium citrate salt. After a few days, sodium citrate recrystallized in the well solution, indicating that it was saturated. Vapor diffusion against 30 ml well solution was allowed to occur at room temperature until useful crystals appeared after ∼2 weeks. Crystal growth was all performed in H2O.
3. Neutron and X-ray data collection and reduction
A candidate crystal of 1.2 mm3 in volume was selected by visual inspection and mounted in a quartz capillary (Fig. 1 ▶), with D2O mother liquor placed at one end of the capillary. The D2O mother liquor was then exchanged each week for two months to ensure H/D exchange of most of the exchangable H atoms. In a recent study by Bennett and coworkers it was reported that, depending on the local fold of the protein and solvent accessibility, most labile H atoms are exchanged by D atoms fairly rapidly; up to 80% amide backbone exchange can occur in some proteins within 30–60 d (Bennett et al., 2008 ▶). Indeed, studies conducted using mass spectometry indicate that some labile and highly acccessible H atoms exchange to D atoms on the second to minute timescale (Truhlar et al., 2006 ▶; Yan & Maier, 2009 ▶). The crystal was then transported to the Protein Crystallography Station (PCS) at the Los Alamos Neutron Science Center (LASNCE; Langan et al., 2004 ▶). A 24 h test exposure was taken from the HCA II crystal depicted in Fig. 1 ▶, yielding diffraction to ∼2.4 Å resolution. The diffraction quality and the signal-to-noise ratio were deemed to be suitable for full data-set collection.
Figure 1.

Crystal of human carbonic anhydrase II used for neutron diffraction data collection. The crystal measured 4.0 × 1.0 × 0.3 mm in size (∼1.2 mm3) and was mounted in a quartz capillary. Each dimension on the ruler in the lower part of the photograph measures 1 mm.
Time-of-flight wavelength-resolved Laue images were collected at room temperature on a Huber κ-circle goniometer at 27 usable settings, with approximately 32 h exposure time per diffraction image (Fig. 2 ▶). The crystal-to-detector distance was kept fixed at 730 mm, which corresponds to the cylindrical radius of the detector. Owing to the limited vertical span of the detector (120° horizontal span and 16° vertical span), the crystal was reoriented seven times using the κ and ω goniometer circles and ϕ scans were performed at each crystal orientation. Each image was processed using a version of d*TREK (Pflugrath, 1999 ▶) modified for wavelength-resolved Laue neutron protein crystallography (Langan & Greene, 2004 ▶). The integrated reflections were wavelength-normalized using LAUENORM (Helliwell et al., 1989 ▶) and then merged using SCALA incorporated into CCP4i (Evans, 2006 ▶; Diederichs & Karplus, 1997 ▶; Weiss & Hilgenfeld, 1997 ▶; Collaborative Computational Project, Number 4, 1994 ▶). The ‘tails’ of the wavelength range were cut off slightly, with a restricted range of 0.7–6.5 Å, from the original 0.6–7.0 Å wavelength distribution of the thermal neutrons so as to eliminate the least accurately measured reflections. The overall completeness was 82% to 2.4 Å resolution, with an R merge of ∼23% and a redundancy of 2.8. The multiplicity value was as expected for the low-symmetry space group P21 (Table 1 ▶). In fact, the diffraction pattern extended to about 2.2 Å resolution and 20 additional frames, requiring a further 30 d of neutron beam time, will be collected to complete the data set to this resolution as beam time becomes available on the PCS.
Figure 2.

Neutron Laue diffraction pattern for the HCA II crystal in two different crystal settings. In each crystal setting the three-dimensional time-of-flight diffraction data were collapsed in the time dimension to produce a conventional two-dimensional Laue pattern.
Table 1. Room-temperature neutron and X-ray diffraction data-collection statistics.
Values in parentheses are for the highest resolution shell.
| Neutron | X-ray | |
|---|---|---|
| Source | PCS, LANSCE, LANL | Rigaku RU-H3R |
| Settings | 27 | 1255 [0.5° oscillation] |
| Space group | P21 | P21 |
| Unit-cell parameters (Å) | [same as X-ray] | a = 42.6, = 41.6, c = 72.8, β = 104.6 |
| Unit-cell volume (Å3) | 125097 | |
| Resolution | 28.9–2.40 (2.53–2.40) | 25.0–1.50 (1.55–1.50) |
| No. of reflections (measured/unique) | 22624/7975 | 959116/38510 |
| Redundancy | 2.8 (2.2) | 9.4 (8.6) |
| Completeness (%) | 82.1 (68.4) | 95.9 (91.9) |
| Rmerge (%)† | 22.9 (36.8) | 5.8 (30.8) |
| Wavelength range (Å) | 0.7–6.5 | 1.54 [monochromatic Cu Kα] |
| Mean I/σ(I) | 4.2 (1.6) | 31.0 (5.7) |
R
merge = 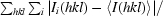
 .
.
A room-temperature X-ray diffraction data set (Table 1 ▶) was collected from a smaller crystal that grew in the same crystallization drop as the larger crystal used for neutron diffraction data collection. The smaller X-ray crystal was subjected to the same intra-capillary H/D-exchange process as the larger neutron crystal prior to X-ray data collection. Data were collected on an in-house R-AXIS IV++ image plate using a Rigaku HU-H3R Cu rotating-anode generator operated at 50 kV and 100 mA. The crystal-to-detector distance was 80 mm and the oscillation steps were 0.5°, with a 2 min exposure per frame. Data processing and reduction were performed using HKL-2000 (Otwinowski & Minor, 1997 ▶). An X-ray data set from an isomorphous crystal is required for the structure determination to enable joint XN refinement of the HCA II structure. Our current neutron data-processing software is unable to refine the unit cell, thus the dimensions are provided by X-ray data processing and unit-cell refinement. Refinement of the structure is in progress utilizing a version of CNS (Brünger et al., 1998 ▶) modified for neutron refinement (nCNS; Mustyakimov & Langan, 2007 ▶) and a starting model generated from PDB entry 2ili, a high-resolution structure determined from a crystal grown under similar conditions (Fisher, Maupin et al., 2007 ▶). Initial neutron and X-ray scattering density maps calculated after one round of rigid-body refinement show clear features of the ordered water leading into the active site. Detailed examination of the active site, its hydration and other structural features will be elucidated during the course of the refinement and reported elsewhere.
4. Discussion
The current neutron diffraction study of HCA II is the first of its kind among the extensive carbonic anhydrase family. A major hurdle in these experiments is the size of the sample. Through careful and exhaustive screening, large (1.2 mm3) crystals of HCA II were obtained that made neutron data collection possible. Our hope is that the results of this study will provide a better understanding of how the enzyme facilitates PT at a rapid rate and that this insight might provide the basis for optimizing the enzyme for use in biomimetic carbon sequestration. Detailed analysis of the active site will also add to the general knowledge base of water networks in macromolecules and how they pertain to catalysis.
Acknowledgments
The PCS is funded by the Office of Biological and Environmental Research of the Department of Energy. MM and PL were partly supported by an NIH–NIGMS-funded consortium (1R01GM071939-01) between LANL and LNBL to develop computational tools for neutron protein crystallography. PL was partly supported by an LANL LDRD grant (20070131ER). AYK was supported by an LANL LDRD grant (20080789PRD3). This work was also partly funded by grants from the National Institutes of Health (GM25154 DNS and RM) and the Thomas Maren Foundation (RM). Finally, RM would like to thank A. Joseph Kalb (Gilboa) and Dean A. A. Myles for introducing him to the possibilities of neutron diffraction studies on HCA II.
References
- Bennett, B. C., Gardberg, A. S., Blair, M. D. & Dealwis, C. G. (2008). Acta Cryst. D64, 764–783. [DOI] [PubMed]
- Bennett, B., Langan, P., Coates, L., Mustyakimov, M., Schoenborn, B. P., Howell, E. E. & Dealwis, C. (2006). Proc. Natl Acad. Sci. USA, 103, 18493–18498. [DOI] [PMC free article] [PubMed]
- Blakeley, M. P., Langan, P., Niimura, N. & Podjarny, A. (2008). Curr. Opin. Struct. Biol.18, 593–600. [DOI] [PMC free article] [PubMed]
- Blakeley, M. P., Ruiz, F., Cachau, R., Hazemann, I., Meilleur, F., Mitschler, A., Ginell, A., Afonine, P., Ventura, O. N., Cousido-Siah, A., Haertlein, M., Joachimiak, A., Myles, D. & Podjarny, A. (2008). Proc. Natl Acad. Sci. USA, 105, 1844–1848. [DOI] [PMC free article] [PubMed]
- Blum, M.-M., Mustyakimov, M., Ruterjans, H., Kehe, K., Schoenborn, B. P., Langan, P. & Chen, J. C.-H. (2009). Proc. Natl Acad. Sci. USA, 106, 713–718. [DOI] [PMC free article] [PubMed]
- Brünger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J.-S., Kuszewski, J., Nilges, M., Pannu, N. S., Read, R. J., Rice, L. M., Simonson, T. & Warren, G. L. (1998). Acta Cryst. D54, 905–921. [DOI] [PubMed]
- Christianson, D. W. & Fierke, C. A. (1996). Acc. Chem. Res.29, 331–339.
- Coates, L., Tuan, H. F., Tomanicek, S., Kovalevsky, A., Mustyakimov, M., Erskine, P. T. & Cooper, J. B. (2008). J. Am. Chem. Soc.130, 7235–7237. [DOI] [PMC free article] [PubMed]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763.
- Cui, Q. & Karplus, M. (2003). J. Phys. Chem. B, 107, 1071-1078.
- Diederichs, K. & Karplus, P. A. (1997). Nature Struct. Biol.4, 269–275. [DOI] [PubMed]
- Domsic, J. F., Avvaru, B. S., Kim, C. U., Gruner, S. M., Agbandje-McKenna, M., Silverman, D. N. & McKenna, R. (2008). J. Biol. Chem.283, 30766–30771. [DOI] [PMC free article] [PubMed]
- Duda, D., Govindasamy, L., Agbandje-McKenna, M., Tu, C., Silverman, D. N. & McKenna, R. (2003). Acta Cryst. D59, 93–104. [DOI] [PubMed]
- Evans, P. (2006). Acta Cryst. D62, 72–82. [DOI] [PubMed]
- Fisher, S. Z., Anderson, S., Henning, R., Moffat, K., Langan, P., Thiyagarajan, P. & Schultz, A. J. (2007). Acta Cryst. D63, 1178–1184. [DOI] [PMC free article] [PubMed]
- Fisher, S. Z., Hernandez Prada, J., Tu, C. K., Duda, D., Yoshioka, C., An, H., Govindasamy, L., Silverman, D. N. & McKenna, R. (2005). Biochemistry, 44, 1097–1105. [DOI] [PubMed]
- Fisher, S. Z., Maupin, C. M., Budayova-Spano, M., Govindasamy, L., Tu, C. K., Agbandje-McKenna, M., Silverman, D. N., Voth, G. A. & McKenna, R. (2007). Biochemistry, 46, 2930–2937. [DOI] [PubMed]
- Fisher, S. Z., Tu, C. K., Bhatt, D., Govindasamy, L., Agbandje-McKenna, M., McKenna, R. & Silverman, D. N. (2007). Biochemistry, 46, 3803–3813. [DOI] [PubMed]
- Gutberlet, T., Heinemann, U. & Steiner, M. (2001). Acta Cryst. D57, 349–354. [DOI] [PubMed]
- Helliwell, J. R., Habash, J., Cruickshank, D. W. J., Harding, M. M., Greenhough, T. J., Campbell, J. W., Clifton, I. J., Elder, M., Machin, P. A., Papiz, M. Z. & Zurek, S. (1989). J. Appl. Cryst.22, 483–497.
- Katz, A. K., Li, X., Carrell, H. L., Hanson, B. L., Langan, P., Coates, L., Schoenborn, B. P., Glusker, J. P. & Bunick, G. J. (2006). Proc. Natl Acad. Sci. USA, 103, 8342–8347. [DOI] [PMC free article] [PubMed]
- Langan, P. & Greene, G. (2004). J. Appl. Cryst.37, 253–257.
- Langan, P., Greene, G. & Schoenborn, B. P. (2004). J. Appl. Cryst.37, 24–31.
- Lindskog, S. & Silverman, D. N. (2000). Carbonic Anhydrases: New Horizons, edited by W. R. Chegwidden, N. D. Carter & Y. H. Edwards, pp. 175–195. Basel: Birkhauser Verlag.
- Maupin, C. M. & Voth, G. A. (2007). Biochemistry, 46, 2938–2947. [DOI] [PMC free article] [PubMed]
- Mustyakimov, M. & Langan, P. (2007). Computational Tools for Macromolecular Neutron Crystallography. http://mnc.lanl.gov.
- Nair, S. K. & Christianson, D. W. (1991). J. Am. Chem. Soc.113, 9455–9458.
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Pflugrath, J. W. (1999). Acta Cryst. D55, 1718–1725. [DOI] [PubMed]
- Shu, F., Ramakrishnan, V. & Schoenborn, B. P. (2000). Proc. Natl Acad. Sci. USA, 97, 3872–3877. [DOI] [PMC free article] [PubMed]
- Silverman, D. N. & Lindskog, S. (1988). Acc. Chem. Res.21, 30–36.
- Silverman, D. N. & McKenna, R. (2007). Acc. Chem. Res.40, 669–675. [DOI] [PubMed]
- Truhlar, S. M. E., Croy, C. H., Torpey, J. W., Koeppe, J. R. & Komives, E. A. (2006). Am. Soc. Mass Spectrom.17, 1490–1497. [DOI] [PubMed]
- Tufts, B. L., Esbaugh, A. & Lund, S. G. (2003). Comput. Biochem. Physiol. A, 136, 259–269. [DOI] [PubMed]
- Weiss, M. S. & Hilgenfeld, R. (1997). J. Appl. Cryst.30, 203–205.
- Yan, X. & Maier, C. S. (2009). Methods Mol. Biol.492, 255–271. [DOI] [PubMed]