

Abstract
Geleophysic dysplasia is an autosomal recessive disorder characterized by short stature, brachydactyly, thick skin and cardiac valvular anomalies often responsible for an early death. Studying six geleophysic dysplasia families, we first mapped the underlying gene to chromosome 9q34.2 and identified five distinct nonsense and missense mutations in ADAMTSL2 (a disintegrin and metalloproteinase with thrombospondin repeats–like 2), which encodes a secreted glycoprotein of unknown function. Functional studies in HEK293 cells showed that ADAMTSL2 mutations lead to reduced secretion of the mutated proteins, possibly owing to the misfolding of ADAMTSL2. A yeast two-hybrid screen showed that ADAMTSL2 interacts with latent TGF-β–binding protein 1. In addition, we observed a significant increase in total and active TGF-β in the culture medium as well as nuclear localization of phosphorylated SMAD2 in fibroblasts from individuals with geleophysic dysplasia. These data suggest that ADAMTSL2 mutations may lead to a dysregulation of TGF-β signaling and may be the underlying mechanism of geleophysic dysplasia.
Geleophysic dysplasia (from the Greek geleos, “happy”, and physis, “nature”; OMIM 231050) is a rare autosomal recessive disorder characterized by short stature, small hands and feet with broad proximal phalanges, cone-shaped epiphyses, delayed bone age and shortened tubular bones1 (Fig. 1). Other features include a ‘happy’ face with upturned corners of the mouth, hepatomegaly, restricted joint mobility, skin thickening and muscle hypertrophy. Affected individuals present with progressive cardiac disease with dilation and thickening of the pulmonary, aortic or mitral valves, often leading to death before 5 years of age. Tracheal stenosis responsible for severe respiratory problems is frequent. Geleophysic dysplasia biopsy material has shown abundant lysosome-like vacuoles in hepatocytes, fibroblasts and macrophages suggestive of a storage disorder2,3 (Supplementary Fig. 1 online). However, biochemical analyses have not detected an enzymatic deficiency or precisely characterized the accumulated material.
Figure 1.
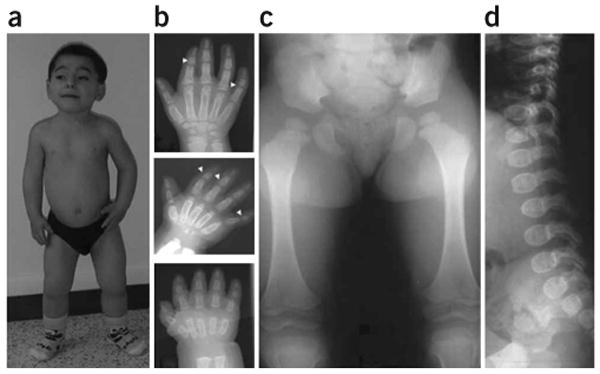
Clinical and radiological manifestations of geleophysic dysplasia in individual 3. (a) Note the facial features, including round face, long philtrum, thin upper lip and very short hands and feet (individual 3 at age 9 years). (b) Hand X-rays of individual 3 at 1 year (bottom), 3 years (middle) and 10 years (top). Note the very small hand with short and plump tubular bones, metacarpal proximal pointing and cone-shaped epiphyses (arrow) at ages 3 and 10 years. Also note the carpal ossification delay, with bone age of a newborn at 1 year (bottom), bone age of 2 years at 3 years (middle) and bone age of 7 years at 10 years (top). (c,d) Hip at age 3 years and spine at age 1 year. Note the small capital femoral epiphyses and the ovoid vertebral bodies. Informed consent was obtained to publish the photographs in this figure.
Homozygosity mapping in four consanguineous geleophysic dysplasia families of French Polynesian, Moroccan, Algerian and Pakistani origins (families 1–4; Fig. 2a) showed linkage of the underlying gene to chromosome 9q34.2–q34.3 in a 619-kb interval (Zmax = 4.52 at θ = 0 at the gt-AL590710 locus). A recombination event in family 4 defined the proximal boundary of the region (gt-AL593848), and a second recombinant in the same family defined the distal boundary (gt-AL593186). Because geleophysic dysplasia belongs to the group of acromelic dysplasias that also includes the autosomal recessive form of Weill-Marchesani syndrome caused by ADAMTS10 mutations, we considered ADAMTSL2, the product of the ADAMTSL2 (ADAMTS-like 2) gene, as a likely candidate among the seven genes located within the critical interval (Fig. 2b). ADAMTSL2 belongs to a large superfamily containing 19 ADAMTS proteases and at least five ADAMTS-like proteins. ADAMTS proteases are secreted enzymes with a conserved organization that includes a metalloprotease domain and an ancillary domain containing one or more thrombospondin type 1 repeats (TSR). Some ADAMTS proteases participate in extracellular matrix (ECM) turnover in arthritis, and others are involved in procollagen and von Willebrand factor maturation or in angiogenesis4. The ADAMTS-like subfamily comprises proteins homologous to the ADAMTS ancillary domains but lacking the protease domain and hence lacking catalytic activity. ADAMTSL-1 and ADAMTSL-3 proteins are closely related secreted glycoproteins5,6, whereas ADAMTSL-2 has a different domain structure7. Their functions are unknown.
Figure 2.

Genetic mapping of the locus involved in geleophysic dysplasia. (a) Pedigrees of geleophysic dysplasia families. (b) The region of homozygosity is located between gt-AL593848 and gt-AL593186; seven genes are located in this region. (c) Exon and intron structure of the ADAMTSL2 with mutations identified in five families with geleophysic dysplasia. Arrows indicate the location of the mutation found in each family. (d) ADAMTSL2 functional domains. The location of the amino acid change found in each family is shown.
The 18 coding exons of ADAMTSL2 encode a 951-residue protein composed of a signal peptide, a TSR, a cysteine-rich module, a spacer module, an N-glycan–rich module, six additional TSRs and a PLAC module7. Direct sequence analysis of the coding region of ADAMTSL2 in families 1–4 and in two additional affected individuals detected four distinct missense mutations and a nonsense mutation (Table 1 and Fig. 2c). The missense mutations consistently involved residues that are conserved across species and across the ADAMTSL family members. The mutations clustered in the regions encoding the cysteine-rich domain (three mutations) and TSR6 (two mutations) (Table 1 and Fig. 2d). We identified the same mutation in the two families from North Africa. All mutations cosegregated with the disease and were absent in chromosomes from ethnicity-matched controls. We did not find any mutations in family 4 (RNA was not available for this family).
Table 1. ADAMTSL2 mutations identified in individuals with geleophysic dysplasia.
| Family | Origin | Consanguinity | Nucleotide change | Amino acid change | Location | Affected domain |
|---|---|---|---|---|---|---|
| 1 | French Polynesia | Yes | 440C>T | P147L | Exon 5 | Cysteine-rich |
| 2 | Morocco | Yes | 338G>A | R113H | Exon 4 | Cysteine-rich |
| 3 | Algeria | Yes | 338G>A | R113H | Exon 4 | Cysteine-rich |
| 4 | Pakistan | Yes | ND | ND | ND | ND |
| 5 | Turkey | Yes | 340G>A | E114K | Exon 4 | Cysteine-rich |
| 6 | France | No | 2431G>A | G811R | Exon 16 | TSR6 |
| 2586G>A | W862X | Exon 16 |
ND, not determined.
To correlate the expression pattern of ADAMTSL2 with the clinical manifestations of geleophysic dysplasia, we performed in situ hybridization experiments on tissue derived from a human fetus at 35 weeks of gestation. In agreement with the clinical manifestations observed in individuals with geleophysic dysplasia, we found ADAMTSL2 mRNA expression in heart, skin and pulmonary arteries (Fig. 3). In the heart, we detected ADAMTSL2 mRNA in cardiomyocytes (Fig. 3a). We also observed strong expression in skin epidermis, dermal blood vessels (Fig. 3b) and in the tracheal wall (Fig. 3g). ADAMTSL2 mRNA was present in developing skeletal muscle (Fig. 3d), and we observed strong expression in the pulmonary arteries and developing bronchioles of the lung (Fig. 3e,f). Because geleophysic dysplasia is a chondrodysplasia, we also performed in situ hybridization of the proximal femoral growth plate. On longitudinal sections, we found a high level of ADAMTSL2 mRNA expression in chondrocyte columns in the hypertrophic and reserve zones (Fig. 3h).
Figure 3.

In situ hybridization analysis of ADAMTSL2 mRNA expression in a human fetus at 35 weeks of gestation. The purple staining indicates sites of RNA hybridization. (a) Transverse section through the heart showing specific signal in cardiomyocytes. (b) In the skin, RNA was expressed in the epidermis (*) and in dermal blood vessels (white arrow). (c) Sense control of b. (d) ADAMTSL2 mRNA was present in the myofibrils (m) of developing skeletal muscles. (e,f) In the lung, ADAMTSL2 expression was present in the developing bronchioles (br), in the wall of the pulmonary artery (a) and in the parenchyma (p). (g) In the trachea, expression was strong in the internal ciliated pseudostratified epithelium (*). (h) Longitudinal section through the femoral end of human fetal growth plate showing high ADAMTSL2 mRNA expression in chondrocytes in the proliferative and hypertrophic zones (PZ and HZ) and in the epiphyseal region (ER). (i) Sense control of h. Scale bars: 50 μm in b,f,h; 20 μm in a,c,e,g,i; 10 μm in d.
We tested the functional consequences of the geleophysic dysplasia mutations using myc-tagged wild-type and mutant ADAMTSL2 (R113H, P147L and G811R) in parallel transfections of HEK293F cells. Protein blot analyses after 48 h of transfection confirmed that wild-type ADAMTSL2 was secreted into the medium7. Although there was not a statistically significant alteration in cellular levels of each mutant protein, we found significantly reduced secretion of each mutant protein compared to wild-type protein (Fig. 4a,b). Thus, the mutant proteins are likely to be synthesized, but it is possible that they are misfolded, which may interfere with their efficient secretion. One can also consider an increased turnover or an altered function of secreted mutant proteins.
Figure 4.

Functional consequences of ADAMTSL2 mutations. (a) Characterization of wild-type and mutant ADAMTSL2 proteins. Conditioned medium (top) from transfected HEK293F cells or cell lysate (bottom) was normalized to either secreted IgG (medium) or actin (cell lysate). Transfections were performed in triplicate using wild-type ADAMTSL2 or the indicated mutant. (b) Protein species were quantified by densitometry, and the ratios of ADAMTSL2 to IgG or actin were compared statistically using Student's t-test. The statistical significance is indicated on the graph. (c) Immunoprecipitation (IP) of ADAMTSL2-LTBP-1S complexes. Anti-myc agarose was used for immunoprecipitation. The blot at left shows immunoblotting with antibody to LTBP-1. The LTBP-1S protein is indicated. The lane on the far right is medium from LTBP-1S–expressing cells used as a positive control for immunoblotting. The blot at right illustrates that ADAMTSL2 was immunoprecipitated successfully in samples used for coimmunoprecipitation. The locations of molecular weight markers (in kDa) are shown between the two panels.
To define the molecular pathway in which ADAMTSL2 might participate, we used full-length ADAMTSL2 lacking its signal peptide as bait to screen for putative ADAMTSL2-binding proteins in a yeast two-hybrid screen of a human muscle cDNA library. Among several positive clones, one clone contained a 783-bp insert that corresponded to residues 673–933 of human latent TGF-β–binding protein 1 (LTBP-1). We verified the interaction of LTBP-1S, the dominant and more widely distributed form, with ADAMTSL2 using immunoprecipitation (Fig. 4c).
LTBP-1 has a major role in the storage of latent TGF-β in the ECM and regulates its availability. TGF-β is secreted from cells either as a dimeric small latent complex (SLC) comprising noncovalently bound latency-associated propeptide and mature TGF-β and/or as a large latent complex (LLC) comprising SLC bound to LTBP-1, LTBP-3 or LTBP-4 through a TGF-β binding motif8. LTBPs are structurally related to fibrillins9, and LTBP-1 is an associated component of tissue microfibrils10. The activation of the TGF-β–SMAD signaling pathway is tightly regulated through various ECM proteins involved in the release of LLC from microfibrils and ECM and in the release of TGF-β from latency-associated propeptide11,12.
To determine the functional significance of this interaction, we quantified active and total TGF-β in the cultured medium of fibroblasts from individuals with geleophysic dysplasia and age- and passage-matched control skin fibroblasts by ELISA. There was a tenfold higher amount of total TGF-β in the cultured medium of geleophysic dysplasia fibroblasts than in cultured medium from control fibroblasts (P < 0.0003) (Fig. 5a). Whereas active TGF-β represented 85% and 92% of total TGF-β in the medium of individuals 6 and 2, respectively, active TGF-β represented only 7% of total TGF-β in control medium (Fig. 5a). Consistent with this observation, protein blot analysis of geleophysic dysplasia cell lysates showed fivefold greater amounts of phosphorylated SMAD2/3 (phospho-SMAD2/3) than control cell lysates (Fig. 5b). Moreover, immunostaining with antibodies to phospho-SMAD2 (anti-phospho-SMAD2) showed an increase in nuclear-localized phospho-SMAD2/3 in two geleophysic dysplasia skin fibroblasts (Fig. 5c).
Figure 5.

Analysis of TGF-β signalling pathway in geleophysic dysplasia fibroblasts. (a) Quantification of total (gray bars) and active (black bars) TGF-β in the conditioned medium of fibroblasts from individuals with geleophysic dysplasia and control fibroblasts. The conditioned medium of geleophysic dysplasia fibroblasts showed a greater amount of total TGF-β (*, P < 0.0003) than conditioned medium of control fibroblasts, which contained only a very small amount of total TGF-β. The active form of TGF-β represented 85% and 92% of total TGF-β in cultured medium from individuals 6 and 2, respectively, but represented only 7% of total TGF-β in control medium. (b) Left panel: enhanced phosphorylation of Smad2 (pSmad2) in control skin fibroblasts and fibroblasts from two individuals with geleophysic dysplasia. Right panel: pSmad2 was normalized to actin for comparison of pSmad2 in fibroblasts from affected and unaffected individuals. (c) Immunostaining for phosphorylated Smad2 in control fibroblasts and fibroblasts from individuals with geleophysic dysplasia. Note the presence of nuclear pSmad2 (arrows) in fibroblasts from affected individuals (center and right) compared to control fibroblasts (left). Scale bar, 10 μm.
TGF-β is a growth factor that regulates cell proliferation, migration, differentiation and survival in a context-dependent fashion and whose activity is tightly regulated through the ECM. TGF-β signaling is crucial in various developmental and homeostatic processes. Enhanced TGF-β signaling has been shown to be a major pathophysiologic factor in Marfan syndrome and related disorders, including Loeys-Dietz syndrome and Camurati-Engelmann disease, all characterized by tall stature and thin habitus13–15. In Marfan syndrome, the diminished ability of microfibrils to bind the LLC presumably results in elevated active TGF-β. Enhanced TGF-β signaling has been demonstrated in various tissues (lung, heart and aorta) in mouse models of Marfan syndrome16,17. In Loeys-Dietz syndrome (which is caused by TGFBR1/R2 loss-of-function mutations), the paradoxical increase of TGF-β signaling has been demonstrated only in the aortic wall14. In addition, the observation of more diffuse and severe arterial disease in Loeys-Dietz syndrome suggests that the disease mechanisms in FBN1 and TGFBR mutations are different18. Similarly, the increased bone density observed only in Camurati-Engelmann disease (due to mutations in the latency-associated peptide of TGF-β) supports bone-specific consequences of enhanced TGF-β signaling in this disorder18. In the context of geleophysic dysplasia, the observation of increased TGF-β signaling in fibroblasts from individuals with geleophysic dysplasia further illustrates the ECM dependence and complexity of the TGF-β–SMAD signaling pathway and demonstrates the existence of new physiological mechanisms regulating TGF-β action. Further-more, the findings of ADAMTS10 and FBN1 mutations in Weill-Marchesani syndrome, a condition related to geleophysic dysplasia, suggests that ADAMTSL2 is a component of a key regulatory network in the ECM that also contains these two proteins. Future studies will clarify the nature of the interactions between LTPB-1, ADAMTS10, FBN1 and ADAMTSL2 and how the lack of functional ADAMTSL2 results in the release of TGF-β. One can hypothesize that ADAMTSL2 acts as a cofactor that enhances or stabilizes binding of LLC to fibrillins. The tissue-specific consequences of ADAMTSL2 mutations also raise the intriguing possibility that other ADAMTS-like proteins may regulate the bioavailability of TGF-β in a site-specific manner dictated by their individual expression profiles. Ongoing studies should contribute to further understanding of the regulatory networks in the ECM and the context-dependent mechanisms leading to the activation of TGF-β signaling.
Methods
Affected individuals
Inclusion criteria for the diagnosis of geleophysic dysplasia were short stature (< −3 s.d.), brachydactyly, restricted joint mobility, characteristic facial features and progressive cardiac involvement. Among the eight affected children, the two from family 1 died of heart disease before 1 year of age. The five other affected individuals, ranging in age from 3 to 18 years, are still alive.
Linkage analysis
We collected blood samples from affected individuals and unaffected siblings after obtaining informed consent. Genomic DNA was extracted from leukocytes using standard procedures. For homozygosity mapping, we used a panel of 400 markers at an average distance of 10 cM. Oligonucleotide sequences of the ADAMTSL2 CA repeat are available on request. We used Linkage software (version M-LINK) package to calculate two-point lod scores between the disease phenotype and each of the markers, assuming a recessive disorder with a mutated allele frequency of 0.01.
Mutation detection
We designed a series of 19 intronic primers to amplify the 18 coding exons of ADAMTSL2 (Supplementary Table 1 online). We purified the amplicons and sequenced them using the fluorescent dideoxy-terminator method on an automatic sequencer (ABI 3100).
In situ hybridization
Probe TSL2 corresponded to nucleotides 1411–1855 of GenBank accession AB011177 (Supplementary Table 1). The PCR product was used to generate antisense and sense cRNA probes with the SP6/T7 DIG RNA labeling kit (Roche) and digoxigenin-11-UTP (Roche) according to the manufacturer's specifications. Paraffin sections of paraformaldehyde-fixed tissues obtained from a human fetus at 35 weeks of gestation (obtained with institutional review board approval) were hybridized to 200 μg/ml DIG-11-UTP–labeled ADAMTSL2 cRNA probe as previously described. After staining with 5-bromo-4-chloro-3′-indoylphosphate p-toluidine salt (BCIP) and nitroblue tetrazolium chloride (NBT) in the dark (Vector Labs) according to the manufacturer's recommendations, we observed the slides using a Zeiss microscope equipped with a Zeiss Axiocam HRc CCD camera with AxioVision acquisition and Cell analysis software.
ADAMTSL2 expression plasmids
Plasmid pcDNAmTSL2, encoding full-length mouse ADAMTSL2 with a C-terminal myc tag, has been previously described7. Clone HG3238a, encoding the ADAMTSL2 cDNA (human ADAMTSL2) was provided by the Kazusa DNA Research Institute. The insert was excised using SalI and NotI and cloned into corresponding sites in pBKCMV (Stratagene) to generate pBKCMV605. A C59F amino acid change present in the HG3238a insert was corrected, and ADAMTSL2 geleophysic dysplasia mutations reported here were introduced in pBKCMV605 using the QuickChange XL site-directed mutagenesis kit (Stratagene) according to the manufacturer's recommendations. The introduced mutations were confirmed by DNA sequencing.
Transfection and protein blotting analysis
HEK293F cells (ATCC) at 80% confluence were transfected (in triplicate) in six-well plates with the full-length wild-type or mutant ADAMTSL2 constructs (plasmid pcDNAmTSL2, 1 μg), using Fugene 6 (Roche Applied Science) according to the manufacturer's instructions. After 48 h, protein blotting with monoclonal anti-myc (9E10; Invitrogen) was used to assess protein expression in the medium and in the cell lysates. Cotransfection of an expression plasmid for human IgG was used for normalization of secretion efficiency, as previously described19,20. Protein species were quantified by densitometry (Kodak 1D image analysis software). For the phospho-Smad2 protein blot, cell lysates were obtained from skin fibroblasts from controls and from individuals with geleophysic dysplasia, and anti-actin (Invitrogen) and anti–phospho-Smad2 (Cell Signaling Technology) were used.
Yeast two-hybrid screening
Full-length human ADAMTSL2 without the signal peptide was used to screen a library of prey proteins from human muscle.
Immunoprecipitation
Human LTBP-1S was purified from the conditioned medium of stably transfected 293 T-Rex cells (Invitrogen)12 using ProBond Ni-chelating resin (Invitrogen). C-terminal myc-tagged mouse ADAMTSL2 was stably transfected in HEK293F cells. The conditioned medium was collected after culture in 293 SFM II medium (Invitrogen) at 37 °C with 8% CO2 for 18 h. After addition of EDTA-free complete mini-protease inhibitor cocktail (Roche), ADAMTSL2 conditioned medium was mixed with purified LTBP-1S (to a final concentration of 5 μg/ml) at 4 °C for 2 h. Anti-myc agarose beads (Sigma) pre-equilibrated with radioimmunoprecipitation assay (RIPA) buffer (5 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.5% (vol/vol) NP-40, 1% (wt/vol) sodium deoxychlorate) were incubated with LTBP-1–supplemented conditioned medium at 4 °C overnight. The beads were washed three times with TBS containing 0.5% NP-40, and bound protein was eluted by boiling in Laemmli sample buffer containing 5% (vol/vol) β-mercaptoethanol for 5 min. The eluted proteins were analyzed by reducing 6% SDS-PAGE followed by protein blotting with either monoclonal anti-human LTBP-1 (R&D) or affinity-purified polyclonal anti-ADAMTSL2 (pAb605-1) using ECL.
ELISA assays for active and total TGF-β1
TGF-β1 present in 100 μl culture medium of confluent fibroblasts from two affected individuals and unaffected controls was quantified using the TGF-β1 EMax Immunoassay kit (Promega). The samples were acidified for measurement of total TGF-β1 (active plus latent). TGF-β1 standard curves were prepared for each assay. All experiments were performed in triplicate, and a t-test was performed.
Immunostaining
Skin fibroblasts from affected individuals and controls were grown in cell culture chambers and fixed in 4% (wt/vol) paraformaldehyde. After blocking with 1% (vol/vol) BSA, 10% (vol/vol) normal goat serum and 0.1% (vol/vol) Triton X-100 in PBS, cells were incubated with pSmad2 overnight at 4 °C, followed by incubation with alkaline phosphatase–coupled anti-mouse IgG at room temperature. The slides were stained with BCIP/NBT (Vector Labs) according to the manufacturer's recommendations.
Transmission electron microscopy
After giving informed consent, an individual with geleophysic dysplasia from family 6 underwent a 4-mm punch biopsy including the dermis. The sample was fixed in 2.5% (vol/vol) glutaraldehyde, post-fixed in 1% (wt/vol) osmium tetroxide, dehydrated in a graded ethanol series and embedded in Epon 812. Semithin sections (1 μm) were stained with toluidine blue. Ultrathin sections were selected, contrasted with uranyl acetate and lead citrate and examined with a Philips EM 300 transmission electron microscope.
Supplementary Material
Acknowledgments
We thank the Kazusa DNA Research Institute for providing the KIAA0605 cDNA. We thank J. Martinovic for her help. We also thank T. Arai and M. Papouin. The work presented here was supported by French National Research Agency (ANR) award R06215KS (to V.C.-D.), the Medical Research Foundation (FRM, to C.L.G.), US National Institutes of Health (NIH) award AR53890 (to S.S.A.), NIH award GM71679 (to D.S.G.) and NIH award HD22657 (to D.K.).
Footnotes
Note: Supplementary information is available on the Nature Genetics website.
Author Contributions
C.L.G. designed the experiments; performed in situ hybridization, protein blot analysis (ADAMTSL2 and SMAD2), TGF-β assays and immunocytochemistry and wrote the manuscript. F.M.-P. and N.D. performed the sequence analysis. L.W.W. performed coimmunoprecipitation studies. C.P., Y.J.C., F.B., E.F., D.K., D.B. and M.L.M. provided clinical data; C.P.-S. performed electron microscopy analysis. G.G. and D.S.G. cloned expression LTBP-1 constructs. A.M. and S.S.A. wrote the manuscript. V.C.-D. provided clinical data, designed the experiments, oversaw all aspects of the research and wrote the manuscript.
References
- 1.Spranger JW, Gilbert EF, Tuffli GA, Rossiter FP, Opitz JM. Geleophysic dwarfism–a “focal” mucopolysaccharidosis? Lancet. 1971;2:97–98. doi: 10.1016/s0140-6736(71)92073-3. [DOI] [PubMed] [Google Scholar]
- 2.Pontz BF, et al. Clinical and ultrastructural findings in three patients with geleophysic dysplasia. Am J Med Genet. 1996;63:50–54. doi: 10.1002/(SICI)1096-8628(19960503)63:1<50::AID-AJMG11>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 3.Shohat M, et al. Geleophysic dysplasia: a storage disorder affecting the skin, bone, liver, heart, and trachea. J Pediatr. 1990;117:227–232. doi: 10.1016/s0022-3476(05)80534-7. [DOI] [PubMed] [Google Scholar]
- 4.Apte SS. A disintegrin-like and metalloprotease (reprolysin type) with thrombospondin type 1 motifs: the ADAMTS family. Int J Biochem Cell Biol. 2004;36:981–985. doi: 10.1016/j.biocel.2004.01.014. [DOI] [PubMed] [Google Scholar]
- 5.Hirohata S, et al. Punctin, a novel ADAMTS-like molecule, ADAMTSL-1, in extracellular matrix. J Biol Chem. 2002;277:12182–12189. doi: 10.1074/jbc.M109665200. [DOI] [PubMed] [Google Scholar]
- 6.Hall NG, Klenotic P, Anand-Apte B, Apte SS. ADAMTSL-3/punctin-2, a novel glycoprotein in extracellular matrix related to the ADAMTS family of metalloproteases. Matrix Biol. 2003;22:501–510. doi: 10.1016/s0945-053x(03)00075-1. [DOI] [PubMed] [Google Scholar]
- 7.Koo BH, et al. ADAMTS-like 2 (ADAMTSL2) is a secreted glycoprotein that is widely expressed during mouse embryogenesis and is regulated during skeletal myogenesis. Matrix Biol. 2007;26:431–441. doi: 10.1016/j.matbio.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 8.Annes JP, Munger JS, Rifkin DB. Making sense of latent TGFβ activation. J Cell Sci. 2003;116:217–224. doi: 10.1242/jcs.00229. [DOI] [PubMed] [Google Scholar]
- 9.Sinha S, Nevett C, Shuttleworth CA, Kielty CM. Cellular and extracellular biology of the latent transforming growth factor-β binding proteins. Matrix Biol. 1998;17:529–545. doi: 10.1016/s0945-053x(98)90106-8. [DOI] [PubMed] [Google Scholar]
- 10.Isogai Z, et al. Latent transforming growth factor β-binding protein 1 interacts with fibrillin and is a microfibril-associated protein. J Biol Chem. 2003;278:2750–2757. doi: 10.1074/jbc.M209256200. [DOI] [PubMed] [Google Scholar]
- 11.ten Dijke P, Arthur HM. Extracellular control of TGFβ signalling in vascular development and disease. Nat Rev Mol Cell Biol. 2007;8:857–869. doi: 10.1038/nrm2262. [DOI] [PubMed] [Google Scholar]
- 12.Ge G, Greenspan DS. BMP1 controls TGFβ1 activation via cleavage of latent TGFβ-binding protein. J Cell Biol. 2006;175:111–120. doi: 10.1083/jcb.200606058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Collod-Beroud G, Boileau C. Marfan syndrome in the third millennium. Eur J Hum Genet. 2002;10:673–681. doi: 10.1038/sj.ejhg.5200876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Loeys BL, et al. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat Genet. 2005;37:275–281. doi: 10.1038/ng1511. [DOI] [PubMed] [Google Scholar]
- 15.Kinoshita A, et al. Domain-specific mutations in TGFB1 result in Camurati-Engelmann disease. Nat Genet. 2000;26:19–20. doi: 10.1038/79128. [DOI] [PubMed] [Google Scholar]
- 16.Neptune ER, et al. Dysregulation of TGF-β activation contributes to pathogenesis in Marfan syndrome. Nat Genet. 2003;33:407–411. doi: 10.1038/ng1116. [DOI] [PubMed] [Google Scholar]
- 17.Jones KB, et al. Toward an understanding of dural ectasia: a light microscopy study in a murine model of Marfan syndrome. Spine. 2005;30:291–293. doi: 10.1097/01.brs.0000152166.88174.1c. [DOI] [PubMed] [Google Scholar]
- 18.Gordon KJ, Blobe GC. Role of transforming growth factor-β superfamily signaling pathways in human disease. Biochim Biophys Acta. 2008;1782:197–228. doi: 10.1016/j.bbadis.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 19.Wang LW, et al. O-fucosylation of thrombospondin type 1 repeats in ADAMTS-like-1/punctin-1 regulates secretion: implications for the ADAMTS superfamily. J Biol Chem. 2007;282:17024–17031. doi: 10.1074/jbc.M701065200. [DOI] [PubMed] [Google Scholar]
- 20.Abbaszade I, et al. Cloning and characterization of ADAMTS11, an aggrecanase from the ADAMTS family. J Biol Chem. 1999;274:23443–23450. doi: 10.1074/jbc.274.33.23443. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.