

XynC, a glycosyl hydrolase family 5 xylanase from B. subtilis, has been purified and crystallized with the primary goal of structural determination and characterization of a second xylanase from this large catalytically diverse glycosyl hydrolase family.
Keywords: xylanase C, Bacillus subtilis, family 5 xylanases
Abstract
The recent biochemical characterization of the xylanases of glycosyl hydrolase family 5 (GH 5) has identified a distinctive endo mode of action, hydrolyzing the β-1,4 xylan chain at a specific site directed by the position of an α-1,2-linked glucuronate moiety. Xylanase C (XynC), the GH 5 xylanase from Bacillus subtilis 168, has been cloned, overexpressed and crystallized. Initial data collection was performed and a preliminary model has been built into a low-quality 2.7 Å resolution density map. The crystals belonged to the primitive monoclinic space group P21. Further screening identified an additive that resulted in large reproducible crystals. This larger more robust crystal form belonged to space group P21212 and a resulting data set has been processed to 1.64 Å resolution. This will be the second structure to be solved from this unique xylanase family and the first from a Gram-positive bacterium. This work may help to identify the structural determinants that allow the exceptional specificity of this enzyme and the role it plays in the biological depolymerization and processing of glucuronoxylan.
1. Introduction
The biodegradation of lignocellulosic biomass for the purpose of bioconversion to fuel ethanol is receiving renewed attention spurred by the unstable cost of petroleum and the desire for energy independence (Demain et al., 2005 ▶; Shanmugam & Ingram, 2008 ▶). Emphasis has been directed toward optimizing physical and chemical disruption and improvement of the enzymatic pretreatment of disrupted biomass to maximize the release of fermentable sugars. New hydrolytic enzyme activities that promise to increase the efficiency of this process are of great interest. The Carbohydrate-Active Enzymes database (http://www.cazy.org/; Cantarel et al., 2009 ▶; Henrissat, 1991 ▶; Henrissat & Bairoch, 1996 ▶), which categorizes enzymes based on primary sequence similarity and hydrophobic cluster analysis, maintains an up-to-date annotation of all glycosyl hydrolases (GH). The xylanases of GH family 5 (GH 5) represent a small subfamily within family 5, which has over 1400 entries. The members of this subfamily are commonly identified by BLASTP (Altschul et al., 1990 ▶; Altschul & Lipman, 1990 ▶) as being more closely related to family GH 30 than to GH 5 enzymes (Keen et al., 1996 ▶; Larson et al., 2003 ▶). Biochemical characterization of GH 5 xylanases have revealed a novel mode of action that may contribute to bioconversion efforts.
To date, two GH 5 xylanases (EC 3.2.1.8 and/or EC 3.2.1.136) with glucuronoxylan endo-1,4-β-xylanase activity have been purified and studied for their biochemical properties. XynA from the Gram-negative bacterium Erwinia chrysanthemi (Hurlbert & Preston, 2001 ▶; Vrsanska et al., 2007 ▶) and XynC from the Gram-positive bacterium Bacillus subtilis (St John et al., 2006 ▶) share 40% sequence identity. Of primary interest in both studies was the specificity and mode of action on the complex substrate glucuronoxylan and the characterization of the xylooligomeric products generated. Both XynA and XynC are glucuronoxylan-specific and cleave the β-1,4-linked main xylan chain in an endo manner directed by the position of an α-1,2-linked glucuronate moiety. The results from several biochemical techniques all support the conclusion that cleavage occurs at the second β-1,4 linkage toward the reducing terminus from a glucuronate-substituted xylose. This mode of action results in signally substituted glucurono-xylooligosaccharides with the substitution penultimate to the reducing terminus (St John et al., 2006 ▶; Vrsanska et al., 2007 ▶). The novel specificity sets these enzymes apart from the more common endo-acting xylanases that cleave the xylan chain in accessible regions. This unique mode of action identifies the first endoxylanase family that degrades polymeric xylan in a directed rather than a random manner.
While many GH 5 xylanases have been identified through protein annotation and sequence analysis (Preston et al., 2003 ▶), XynC is only the second GH 5 xylanase to be structurally studied (Barba de la Rosa et al., 1997 ▶; Larson et al., 2003 ▶). This work reports initial efforts in protein preparation, crystallization, crystal optimization, data collection and preliminary structural characterization.
2. Materials and methods
2.1. Cloning, expression and purification of XynC
The gene encoding XynC (ynfF) of B. subtilis subsp. subtilis strain 168 (Bacillus Genetic Stock Center accession number 1A1) was cloned by PCR using a previous expression construct as template (St John et al., 2006 ▶). Primers were designed to truncate the SignalP (Bendtsen et al., 2004 ▶) determined secretion signal peptide (…VLA-AS…) on the N-terminus and create a C-terminal in-frame fusion with the vector-encoded 8×His tag. The primers also included the restriction sites for subcloning the PCR product into pET41. The N-terminal restriction site incorporated in the primer was AseI, which offers a compatible sticky end for cloning into the NdeI start codon restriction site of pET41 and the C-terminal restriction site was XhoI for cloning with an in-frame fusion into the same restriction site of pET41. Cloning was performed as described elsewhere (St John et al., 2006 ▶). The new construct was verified by DNA sequencing to express a protein of 401 amino acids with a C-terminal 8×His tag having a theoretical molecular weight of 45.4 kDa.
Protein expression was performed as described previously (St John et al., 2006 ▶). Harvested cultures were processed for XynC purification as recommended in the pET System Manual (10th ed., EMD Biosciences, Gibbstown, New Jersey, USA). Cell pellets were resuspended in 50 mM NaH2PO4 pH 7.2 using 5 ml buffer per gram of cell pellet. Halt Protease Inhibitor (Promega Corporation, Madison, Wisconsin, USA), Lysonase (EMD Biosciences) and MgCl2 (2 mM final concentration) were added to the cell suspension. The suspension was incubated at room temperature for 45 min on a rocking platform shaker. The cells were then subjected to further lysis by sonication on ice. The solution was then centrifuged for 20 min at 27 000g and the resulting supernatant was placed on ice. Expressed fusion protein was initially purified using a 1 ml HisTrap HP metal-chelating column in the nickel form (GE Healthcare, Pittsburgh, Pennsylvania, USA). The lysate was passed through the column using a peristaltic pump at a flow rate of 25 ml h−1. The column was then washed with 20 ml 50 mM NaH2PO4, 50 mM imidazole, 500 mM NaCl pH 7.2 and the fusion protein was eluted using 20 ml 50 mM NaH2PO4, 250 mM imidazole, 500 mM NaCl pH 7.4 buffer. The metal-affinity column elution fraction was dialyzed and concentrated (3×) using an Amicon stirred-cell filtration unit with a 10 kDa polyethersulfone membrane into gel-filtration buffer (20 mM HEPES, 150 mM NaCl pH 7.2) to a final volume of less than 4 ml. Concentrated XynC was then loaded onto a Superdex 200 gel-filtration column. Single injections of 500 µl were made and the protein was eluted with the gel-filtration buffer at 0.5 ml min−1. Purified XynC from gel filtration was concentrated to 5 mg ml−1 for crystal screens.
2.2. Crystal screens
The first crystal screens were performed in 24-well NeXtal NCK-24 plates (Qiagen, Valencia, California, USA) and the original crystal-positive condition resulted using the Hampton PEG/Ion Screen (Hampton Research, Aliso Viejo, California, USA). Wells contained 500 µl precipitant solution and 2 µl crystallization drops were prepared from equal volumes of mother liquor and XynC using the hanging-drop vapor-diffusion method. Additional screens including the NeXtal Classic I, Classic II, PEG, MPD and JCSG Suites (Qiagen) as well as the Additive Screen from Hampton Research (Hampton Research) were set up with an OryxNano crystallization robot (Douglas Instruments Ltd, Berkshire, England) using Intelli-Plate 96 crystallization trays from Art Robbins (Art Robbins Instruments, Sunnyvale, California, USA). In these trays, the drop volumes were 500 nl (50% and 66.6% XynC). Additional crystal refinement trays were set up using either an OryxNano crystallization robot with 24-well SBS-formatted sitting-drop Supper plates (Charles Supper Company, Natick, Massachusetts, USA) or by hand using Cryschem 24-well sitting-drop trays. Seeding studies were performed using the Seed Bead Kit (Hampton Research). Crystals were cryoprotected in mother liquor containing 12% glycerol. Xylobiose, xylotriose, glucuronate and aldotetrauronate were used as possible ligands. Ligand cocrystallizations and crystal soaks were performed with initial ligand concentrations in the drop ranging from 3 to 6 mM. Cocrystallization screens with these ligands were set as described above using the OryxNano robot and several of the same NeXtal screens.
2.3. Data collection and processing
The initial low-resolution model (XynC_D6; PDB code 3gtn) was built from data collected at the Stanford Synchrotron Radiation Lightsource (SSRL; beamline 9-1) at 105 K. Images were processed and scaled in HKL-2000 (Otwinowski & Minor, 1997 ▶). Phases were derived by molecular replacement with the program Phaser (McCoy et al., 2007 ▶) using XynA (PDB code 1nof; Larson et al., 2003 ▶), the GH 5 xylanase from E. chrysanthemi, which shares 40% identity with XynC, as a structure model. PHENIX (Adams et al., 2002 ▶) and REFMAC (Murshudov et al., 1997 ▶; Vagin et al., 2004 ▶) were used for refinement, with Coot used for iterative model building as part of the CCP4 suite of programs (Collaborative Computational Project, Number 4, 1994 ▶; Emsley & Cowtan, 2004 ▶). The quality of the XynC_D6 model used for molecular replacement was analyzed using MolProbity (Davis et al., 2007 ▶). A data set (XynC_A5) from the improved crystal form was collected at SSRL (beamline 7-1) and processed in HKL-2000 (Otwinowski & Minor, 1997 ▶; Table 1 ▶). The low-resolution (2.7 Å) XynC_D6 model will be used to determine phases by molecular replacement for the XynC_A5 data set.
Table 1. Data-collection statistics and final model quality.
Values in parentheses are for the highest resolution shell (2.82.7).
| XynC_D6 (PDB code 3gtn) | XynC_A5 | |
|---|---|---|
| Data collection | ||
| Crystal temperature (K) | 105 | 105 |
| Wavelength | 0.900 | 0.976 |
| Space group | P21 | P21212 |
| Unit-cell parameters (, ) | a = 61.34, b = 82.11, c = 96.65, = 104.70 | a = 138.49, b = 195.80, c = 66.25 |
| Resolution range () | 50.02.7 | 50.01.64 |
| Redundancy | 3.4 (3.3) | 7.2 (6.7) |
| No. of measured reflections | 2589484 | 4156029 |
| No. of unique reflections | 25003 (2514) | 220570 (21814) |
| R merge † (%) | 23.7 | 12.4 |
| Completeness (%) | 96.4 (97.8) | 100 (100) |
| Mean I/(I) | 4.7 (1.2) | 15.1 (1.0) |
| Matthews coefficient (3Da1) | 2.80 | 2.61 |
| Molecules of XynC per unit cell | 2 | 4 |
| Refinement statistics | ||
| Resolution range | 33.32.7 | |
| No. of reflections | 23709 | |
| R cryst ‡ (%) | 23.6 | |
| R free (%) | 28.7 | |
| No. of reflections used for R free | 1281 | |
| No. of protein atoms | 6308 | |
| No. of water molecules | 0 | |
| Average B factor (2) | 26.9 | |
| Ramachandran statistics | ||
| Favored (%) | 90.5 | |
| Outliers (%) | 1.5 | |
| R.m.s.d. from ideality | ||
| Bond lengths () | 0.015 | |
| Bond angles () | 1.656 |
R
merge = 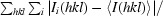
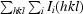 .
.
R
cryst = 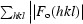
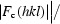
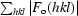 .
.
3. Results
3.1. Protein purification and crystallization
Native XynC was previously reported to have limited solubility (St John et al., 2006 ▶). The protein-expression construct described in this work was made specifically to prepare XynC for crystallization studies. The resulting XynC expression product was also limited in solubility even with the C-terminal 8×His tag. The preparation was optimized to obtain a 5 mg ml−1 XynC stock and this was used for all crystal screens.
Initial crystal screens using the Hampton Research PEG/Ion Screen resulted in clusters of very small rod-shaped crystals after many weeks of growth [0.2 M sodium tartrate dibasic dihydrate (sodium tartrate), 20% PEG 3350]. This condition was reproducible, but crystal quality did not improve in subsequent optimization or by microseeding (D’Arcy et al., 2007 ▶; St John et al., 2008 ▶). After setting up many trays attempting to reproduce and refine crystal growth, one well containing the original condition resulted in a crystal that was large enough for data collection (shown in Fig. 1 ▶ a). The resulting data set (XynC_D6) was sufficient to build a preliminary model of XynC. This low-resolution model has been deposited in the PDB with accession code 3gtn.
Figure 1.

(a) Crystals of XynC from B. subtilis 168 in the original crystallization condition of 200 mM sodium tartrate and 20% PEG 3350. Crystals from this well were used to collect the XynC_D6 data set. (b) Crystals of XynC in the refined crystallization condition which was identified not for general crystal quality but rather for reproducibility. This condition consisted of 0.2 M sodium tartrate, 0.2 M sodium malonate pH 7.0 in 19% PEG 3350. Screens of this condition in small volumes set up using the OryxNano crystallization robot led to large rods (c). This condition was almost 100% reproducible, with a large percentage of additive conditions resulting in identical crystals with similar unit-cell parameters. The XynC_A5 data set was collected from the crystal pictured. It was grown in 0.2 M sodium malonate pH 7.0, 0.2 M sodium tartrate and 19% PEG 3350 containing 300 mM nondetergent sulfobetaine 195 as an additive.
Further screening resulted in the identification of a condition from the NeXtal JCSG screen (0.2 M sodium malonate pH 7.0, 20% PEG 3350) that yielded low-quality twisted rod clusters. Combination of the two known crystallization conditions was inadvertently performed while using the Additive Screen in conjunction with the original condition of 0.2 M sodium tartrate and 20% PEG 3350, which resulted in a crystal-positive condition with 0.1 M sodium malonate pH 7.0 as an additive. These crystals were still not suitable for diffraction data collection, but the PEG 3350, sodium tartrate and sodium malonate condition was refined and shown to give reproducible twisted rod clusters in a 24-well sitting-drop crystallization tray, with the best condition being 0.2 M sodium tartrate, 0.2 M sodium malonate pH 7.0 and 19% PEG 3350 (Fig. 1 ▶ b). This condition was subsequently used in a second Additive Screen. The results from this screen supported the observation that this new condition was very robust, yielding single crystals for many of the 96 additives (Fig. 1 ▶ c). Likewise, crystal formation was not disrupted during cocrystallization experiments with ligand xylooligosaccharides and aldouronates nor were native crystals disrupted during ligand-soaking experiments. Cocrystallization screens with these ligands were set up as described above using the OryxNano robot and several of the same NeXtal screens. No new crystal-positive conditions were observed. In the optimized crystallization condition, small crystals could first be observed within 7 d and could grow to be as large as 300 × 70 × 70 µm.
3.2. Crystal diffraction and data analysis
The initial XynC_D6 crystal data set was collected from a crystal as shown in Fig. 1 ▶(a) that belonged to the primitive monoclinic space group P21. Poor model data quality is suggested from the overall R merge of approximately 24%. The data had a redundancy of 3.4 and were 97.8% complete in the highest resolution shell. The resolution for acceptable data quality was cut at 2.7 Å. Scaling and model statistics for XynC_D6 are presented in Table 1 ▶. An acceptable difference between R cryst and R free was obtained by applying NCS restraints in refinement.
Several crystals prepared with the optimized growth condition were screened and all diffracted to better than 2 Å resolution. The XynC_A5 crystal diffracted to 1.6 Å resolution and was found to belong to the primitive orthorhombic space group P21212 (Table 1 ▶).
4. Discussion
XynC represents a new class of xylanase that cleaves glucuronoxylan chains at sites of glucuronate substitution. The novel function of XynC may increase the efficiency of enzymatic pretreatments of lignocellulosic biomass for the purpose of bioconversion to fuels and value-added products. Further, the unique mode of action of XynC and similar enzymes such as XynA from E. chrysanthemi highlight a specialized application in the microbial world. In part, this is supported by its narrow phylogenetic distribution when compared with the more common xylanases such as those from families GH 10 and 11.
While the original crystallization condition that led to the XynC_D6 crystal and eventually to the first structure of XynC was not robust in any way, with crystallization in the mother liquor being highly sensitive to slight batch-to-batch variations in the protein preparation, the condition, crystal space group and unit-cell parameters are very similar to those originally reported for XynA from E. chrysanthemi (Barba de la Rosa et al., 1997 ▶; Larson et al., 2003 ▶). Observing similar crystallization conditions and unit-cell properties between two proteins that share only 40% identity seems serendipitous even with the expected structural similarity. A major difference occurs in the unit-cell packing. XynA contained only a single chain, while XynC contained two chains. This may be worth revisiting since the authors also described efforts to improve their poor reproducibility and they had success in cocrystallization with polymeric xylan, which could possibly lead to a XynC–substrate complex. While a XynA–substrate complex crystal was reported (Barba de la Rosa et al., 1997 ▶), the subsequent structural analysis paper of XynA did not include such a structure (Larson et al., 2003 ▶). Even with their similarities, the actual crystal forms are different. XynA grew as parallelepiped-shaped crystals (Barba de la Rosa et al., 1997 ▶; Larson et al., 2003 ▶), whereas XynC grows in this crystal form as a small rod (clusters), indicating that it grows faster in one direction when compared with the other two directions (Fig. 1 ▶ a).
The refined crystallization condition of XynC seemed to result from the optimization of the tartrate and malonate salt concentrations and also seemed to benefit from the small drop volumes (500 nl) possible using the OryxNano robot. Crystals from this condition are almost 100% reproducible and mechanically sturdy in handling and in crystal ligand-soaking experiments. Crystallization in this condition is also robust in the sense that it seems difficult to disrupt the nucleation and crystal-growth phenomena as determined by the additive screen and cocrystallizations with probable ligands.
Research is currently aimed at obtaining and refining the structure of XynC_A5 and obtaining data sets of XynC in ligand-bound forms. Obtaining a GH 5 xylanase structure in complex with the putative ligand will help to elucidate the substrate-binding mechanism and specificity of this novel xylanase family.
Acknowledgments
Support for this research was shared between the laboratories of JP at the University of Florida, JH at Winthrop University and EP at the University of Maryland, Baltimore. XynC protein-expression constructs were made and the purified xylooligosaccharides were supplied by the laboratory of JP. Protein expression, crystal screening and the majority of model building were performed by the laboratory of JH. In this laboratory, work was supported by NIH Grant No. P20 RR-016461 from the National Center for Research Resources. Large-scale crystal screens, crystal growth and data collection was performed in the laboratory of EP. We would like to thank the X-ray Crystallography Core Facility and the Department of Pharmaceutical Sciences at the University of Maryland, Baltimore for support in crystallographic equipment and general crystallographic resources. Portions of this research were carried out at the Stanford Synchrotron Radiation Lightsource, a national user facility operated by Stanford University on behalf of the US Department of Energy, Office of Basic Energy Sciences. The SSRL Structural Molecular Biology Program is supported by the Department of Energy, Office of Biological and Environmental Research and by the National Institutes of Health, National Center for Research Resources, Biomedical Technology Program and the National Institute of General Medical Sciences.
References
- Adams, P. D., Grosse-Kunstleve, R. W., Hung, L.-W., Ioerger, T. R., McCoy, A. J., Moriarty, N. W., Read, R. J., Sacchettini, J. C., Sauter, N. K. & Terwilliger, T. C. (2002). Acta Cryst. D58, 1948–1954. [DOI] [PubMed]
- Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. (1990). J. Mol. Biol. 215, 403–410. [DOI] [PubMed]
- Altschul, S. F. & Lipman, D. J. (1990). Proc. Natl Acad. Sci. USA, 87, 5509–5513. [DOI] [PMC free article] [PubMed]
- Barba de la Rosa, A. P., Day, J., Larson, S. B., Keene, N. & McPherson, A. (1997). Acta Cryst. D53, 256–261. [DOI] [PubMed]
- Bendtsen, J. D., Nielsen, H., von Heijne, G. & Brunak, S. (2004). J. Mol. Biol. 340, 783–795. [DOI] [PubMed]
- Cantarel, B. L., Coutinho, P. M., Rancurel, C., Bernard, T., Lombard, V. & Henrissat, B. (2009). Nucleic Acids Res. 37, D233–D238. [DOI] [PMC free article] [PubMed]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763.
- D’Arcy, A., Villard, F. & Marsh, M. (2007). Acta Cryst. D63, 550–554. [DOI] [PubMed]
- Davis, I. W., Leaver-Fay, A., Chen, V. B., Block, J. N., Kapral, G. J., Wang, X., Murray, L. W., Arendall, W. B. III, Snoeyink, J., Richardson, J. S. & Richardson, D. C. (2007). Nucleic Acids Res. 35, W375–W383. [DOI] [PMC free article] [PubMed]
- Demain, A. L., Newcomb, M. & Wu, J. H. (2005). Microbiol. Mol. Biol. Rev. 69, 124–154. [DOI] [PMC free article] [PubMed]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Henrissat, B. (1991). Biochem. J. 280, 309–316. [DOI] [PMC free article] [PubMed]
- Henrissat, B. & Bairoch, A. (1996). Biochem. J. 316, 695–696. [DOI] [PMC free article] [PubMed]
- Hurlbert, J. C. & Preston, J. F. III (2001). J. Bacteriol. 183, 2093–2100. [DOI] [PMC free article] [PubMed]
- Keen, N. T., Boyd, C. & Henrissat, B. (1996). Mol. Plant Microbe Interact. 9, 651–657. [DOI] [PubMed]
- Larson, S. B., Day, J., Barba de la Rosa, A. P., Keen, N. T. & McPherson, A. (2003). Biochemistry, 42, 8411–8422. [DOI] [PubMed]
- McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C. & Read, R. J. (2007). J. Appl. Cryst. 40, 658–674. [DOI] [PMC free article] [PubMed]
- Murshudov, G. N., Vagin, A. A. & Dodson, E. J. (1997). Acta Cryst. D53, 240–255. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol. 276, 307–326. [DOI] [PubMed]
- Preston, J. F., Hurlbert, J. C., Rice, J. D., Ragunathan, A. & St John, F. J. (2003). Applications of Enzymes to Lignocellulosics, edited by S. D. Mansfield & J. N. Saddler, pp. 191–210. Washington DC: American Chemical Society.
- Shanmugam, K. T. & Ingram, L. O. (2008). J. Mol. Microbiol. Biotechnol. 15, 8–15. [DOI] [PubMed]
- St John, F. J., Feng, B. & Pozharski, E. (2008). Acta Cryst. D64, 1222–1227. [DOI] [PubMed]
- St John, F. J., Rice, J. D. & Preston, J. F. (2006). J. Bacteriol. 188, 8617–8626. [DOI] [PMC free article] [PubMed]
- Vagin, A. A., Steiner, R. A., Lebedev, A. A., Potterton, L., McNicholas, S., Long, F. & Murshudov, G. N. (2004). Acta Cryst. D60, 2184–2195. [DOI] [PubMed]
- Vrsanska, M., Kolenova, K., Puchart, V. & Biely, P. (2007). FEBS J. 274, 1666–1677. [DOI] [PubMed]