

Human ADP-ribosylhydrolase 1, which cleaves the glycosidic bond between ADP-ribose and specific Arg residues in proteins, has been cloned, expressed, purified and crystallized.
Keywords: ADP-ribosylation, ADP-ribosylhydrolase, ADP-ribosylarginine hydrolase, X-ray fluorescence, ARH1, ADPRH
Abstract
Human ADP-ribosylhydrolase 1 (hARH1, ADPRH) cleaves the glycosidic bond of ADP-ribose attached to an Arg residue of a protein. hARH1 has been cloned, expressed heterologously in Escherichia coli, purified and crystallized in complex with K+ and ADP. The orthorhombic crystals contained one monomer per asymmetric unit, exhibited a solvent content of 43% and diffracted X-rays to a resolution of 1.9 Å. A prerequisite for obtaining well diffracting crystals was the performance of X-ray fluorescence analysis on poorly diffracting apo hARH1 crystals, which revealed the presence of trace amounts of K+ in the crystal. Adding K-ADP to the crystallization cocktail then resulted in a crystal of different morphology and with dramatically improved diffraction properties.
1. Introduction
ADP-ribosylation is a reversible post-translational modification of proteins and DNA. The attachment of ADP-ribose to an acceptor molecule is catalysed by mono-ADP-ribosyltransferases (mARTs) or poly-ADP-ribosyltransferases (pARTs or PARPs). ADP-ribosylation often modulates the specific activity of its target molecules. The opposite reaction, i.e. the release of ADP-ribose from the acceptor molecule, is catalysed by ADP-ribosylhydrolases (ARHs) or poly-ADP-ribosylglycohydrolases (PARGs).
The first reported biological effect of mono-ADP-ribosylation was that caused by diphtheria toxin (DT). DT ADP-ribosylates a diphthamide residue of elongation factor 2, thus inhibiting protein biosynthesis (Honjo et al., 1968 ▶). Other bacterial toxins also function as ADP-ribosyltransferases. In many cases these toxins originate from bacteria which have a significant impact on human health. Examples are pseudomonas, salmonella, clostridial, pertussis and cholera toxins (see, for example, Aktories & Just, 2000 ▶; Koch-Nolte et al., 2008 ▶). Besides its toxic function in certain pathogens, ADP-ribosylation and de-ADP-ribosylation is also utilized for regulatory purposes. One of the best-studied systems for which a complete ADP-ribosylation/de-ADP-ribosylation cycle has been described is the enzyme dinitrogenase reductase (DR) from the phototrophic bacterium Rhodospirillum rubrum (Lowery & Ludden, 1988 ▶; Ludden, 1994 ▶), which controls the fixation of molecular nitrogen. In the presence of light or a high concentration of NH4 +, DR is inactivated by the mono-ADP-ribosylation of a specific Arg residue, while the opposite reaction, the hydrolysis of the Arg-ADP-ribose, restores the protein’s enzymatic activity. ADP-ribosylation/de-ADP-ribosylation cycles have been described in prokaryotes and eukaryotes. For instance, in human and murine cells the reversible ADP-ribosylation of the Gβ subunit on Arg129 modulates the interaction of the Gβγ subunit and effector proteins (Lupi et al., 2000 ▶, 2002 ▶) and in human liver mitochondria the reversible ADP-ribosylation of Cys119 controls the enzymatic activity of glutamate dehydrogenase, a key enzyme in amino-acid metabolism (Herrero-Yraola et al., 2001 ▶; Choi et al., 2005 ▶).
The human genome contains a gene family of ecto-ARTs consisting of four functional genes (ART1, ART3, ART4 and ART5; Glowacki et al., 2002 ▶; Koch-Nolte et al., 2008 ▶) and a family of cytosolic PARPs with 17 functional members (Otto et al., 2005 ▶; Hakmé et al., 2008 ▶). The ARH gene family comprises ARH1, ARH2, ARH3 (Glowacki et al., 2002 ▶) and one PARG (Koch-Nolte et al., 2008 ▶). ARH1 has been described as an Arg-specific mono-ADP-ribosylhydrolase which can de-ADP-ribosylate many different Arg-ADP-ribosylated target proteins (Moss et al., 1992 ▶). Genetic deletion of ARH1 enhances the sensitivity of cells to cholera toxin (Kato et al., 2007 ▶), which ADP-ribosylates Arg187 in the Gs α-subunit of the guanine nucleotide-binding regulatory protein that stimulates adenylyl cyclase (Freissmuth & Gilman, 1989 ▶), although the natural substrate of ARH1 is still unknown. Neither the substrate specificity nor the enzymatic activity of ARH2 is known. ARH3 was recently shown to de-ADP-ribosylate poly-ADP-ribosylated substrates in mitochondria (Mueller-Dieckmann et al., 2006 ▶; Oka et al., 2006 ▶; Niere et al., 2008 ▶) and to hydrolyse O-acetyl-ADP-ribose, a side product of sirtuin-dependent deacetylation (Ono et al., 2006 ▶).
The structures of several bacterial mono-ADP-ribosyltransferases and of several eukaryotic PARPs are known to date (see, for example, Deng & Barbieri, 2008 ▶; Koch-Nolte et al., 2008 ▶) as well as the three-dimensional structure of one mammalian mART (Mueller-Dieckmann et al., 2002 ▶). Even less structural information is available for ADP-ribosylhydrolases. To date, only the three-dimensional structures of human and mouse ADP-ribosylhydrolase 3 (Mueller-Dieckmann et al., 2006 ▶, 2007 ▶, 2008 ▶) have been determined. Moreover, based on fold comparisons, two other proteins which had previously been annotated as proteins of unknown function were identified as likely ADP-ribosylhydrolases (Mueller-Dieckmann et al., 2008 ▶): one from Methanocaldococcus jannaschii (PDB code 1t5j; A. Gogos, J. Gorman & L. Shapiro, unpublished work) and another from Thermus thermophilus (PDB code 2cwc; A. Ebihara, S. Yokoyama & S. Kuramitsu, unpublished work).
Here, we report the cloning, expression, purification and crystallization of the 357 amino-acid residue Arg-specific ADP-ribosylhydrolase 1 from Homo sapiens (hARH1) as well as an X-ray fluorescence-guided improvement of crystallization and the characterization of the resulting crystals by X-ray diffraction experiments.
2. Experimental methods
2.1. Cloning
A cDNA clone containing the coding sequence for hARH1 was obtained from the IMAGE consortium (IMAGE Consortium Clone ID 4419930). The ARH1-coding region was amplified by PCR using the appropriate primers (5′-CAAGAGACATATGGAGAAGTATGTGGCTGCT-3′ and 5′-ACGCACTCGAGGGAAATTACAGTGTCTTCTTTTG-3′; MWG Biotech) and cloned into the NdeI and XhoI sites of the prokaryotic expression plasmid pET26b (Novagen). This cloning strategy resulted in the removal of the pelB leader sequence from pET26b and a C-terminal extension of one amino acid (Glu) encoded by the XhoI recognition sequence followed by a His6 tag. The correct cloning of the gene was verified by DNA sequencing.
2.2. Protein expression
Escherichia coli BL21-Gold (DE3) cells (Stratagene) were used for hARH1 expression experiments. A 10 ml overnight preculture was grown at 310 K in non-inducing MDG medium (Studier, 2005 ▶) containing 100 µg ml−1 kanamycin. The cells were harvested and resuspended in expression medium. Five cultures of 400 ml autoinducing Zym5052 medium (Studier, 2005 ▶) in baffled 2 l flasks were inoculated at a ratio of 1:200 and grown at 310 K until the cells reached an optical density at 600 nm of about 0.7. The temperature was decreased to 297 K and after 17 h 1 mM isopropyl β-d-1-thiogalactopyranoside (IPTG) was added to ensure complete induction. 4 h later, the cells were harvested and were stored at 253 K until further processing.
2.3. Protein purification
The cells were suspended in 6 ml lysis buffer [50 mM NaH2PO4 pH 8.0, 500 mM NaCl, 10 mM MgCl2, 3 mM β-mercaptoethanol (β-ME), 1 mM 4-(2-aminoethyl)benzenesulfonylfluoride, 0.1%(v/v) Triton X-100, 0.1 µg ml−1 lysozyme and 12.5 U ml−1 Benzonase nuclease (Novagen)] per gram wet weight and incubated at 294 K for 30 min. Lysis was performed by sonication for 5 min in 15 s pulses at 277 K. Cell debris was removed by centrifugation at 15 500g for 20 min at 277 K. 1 g Protino Ni–IDA matrix (Macherey-Nagel) was added per 50 ml crude lysate and the slurry was incubated for 1–2 h at 277 K in order to allow complete binding. The matrix was washed four times with eight bed volumes of washing buffer (50 mM NaH2PO4 pH 8.0, 500 mM NaCl, 10 mM MgCl2 and 3 mM β-ME), transferred to a column and washed with four bed volumes of washing buffer and five bed volumes of washing buffer containing 2.5 mM imidazole. The protein was eluted with two column volumes of elution buffer (50 mM NaH2PO4 pH 8.0, 500 mM NaCl, 10 mM MgCl2, 3 mM β-ME and 250 mM imidazole). For further purification, the eluate was concentrated by ultrafiltration (Vivaspin 20, molecular-weight cutoff 30 000; Sartorius) to a volume of less than 1 ml and, analogously to the purification of hARH3 (Kernstock et al., 2006 ▶), applied onto a Superdex S75 16/60 gel-filtration column (GE Healthcare) pre-equilibrated with filtration buffer [50 mM Tris–HCl pH 8.0, 150 mM NaCl, 3 mM MgCl2 and 1 mM dithiothreitol (DTT)]. Pure protein fractions were concentrated to <500 µl and the buffer was exchanged to storage buffer (20 mM HEPES pH 7.0, 150 mM NaCl, 1 mM MgCl2 and 3 mM DTT) by repeated ultrafiltration (Vivaspin 20 with diafiltration cup, molecular-weight cutoff 30 000) and adjusted to 15 mg ml−1. The purity of the protein sample was verified by SDS–PAGE.
2.4. Crystallization and X-ray fluorescence analysis
Initial crystallization trials were performed by vapour diffusion in sitting drops at the high-throughput crystallization facility at the EMBL Hamburg outstation (Mueller-Dieckmann, 2006 ▶). 600 nl droplets (300 nl sample + 300 nl reservoir solution) were equilibrated against reservoirs from five complete crystallization screens: Crystal Screen, Index, SaltRx and Grid from Hampton Research and Jena Screens 1–4 from Jena Biosciences. A promising lead condition containing lithium acetate and PEG 3350 was confirmed in two customized follow-up 96-well plates also at the EMBL Hamburg facility. Further refinement of the conditions was performed manually.
Optimized crystallization experiments were performed using the hanging-drop method. Equal volumes of protein solution (15–20 mg ml−1 protein in 20 mM HEPES pH 7.0, 100 mM NaCl and 3 mM DTT) and reservoir solution [150 mM lithium acetate and 25%(w/v) PEG 3350] at 293 K were mixed and equilibrated over 750 µl reservoir solution. Crystals of bipyramidal shape appeared after about two weeks and typically grew to dimensions of 150 × 150 × 150 µm. For diffraction data collection, they were cryoprotected in a solution of 20–25%(v/v) PEG 200 in reservoir solution. These crystals (Fig. 1 ▶ a) diffracted X-rays very anisotropically to about 15 Å resolution only. The diffraction pattern was not of sufficient quality to give any information on the cell symmetry or cell constants. The addition of Na-ADP, which has been reported to act as an inhibitor of ARH1 (Moss et al., 1986 ▶), yielded crystals of the same morphology and with the same diffraction properties as the apo crystals. X-ray fluorescence (XRF) analysis (Leonard et al., 2009 ▶) of the cryoprotected apo crystals of hARH1 performed with an incident beam energy of 12.7 keV and an exposure time of 30 s (with 0.7% transmitted beam intensity) revealed a small but reproducible peak at the potassium Kα emission line (Fig. 2 ▶), although no potassium had been added during purification and crystallization. In order to explore this observation further, crystallization was attempted in the presence of K-ADP instead of Na-ADP. The addition of 15 mM K-ADP to the crystallization drop [20 mg ml−1 protein in 20 mM HEPES pH 7.0, 100 mM NaCl and 3 mM DTT mixed with an equal volume of reservoir solution consisting of 150 mM lithium acetate and 25%(w/v) PEG 3350] yielded rod-shaped crystals. These crystals appeared from a very heavy precipitate within about two weeks (Fig. 1 ▶ b) and grew to maximum dimensions of 50 × 50 × 200 µm. Most of the crystals were multiple crystals that were badly grown together. The few which grew as single crystals diffracted X-rays to about 1.9 Å resolution. XRF analysis at the same incident beam energy and for the same exposure time of carefully back-soaked cryocooled crystals (the same cryoprotectant was used as for the apo crystals) revealed a significant increase in the K fluorescence signal compared with the apo ARH1 crystals (Fig. 2 ▶ a).
Figure 1.

Crystals of hARH1. (a) Crystal of apo ARH1 and (b) crystal of hARH1–K-ADP mounted into a nylon loop from heavy precipitate. The red cross indicates the location of the incident X-ray beam.
Figure 2.

Section of the X-ray fluorescence spectrum from a hARH1 crystal crystallized without (red line) and in the presence of (blue line) K-ADP. The Kα emission lines for S (2.3 keV), Cl (2.6 keV) and K (3.3 keV) are clearly visible, as well as the Kβ emission line of K (3.6 keV).
2.5. Diffraction data collection and processing
Diffraction data were collected on beamline ID29 (ESRF, Grenoble, France) at 100 K in an N2 cryostream. As described in §2.3, crystals were cryoprotected for about 10 s in a solution consisting of 20–25%(v/v) PEG 200 in reservoir solution. Data were collected at an energy of 6.7 keV (λ = 1.85 Å) to a maximum resolution of 1.92 Å using an ADSC Q315 3 × 3 CCD detector. Data collection was carried out at this long wavelength in order to attempt structure determination by the sulfur-SAD (single anomalous dispersion) method (S-SAD; Hendrickson & Teeter, 1981 ▶; Dauter et al., 1999 ▶) should molecular replacement fail. The wavelength of 1.85 Å is close to the wavelength at which the maximum anomalous signal-to-noise ratio can be expected (Mueller-Dieckmann et al., 2005 ▶). An optimal data-collection strategy was calculated using the programme BEST (Bourenkov & Popov, 2006 ▶). All data were indexed and integrated using XDS (Kabsch, 1993 ▶) and scaled using XSCALE. Intensities were converted to structure-factor amplitudes using the program TRUNCATE (French & Wilson, 1978 ▶; Collaborative Computational Project, Number 4, 1994 ▶). The redundancy-independent merging R factor (R r.i.m.) and the precision-indicating merging R factor (R p.i.m.) were calculated using the program RMERGE (Weiss, 2001 ▶). Data-collection and processing parameters are given in Table 1 ▶. The optical resolution was calculated using SFCHECK (Vaguine et al., 1999 ▶).
Table 1. Data-collection and processing statistics.
Values in parentheses are for the highest resolution bin.
| No. of crystals | 1 |
| Wavelength (Å) | 1.8505 |
| Crystal-to-detector distance (mm) | 106.7 |
| Rotation range per image (°) | 1.75 |
| Total rotation range (°) | 875 |
| Beam attenuation (%) | 70 |
| Exposure time per image (s) | 0.7 |
| Resolution range (Å) | 99.0–1.92 (2.04–1.92) |
| Space group | P212121 |
| Unit-cell parameters (Å) | a = 45.28, b = 52.69, c = 140.41 |
| Mosaicity (°) | 0.42 |
| Total No. of reflections | 827238 |
| Unique reflections | 49084 |
| Redundancy | 17.0 (13.6) |
| 〈I/σ(I)〉 | 27.0 (3.6) |
| Completeness (%) | 98.8 (94.4) |
| Rmerge† (%) | 9.0 (68.3) |
| Rr.i.m.‡ (%) | 9.7 (80.9) |
| Rp.i.m.§ (%) | 1.7 (15.7) |
| Ranom (%) | 3.4 (68.0) |
| Overall B factor from Wilson plot (Å2) | 27.3 |
| Optical resolution (Å) | 1.88 |
R
merge = 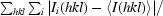
 .
.
R
r.i.m. = 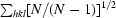
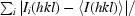 .
.
R
p.i.m. = 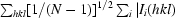 −
− 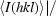 .
.
3. Results and discussion
Human ADP-ribosylhydrolase 1 has been cloned, overexpressed and purified. From 2 l of culture, corresponding to about 24 g of wet cells, approximately 20 mg protein could be purified. After purification, the protein was >99% pure as judged by SDS–PAGE. The initial crystals (Fig. 1 ▶ a) exhibited very poor diffraction properties with respect to the maximum resolution to which diffraction could be observed and to anisotropy of diffraction. XRF analyses performed on these crystals indicated the presence of small but significant and reproducible amounts of potassium ions (Fig. 2 ▶), although no K+ had been added during the purification or crystallization of the protein. Similarly, all attempts to cocrystallize hARH1 with the inhibitor Na-ADP yielded crystals of the same morphology and diffraction quality as those which could be obtained from the apo protein. However, cocrystallization in the presence of K-ADP resulted in the initial formation of a heavy precipitate, from which small rod-shaped crystals appeared within 1–2 weeks (Fig. 1 ▶ b). These crystals diffracted X-rays well and to a maximal resolution of 1.92 Å and they belonged to the orthorhombic space group P212121 (Table 1 ▶). Based on the Matthews parameter (Matthews, 1968 ▶), the asymmetric unit contains one protein molecule (V M = 2.16 Å3 Da−1 and 43.0% solvent content).
All attempts to date to solve the structure by molecular replacement using the full or a partial model of human ADP-ribosylhydrolase 3 (PDB code 2foz; Mueller-Dieckmann et al., 2006 ▶) as a search model were unsuccessful. Thus, structure determination will be attempted using the long-wavelength S-SAD approach. While the low Laue symmetry of the crystals and the relatively large protein may seem unfavourable at first sight, the comparatively high sulfur content (12 Met and four Cys residues) of hAHR1 as well as the presence of K+ may make this approach feasible.
In summary, hARH1 is the first eukaryotic mono-ADP-ribosyl-Arg hydrolase to be crystallized to date. The use of X-ray fluorescence analysis on the initial poorly diffracting crystals was crucial to obtain well diffracting crystals. To our knowledge, this is the first time that this method has been deployed to improve crystallization conditions. Once the three-dimensional structure of hARH1 in complex with ADP becomes available, a better understanding of the enzymatic mechanism of hARH1 and other mono-ADP-ribosylhydrolases will be possible. A comparison with the recently solved structures of ARH3 from human and mouse (Mueller-Dieckmann et al., 2006 ▶, 2008 ▶), with which ARH1 shows approximately 20% sequence identity, will allow a detailed analysis of the structural differences between mono- and poly-ADP-ribosylhydrolases.
Acknowledgments
We thank Fabienne Seyfried, Institute of Immunology, UKE Hamburg for excellent technical assistance. This work was supported by grants from the Deutsche Forschungsgemeinschaft (to FK-N and MSW) and the Studienstiftung des deutschen Volkes (to SK).
References
- Aktories, K. & Just, I. (2000). Bacterial Protein Toxins. Berlin: Springer Verlag.
- Bourenkov, G. P. & Popov, A. N. (2006). Acta Cryst. D62, 58–64. [DOI] [PubMed]
- Choi, M.-M., Huh, J.-W., Yang, S.-J., Cho, E. H., Choi, S. Y. & Cho, S.-W. (2005). FEBS Lett.579, 4125–4130. [DOI] [PubMed]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763.
- Dauter, Z., Dauter, M., de La Fortelle, E., Bricogne, G. & Sheldrick, G. M. (1999). J. Mol. Biol.289, 83–92. [DOI] [PubMed]
- Deng, Q. & Barbieri, J. T. (2008). Annu. Rev. Microbiol.62, 271–288. [DOI] [PubMed]
- Freissmuth, M. & Gilman, A. G. (1989). J. Biol. Chem.264, 21907–21914. [PubMed]
- French, S. & Wilson, K. (1978). Acta Cryst. A34, 517–525.
- Glowacki, G., Braren, R., Firner, K., Nissen, M., Kühl, M., Reche, P., Bazan, F., Cetkovic Cvrlje, M., Leiter, E., Haag, F. & Koch-Nolte, F. (2002). Protein Sci.11, 1657–1670. [DOI] [PMC free article] [PubMed]
- Hakmé, A., Wong, H. K., Dantzer, F. & Schreiber, V. (2008). EMBO Rep.9, 1094–1100. [DOI] [PMC free article] [PubMed]
- Hendrickson, W. A. & Teeter, M. M. (1981). Nature (London), 290, 107–113. [DOI] [PMC free article] [PubMed]
- Herrero-Yraola, A., Bakhit, S. M. A., Franke, P., Weise, C., Schweiger, M., Jorcke, D. & Ziegler, M. (2001). EMBO J.20, 2404–2412. [DOI] [PMC free article] [PubMed]
- Honjo, T., Nishizuka, Y. & Hayaishi, O. (1968). J. Biol. Chem.243, 3553–3555. [PubMed]
- Kabsch, W. (1993). J. Appl. Cryst.26, 795–800.
- Kato, J., Zhu, J., Liu, C. & Moss, J. (2007). Mol. Cell. Biol.27, 5534–5543. [DOI] [PMC free article] [PubMed]
- Kernstock, S., Koch-Nolte, F., Mueller-Dieckmann, J., Weiss, M. S. & Mueller-Dieckmann, C. (2006). Acta Cryst. F62, 224–227. [DOI] [PMC free article] [PubMed]
- Koch-Nolte, F., Kernstock, S., Mueller-Dieckmann, C., Weiss, M. S. & Haag, F. (2008). Front. Biosci.13, 6716–6729. [DOI] [PubMed]
- Leonard, G. A., Solé, V. A., Beteva, A., Gabadinho, J., Guijarro, M., McCarthy, J., Marrocchelli, D., Nurizzo, D., McSweeney, S. & Mueller-Dieckmann, C. (2009). J. Appl. Cryst.42, 333–335.
- Lowery, R. G. & Ludden, P. W. (1988). J. Biol. Chem.263, 16714–16719. [PubMed]
- Ludden, P. W. (1994). Mol. Cell. Biochem.138, 123–129. [DOI] [PubMed]
- Lupi, R., Corda, D. & DiGirolamo, M. (2000). J. Biol. Chem.275, 9418–9424. [DOI] [PubMed]
- Lupi, R., Dani, N., Dietrich, A., Marchegiani, A., Turacchio, S., Berrie, C. P., Moss, J., Gierschik, P., Corda, D. & DiGirolamo, M. (2002). Biochem. J.367, 825–832. [DOI] [PMC free article] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol.33, 491–497. [DOI] [PubMed]
- Moss, J., Oppenheimer, N. J., West, R. E. Jr & Stanley, S. J. (1986). Biochemistry, 25, 5408–5414. [DOI] [PubMed]
- Moss, J., Stanley, S. J., Nightingale, M. S., Murtagh, J. J., Monaco, L., Mishima, K., Chen, H.-C., Williamson, K. C. & Tsai, S.-C. (1992). J. Biol. Chem.267, 10481–10488. [PubMed]
- Mueller-Dieckmann, C., Kernstock, S., Lisurek, M., von Kries, J. P., Haag, F., Weiss, M. S. & Koch-Nolte, F. (2006). Proc. Natl Acad. Sci. USA, 103, 15026–15031. [DOI] [PMC free article] [PubMed]
- Mueller-Dieckmann, C., Kernstock, S., Mueller-Dieckmann, J., Weiss, M. S. & Koch-Nolte, F. (2008). Acta Cryst. F64, 156–162. [DOI] [PMC free article] [PubMed]
- Mueller-Dieckmann, C., Panjikar, S., Schmidt, A., Mueller, S., Kuper, J., Geerlof, A., Wilmanns, M., Singh, R. K., Tucker, P. A. & Weiss, M. S. (2007). Acta Cryst. D63, 366–380. [DOI] [PubMed]
- Mueller-Dieckmann, C., Panjikar, S., Tucker, P. A. & Weiss, M. S. (2005). Acta Cryst. D61, 1263–1272. [DOI] [PubMed]
- Mueller-Dieckmann, C., Ritter, H., Koch-Nolte, F. & Schulz, G. E. (2002). J. Mol. Biol.322, 687–696. [DOI] [PubMed]
- Mueller-Dieckmann, J. (2006). Acta Cryst. D62, 1446–1452. [DOI] [PubMed]
- Niere, M., Kernstock, S., Koch-Nolte, F. & Ziegler, M. (2008). Mol. Cell. Biol.28, 814–824. [DOI] [PMC free article] [PubMed]
- Oka, S., Kato, J. & Moss, J. (2006). J. Biol. Chem.281, 705–713. [DOI] [PubMed]
- Ono, T., Kasamatsu, A., Oka, S. & Moss, J. (2006). Proc. Natl Acad. Sci. USA, 103, 16687–16691. [DOI] [PMC free article] [PubMed]
- Otto, H., Reche, P. A., Bazan, F., Dittmar, K., Haag, F. & Koch-Nolte, F. (2005). BMC Genomics, 6, 139. [DOI] [PMC free article] [PubMed]
- Studier, W. (2005). Protein Expr. Purif.1, 207–234. [DOI] [PubMed]
- Vaguine, A. A., Richelle, J. & Wodak, S. J. (1999). Acta Cryst. D55, 191–205. [DOI] [PubMed]
- Weiss, M. S. (2001). J. Appl. Cryst.34, 130–135.