

Abstract
We investigated the influence of membrane cholesterol content on preferential and non-preferential signaling through the M2 muscarinic acetylcholine receptor expressed in CHO cells. Cholesterol depletion by 39% significantly decreased the affinity of M2 receptors for [3H]-N-methylscopolamine ([3H]-NMS) binding and increased Bmax in intact cells and membranes. Membranes displayed two-affinity agonist binding sites for carbachol and cholesterol depletion doubled the fraction of high-affinity binding sites. In intact cells it also reduced the rate of agonist-induced receptor internalization and changed the profile of agonist binding from a single site to two affinity states. Cholesterol enrichment by 137% had no effects on carbachol Emax of cAMP synthesis inhibition and on cAMP synthesis stimulation and inositolphosphates (IP) accumulation at higher agonist concentrations (non-preferred pathways). On the other hand, cholesterol depletion significantly increased Emax of cAMP synthesis inhibition or stimulation without change in potency, and decreased Emax of IP accumulation. Noteworthy, modifications of membrane cholesterol had no effect on membrane permeability, oxidative activity, protein content, or relative expression of Gs, Gi/o, and Gq/11 alpha subunits. These results demonstrate distinct changes of M2 receptor signaling through both preferential and non-preferential G-proteins consequent to membrane cholesterol depletion that occur at the level of receptor/G-protein/effector protein interactions in the cell membrane. The significant decrease of IP accumulation by cholesterol depletion was also observed in cells expressing M3 receptors and by both cholesterol depletion and enrichment in cells expressing M1 receptors indicating relevance of reduced Gq/11 signaling for the pathogenesis of Alzheimer’s disease.
Keywords: muscarinic receptors, cholesterol, G-proteins coupling, inositolphosphates, cAMP, agonist binding
1. Introduction
The muscarinic acetylcholine receptor family consists of five subtypes denoted M1-M5 (Bonner, 1989; Caulfield and Birdsall, 1998). Each of these subtypes has distinct tissue distribution and serves a specific physiological function (Hulme et al., 1990). Muscarinic receptors belong to the family of G-protein-coupled receptors with seven segments spanning the cell membrane (Fredriksson et al., 2003). Conventionally, individual G-protein coupled receptors selectively interact with distinct subclasses of G-proteins to preferentially activate different intracellular signaling pathways. In line with this concept, M1/3/5 muscarinic receptors preferentially couple with effector molecules through the Gq/11 subclass of G-proteins and M2/4 receptors favor coupling via the Gi/o G-proteins. However, their coupling specificity is not absolute. Muscarinic receptor interaction with nonpreferential G-proteins and stimulation of their signaling pathways has been demonstrated in many studies (Migeon and Nathanson, 1994; Vogel et al., 1995; Michal et al., 2001; Jakubík et al., 2006) and the direct interaction of muscarinic M2 receptor with nonpreferential G-proteins using RNA interference knockdown has been recently demonstrated (Michal et al., 2007). These observations strongly support the concept of multiple agonist-induced receptor conformations (Kenakin, 2003; Kobilka, 2007).
Efficiency of signal transduction through muscarinic receptors depends not only on the concentration of agonist in the extracellular fluid but can also be both increased or decreased by substances acting as allosteric modulators (Tuček et al. 1990; Jakubík et al., 1995, 1997, and 2002; Lazareno and Birdsall 1995; Doležal and Tuček, 1998, Lazareno et al., 2004). Another factor that likely plays an important role in signal transduction through muscarinic receptors is lipid composition of the cell membrane in which the receptor is incorporated. Investigations of rhodopsin, a prototypic and best molecularly-characterized G-protein coupled receptor (activated by light), and the oxytocin receptor have indicated that cholesterol content in membranes has important influence on the transfer of information by these receptors. In the case of rhodopsin, high membrane content of cholesterol completely blocks its activation (Mitchell et al., 1990) and in the case of oxytocin receptor high cholesterol content converts receptors to a low-affinity conformation (Klein et al., 1995).
Lipid composition of the cell membrane is not homogenous. There are domains with high content of cholesterol denominated „lipidic rafts“ (Simons and Toomre, 2000). G-protein-coupled receptors are often associated with these rafts and disruption of lipidic rafts may lead to impairment of signal transduction (Pike, 2003). It has been demonstrated that stimulation of luteinizing hormone receptors leads to their translocation into rafts and serves to fine tune cellular responses, but this translocation is not necessary for hormone-induced signaling (Smith et al., 2006). In case of muscarinic M2 and M3 receptors expressed in Chinese hamster ovary (CHO) cells, studies of fluorescence resonance energy transfer of fluorescent protein-tagged G-protein subunits have established their free diffusion in the cell membrane and interactions with G-proteins (Azpiazu and Gautam, 2004).
Direct inhibitory influence of endogenous steroids derived from progesterone on the binding of N-methylscopolamine to M2 muscarinic receptors was observed in intact rat cardiac tissue. This effect was not due to binding of these agents to either the orthosteric or allosteric sites of the muscarinic M2 receptor and it was hypothesized that it could involve a mechanism at the level of cell membrane (Wilkinson et al., 1995). Moreover, the influence of changes of membrane cholesterol content induced by growth in medium supplemented with lipoprotein-deficient serum on the binding characteristics and functional outcome of muscarinic M2 receptors stimulation was studied in non-differentiated chick embryonic cardiocytes. The increase of membrane cholesterol content induced by the treatment was associated with the appearance of a characteristic muscarinic receptor-evoked negative chronotropic response (Renaud et al., 1982). However, this treatment also increased expression levels of muscarinic receptors and G-proteins (Haigh et al., 1988). Recently it has been proposed that cholesterol may bind to muscarinic M2 receptors and influence its properties in a fashion similar to that of allosteric modulators (Colozo et al., 2007).
The main objective of our work presented here was to determine the effects of immediate manipulation of membrane cholesterol content on M2 receptor binding properties and efficacy of signal transduction via both preferential and non-preferential G-proteins. To achieve this aim we used human M2 muscarinic receptors heterologously expressed in CHO cells. We report that modification of cell membrane cholesterol content using methyl-β-cyclodextrin (MBCD) in intact cells leads to acute changes of the binding affinity of the muscarinic antagonist N-methylscopolamine (NMS) and causes various alterations in both the potency and efficacy of the acetylcholine analogue carbachol in activating signaling through the Gi/o, Gs, and Gq/11 subclasses of G-proteins.
2. Methods
2.1. Cell culture and chemicals
Experiments were performed on CHO cells stably transfected with the human genes of the muscarinic M1, M2, and M3 receptor subtypes (CHO-M1, CHO-M2, and CHO-M3 cells, respectively) kindly supplied by Prof. Tom Bonner. Cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) with 10% fetal calf serum and 0.005% geneticin, and used for experiments three to four days after seeding. In some experiments, pertussis toxin was present at a concentration of 100 ng/ml during the last 24 h of cultivation to inactivate Gi/o G-proteins. Cells were grown in 10 cm diameter Petri dishes for radioligand saturation and competition binding analysis or in 24-well plates for functional assays. Chemicals were obtained from Sigma (Prague, Czech Republic) unless indicated otherwise.
2.2. Saturation and competition binding assays
Density and affinity of muscarinic receptors were determined in saturation binding experiments with the membrane-impermeable quaternary amine muscarinic antagonist [3H]-N-methylscopolamine (3H-NMS) (GE Healthcare, UK) in cells in suspension and cell membranes (Michal et al., 2001). Briefly, cells were grown to confluency in 10 cm diameter Petri dishes. Before experiments, cells from the same Petri dish were detached by mild trypsinization and incubated for 1 h at 37 °C in 15 ml of DMEM containing either 10 mM methyl-β-cyclodextrin (MBCD) to deplete cholesterol content or 2 mM MBCD saturated with cholesterol (Ch-MBCD) to increase cholesterol content. Medium containing cholesterol-modifying agents was always washed off before experiments. Membranes were prepared as described by Jakubík et al. (2006). In control experiments, membranes prepared from naïve cells (control membranes) were treated with cholesterol-modifying agents as described above and washed before radioligand saturation binding analysis. Non-specific binding was determined in the presence of 10 μM atropine. Binding characteristics of the muscarinic agonist carbachol were determined in competition experiments with 0.6 nM [3H]-NMS as a tracer.
2.3. Functional assays
Inositolphosphates (IP) accumulation and cAMP production were assayed in attached cells grown in 24-well-plates. For cAMP synthesis measurements, cells were loaded in 0.2 ml of DMEM with [3H]-adenine (10 μCi/ml; GE Healthcare, UK) for 4 h at 37 °C, and afterwards treated with cholesterol-modifying agents for 1 h in 1.2 ml of DMEM containing either 10 mM methyl-β-cyclodextrin (MBCD) to deplete cholesterol content or 2 mM MBCD saturated with cholesterol (Ch-MBCD) to increase cholesterol content. Medium containing cholesterol-modifying agents was always washed off before experiments. Cells were then preincubated in 0.4 ml of DMEM containing 1.2 mM isobutylmethylxanthine for 15 min at 37 °C. After preincubation, cells were incubated with 10 μM forskolin and increasing concentrations of carbachol for 10 min at 37 °C in a final volume of 0.5 ml. The reaction was stopped by adding 0.2 ml of 20% trichloroacetic acid (TCA), 0.1 ml of cyclic [14C]-AMP (GE Healthcare, UK) that was used as recovery standard, and 1.2 ml of water. Aliquots of TCA extracts were used for determination of TCA-soluble radioactivity and separation of [3H]-AMP from other labeled metabolites (Michal et al., 2001). TCA precipitates were dissolved in 1 ml of 200 mM NaOH and aliquots of these lysates were used for determination of protein content. The concentration of forskolin used in these experiments (10 μM) induced less than half-maximal stimulation of cAMP synthesis in the absence of carbachol and thus allowed detection of either decreases or increases of the response by activation of muscarinic receptors. A time interval of 10 min was chosen as the shortest incubation that allows reliable quantification of labeled cAMP as assessed previously (Michal et al., 2001 and 2007). Basal synthesis of cAMP (shown in Table 4) was determined as a difference between samples incubated without carbachol in the presence and absence of the phosphodiesterase inhibitor.
Table 4.
Influence of changes in membrane cholesterol content on resting values of cAMP synthesis and inositol phosphates accumulation
| Treatment | |||
|---|---|---|---|
| control | MBCD | Ch-MBCD | |
| cAMP synthesis | |||
| CHO-M2 native cells | 1.27±0.03 | 1.71±0.04a | 1.18±0.02 |
| CHO-M2 PTX-treated cells | 1.09±0.01 | 1.33±0.02a | 0.97±0.02a |
|
| |||
| IP accumulation | |||
| CHO-M2 cells | 1.78±0.16 | 0.83±0.15a | 3.42±0.21a |
Resting values of cAMP synthesis and IP accumulation in control, cholesterol-depleted (MBCD), and cholesterol-enriched (Ch-MBCD) native or pertussis toxin-pretreated cells are expressed in percent of tissue content of radioactivity as mean±S.E.M. of number of observations indicated in Fig. 2 and 3.
P<0.01, significantly different from controls by Anova and Tukey’s test.
IP formation was determined in cells that were prelabeled for 4 h by [3H]myo-inositol (10 μCi/ml; GE Healthcare, UK) in 0.2 ml of DMEM at 37 °C and treated with cholesterol-modifying agents as described for cAMP synthesis measurement. Cells were then washed with fresh medium and preincubated in 0.4 ml of DMEM containing 12 mM LiCl for 15 minutes at 37 °C. The indicated concentrations of carbachol were added and samples were incubated for additional 20 min (CHO-M2 cells) or 10 min (CHO-M1 and CHO-M3 cells) in a final incubation volume of 0.5 ml. Incubation was stopped on ice by adding 0.2 ml of 20% TCA and 1.2 ml of water. In case of CHO-M2 cells aliquots of TCA extracts were used for determination of TCA-soluble radioactivity and separation of total inositolphosphates using [14C]-inositolphosphate as recovery standard (Jakubík et al., 1995). TCA precipitates were dissolved in 1 ml of 200 mM NaOH and aliquots of these lysates were used for determination of protein content and incorporated radioactivity. In case of cells expressing M1 and M3 muscarinic receptors that preferentially couple with Gq/11 G-proteins and induce a large response, accumulated TCA-soluble radioactivity was used for estimation of formation of inositol phosphates without further separation. Sensitivity of carbachol-evoked accumulation of TCA-soluble radioactivity to a muscarinic antagonist was verified in attached CHO-M3 cells. Atropine (10 μM) inhibited accumulation of TCA-soluble radioactivity evoked by 0.1–100 μM carbachol by more than 97%. Comparison of the concentration-responses of carbachol-induced increases of IP accumulation in CHO-M1 cells in suspension after separation of inositolphosphates and in TCA-soluble radioactivity without separation in attached cells showed similar pEC50 (5.74±0.05 and 5.95±0.05, respectively, in three experiments in triplicates). However, measurement of TCA-soluble radioactivity without separation does not allow quantification of basal levels of inositolphosphates that unlike carbachol-iduced increase of TCA-soluble radioactivity represent only a small fraction of total radioactivity in sample. Basal accumulation of inositolphosphates (shown in Table 4) was determined as the difference between samples incubated without carbachol in the presence and absence of lithium.
Radioactivity of samples was measured by liquid scintillation counting. The muscarinic receptor specificity of carbachol effects on cAMP synthesis and IP accumulation was verified by abolishing the response with 10 μM atropine in parallel experiments.
Internalization of muscarinic receptors was measured in attached cells as a decrease of [3H]-NMS specific binding in cells grown in 24-well-plates. After cholesterol-modifying treatments and washing as described above, CHO-M2 cells were preincubated with various concentrations of carbachol for various time intervals at 37 °C in 1 ml of DMEM, washed with fresh medium, and incubated in 0.5 ml of medium containing 2 nM [3H]-NMS for 12 h at 4 °C to prevent further internalization. Incubation medium with free [3H]-NMS was then removed, cells were quickly washed with 1.5 ml of ice-cold phosphate buffered saline, and dissolved in 1.5 ml of 200 mM NaOH. Aliquots of lysates were used for scintillation counting and protein determination. Non-specific binding was measured in the presence of 10 μM atropine.
2.4. Miscellaneous
Metabolic activity and membrane permeability of attached cells grown in 24-well-plates after treatments was deduced from fluorescein oxidation and calcein retention as described in Nováková et al. (2005). Western blotting and immunodetection of individual G-protein α-subunits was done as described by Rudajev et al. (2005) with 20 μg of membrane proteins per sample. Protein content was determined in TCA precipitates by Peterson’s modification of the Lowry’s method (Peterson, 1977). Effects of treatments on cholesterol levels were determined using Amplex® Red Cholesterol Assay Kit (Molecular Probes, Eugene, OR) according to manufacturer’s protocol.
2.5. Data treatment
Curve fitting and statistical evaluation of data was done using Prism 5 (GraphPad Software Inc., CA). A rectangular hyperbola Y=Bmax*X/(Kd+X) was fitted to data shown in Fig. 1A, B. Y is the specific binding of [3H]-NMS, Bmax maximum number of binding sites, X concentration of free [3H]-NMS, and Kd equilibrium dissociation constant.
Fig. 1.

Binding of [3H]-NMS and carbachol in cells and membranes after cholesterol depletion or cholesterol enrichment. A and B: Specific [3H]-NMS binding in intact cells (A) and membranes (B) is fitted as a saturation isotherm (abscissa, concentration of [3H]-NMS; ordinate, specific binding in pmol/mg protein). Shown is a representative experiment. Each point is the mean±S.E.M. of triplicate values. When S.E.M. is not depicted it was smaller than the symbol. Parameters of non-linear regression fits are shown in Table 2. C and D: Displacement of 0.6 nM [3H]-NMS by carbachol (abscissa, log M) is expressed as percent of control (ordinate) in intact cells (C) and membranes (D) without or with added GTP as indicated. Each point is the mean±S.E.M. of six values from two independent experiments. One- or two-site competition equation is fitted to data as appropriate. Parameters of fits are shown in Table 3. Control, cells or membranes derived from untreated cells; MBCD, cholesterol-depleted cells or membranes prepared from these cells; Cholesterol-MBCD, cholesterol-enriched cells or membranes derived from these cells. Dashed lines in (D) denote competition curves in membranes incubated in the presence of 10 mM GTP.
Sigmoidal concentration-response curve equations with slope of unity (Y=(Bottom+(Top-Bottom))/(1+10(logEC50−X))) or variable slope (Y=(Bottom+(Top-Bottom))/(1+10(logEC50−X)*Hill slope))) were fitted to the data shown in Figs. 2A (pertussis toxin-treated cells, dashed lines) and B, 3A–D, and 4A as appropriate. X is log of agonist concentration, Y measured effect (cAMP synthesis or IP accumulation), and EC50, concentration of agonist required to produce half-maximal effect. In case of bell-shaped concentration-response curve of carbachol effects on cAMP synthesis, a logistic equation that describes the sum of two concentration-response curves Y=Dip+Span1/(1+10(LogEC501−X)*nH1))+ Span2/(1+10(X−LogEC502)*nH2)) was fitted to data (Fig. 2A, full lines) to confirm correctness of parameters obtained by fitting a sigmoidal concentration-response equation to data after subtraction of basal values as described in Fig. 2B. nH1 and nH2 are Hill slopes, Span1, Dip, and Span2 are plateau levels.
Fig. 2.

Influence of changes in membrane cholesterol content on carbachol-induced modulation of cAMP synthesis in intact cells. A: Raw data of concentration-response relationship of carbachol effects (abscissa, log M) on forskolin-stimulated cAMP synthesis are expressed as percent of incorporated radioactivity (ordinate). Bell-shaped concentration-response equation is fitted to data obtained in native cells (full lines) and Sigmoidal concentration-response equation with slope of unity to data obtained in pertussis toxin-pretreated cells (dashed lines). Statistical evaluation of the effect of treatments on resting values is shown in Table 4. B: For better clarity, effects of cholesterol depletion or enrichment on carbachol-induced changes of forskolin-stimulated cAMP synthesis is expressed as a net change (ordinate; in percent of incorporated radioactivity). The net inhibition of cAMP synthesis was obtained by subtracting basal values from values measured in the presence of carbachol up to a concentration of 3 μM shown in A. The net stimulation of cAMP synthesis in native cells by carbachol from a concentration of 10 μM up was obtained by subtracting calculated maximal inhibition values. The net stimulation of cAMP synthesis in pertussis toxin-treated cells (dashed lines) was obtained by subtracting measured basal values from those in the presence of carbachol. Sigmoidal concentration-response equation with slope of unity was fitted to the data. Parameters of fits are shown in Table 5. Symbols are as described in Fig. 1.
One-site displacement (log EC50=log(10logKi*(1+HotnM/HotKdnM)); Y=Bottom + (Top-Bottom)/(1+10(X−LogEC50)) or two-sites displacement curve equations (ColdnM=10(X+9); KiHinM = 10(LogKiHi+9); KiLonM = 10(LogKiLo+9); SITE1= HotnM*(Top-Bottom)/(HotnM + KdHotnMKi*(1+coldnM/KiHinM)); SITE2= HotnM*(Top-Bottom)/(HotnM + KdHotnMLo*(1+coldnM/KiLonM)); Y = (SITE1*FractionHi) + (SITE2*(1-FractionHi)) + Bottom) were fitted to data shown in Fig. 1C, D. Ki is the equilibrium dissociation constant, HotnM, the concentration of tracer [3H]-NMS, HotKdnM, equilibrium dissociation constant of tracer antagonist, coldnM, concentration of carbachol, X log M, concentration of carbachol, Y binding of tracer, Ki Hi and Ki Lo, equilibrium dissociation constant of carbachol for high (Hi) and low (Lo) affinity binding sites, respectively.
One-phase exponential decay equation Y=Span*e(−K*X) + Plateau was fitted to data shown in Fig. 4B, C. Y is number of binding sites, K is rate constant, and X is time.
Fig. 4.
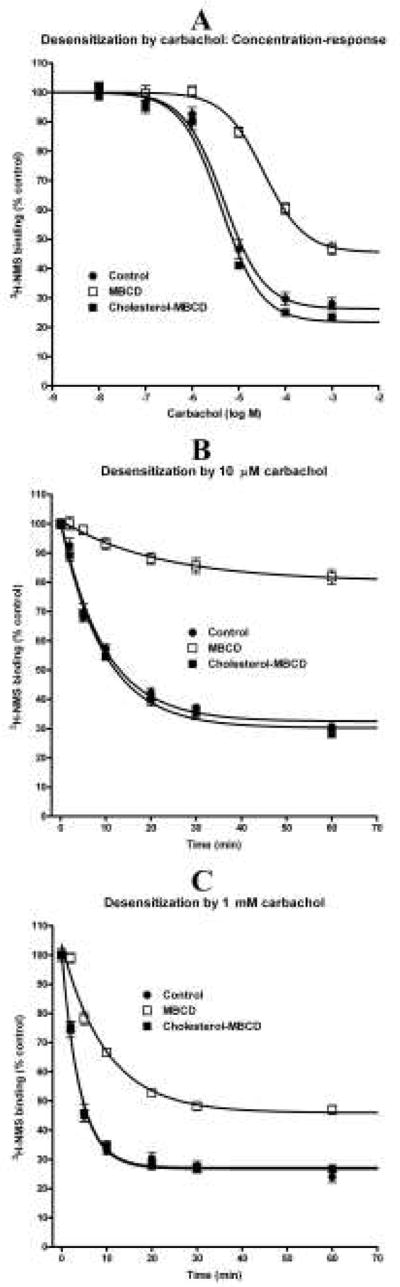
Effects of changes in membrane cholesterol content on internalization of M2 receptors induced by carbachol stimulation. A: Concentration-response of carbachol during 20 min incubations (abscissa log M) on the number of plasma membrane M2 receptors in intact cells. B and C: Time-course (abscissa, time in min) of 10 μM (B) and 1 mM (C) carbachol effects on the number of plasma membrane M2 receptors in intact cells is expressed as percent of control values (ordinates). Points are mean±S.E.M. when bigger than symbols, of six values from two independent experiments. Symbols and descriptions are the same as in Figure 1. Sigmoidal concentration-response equation with slope of unity (A) or single phase exponential decay equation (B and C) was fitted to data. Parameters of fits are shown in Table 6.
Better fits were determined using F-test. Significance of differences among groups was estimated by Anova and Tukey’s. Results are shown as mean±S.E.M.
3. Results
Total cholesterol content in control intact CHO-M2 cells and in cell membranes was 56.0 and 129.4 pmol/μg protein, respectively (Table 1). Free cholesterol represented 96.5±1.2% and 99.0±3.2% of total cholesterol in cells and membranes, respectively (mean±range of two experiments done in triplicates). Treatment of cells with MBCD resulted in a significant decrease in membrane cholesterol content by 39%. Incubation of cells in the presence of Ch-MBCD increased membrane cholesterol levels by 137%. As expected, changes in total cellular cholesterol content were more pronounced and amounted to 74% decrease and 169% increase after MBCD and Ch-MBCD treatment, respectively (Table 1).
Table 1.
Cholesterol content
| Cholesterol content (pmol/μg protein) | ||
|---|---|---|
| Treatment | Intact cells | Membranes |
| No treatment-control | 56.0±1.1 (6) | 129.4±10.1 (4) |
| MBCD | 14.5±1.4 (6)a | 79.4±5.6 (4)a |
| Cholesterol-MBCD | 150.5±4.2 (5)a | 306.7±28.3 (4)a |
Total cholesterol (cholesterol+cholesterol esters) content values are mean±S.E.M. of the number of independent experiments given in parentheses.
P<0.01, significantly different from controls by Anova and Tukey’s test.
Saturation analysis of [3H]-NMS specific binding with intact cells and cell membranes confirmed binding to a single affinity site in all conditions (Fig 1A, B; Table 2). Cholesterol-depleted intact cells demonstrated significant augmentation (by 68%) of cell surface muscarinic receptor number (Bmax). This effect was accompanied by a significant two-fold decrease in [3H]-NMS affinity (Kd). Similar changes (35% increase in Bmax and 50% decrease in affinity) were found in membranes prepared from MBCD-treated cells. We performed two types of control experiments. Treatment of membranes prepared from control cells with MBCD showed similar increases in Bmax (from 1.43±0.03 to 1.91±0.05 pmol/mg protein; mean±S.D. of triplicate measurements) and Kd (from 410±23 to 675±41 pM). In another experiment we used the membrane permeable muscarinic antagonist [3H]-quinuclidinylbenzilate (GE Healthcare, UK) that labels both intracellular and plasma membrane receptors to see whether the observed increase in plasma membrane M2 receptor density following MBCD treatment is due to incorporation of preformed intracellular receptors into the plasma membrane. Similar to the observed changes in [3H]-NMS binding, treatment of cells with MBCD increased specific [3H]-quinuclidinylbenzilate binding to intact cells (from 0.72±0.01 to 1.05±0.01 pmol/mg protein; mean±S.D. of triplicate measurements) and membranes prepared from MBCD-treated cells (from 1.05±0.04 to 1.54±0.02 pmol/mg protein; mean±S.D. of triplicate measurements). Unlike [3H]-NMS binding, MBCD treatment had no effect on affinity of [3H]-quinuclidinylbenzilate binding (97±12, 95±5, 94±8, and 87±3 pM in control cells, MBCD-treated cells, membranes prepared from control cells, and membranes prepared from MBCD-treated cells, respectively). Cholesterol enrichment had no effect on [3H]-NMS binding parameters (Table 2). Effects of treatments on the affinity of carbachol were derived from competition [3H]-NMS binding assays summarized in Fig. 1C, D and Table 3. In intact cells (Fig. 1C, Table 3), one-site competition equation adequately fit carbachol binding in control and cholesterol-enriched cells with comparable Ki values (4.9 and 4.3 μM in control and cholesterol-enriched cells, respectively). In contrast, a two-site competition equation provided a better fit in cholesterol-depleted cells (P<0.0001; F-test). These cells demonstrated 24% of high affinity sites with Ki of 3.6 μM and low affinity sites with Ki of 72.6 μM.
Table 2.
Influence of changes in membrane cholesterol content on parameters of [3H]-NMS binding
| Intact cells | Membranes | |||
|---|---|---|---|---|
| Treatment | Bmax (pmol/mg protein) | Kd (pM) | Bmax (pmol/mg protein) | Kd (pM) |
| Control | 1.18±0.07 | 243±31 | 1.87±0.30 | 334±54 |
| MBCD | 1.98±0.18a | 511±90a | 2.53±0.37a | 500±43a |
| Cholesterol-MBCD | 0.83±0.08 | 205±28 | 1.60±0.31 | 316±48 |
Parameters of [3H]-NMS saturation analysis are mean±S.E.M. of five (intact cells) or six (membranes) independent experiments in triplicates as that shown in Fig. 1A, B. Bmax, maximal binding; Kd, equilibrium dissociation constant of [3H]-NMS;
P<0.01, significantly different from controls by two-way Anova and Tukey’s test.
Table 3.
Affinity of carbachol in intact CHO-M2 cells and membranes after cholesterol depletion or enrichment.
| Treatment | |||
|---|---|---|---|
| control | MBCD | Ch-MBCD | |
| Cells one-site fit | |||
| Ki (log M) | −5.30±0.01 | - | −5.36±0.01 |
|
| |||
| Cells two-site fit | |||
| Ki high (log M) | - | −5.45±0.08 | - |
| Ki low (log M) | - | −4.14±0.03 | - |
| Fraction high | - | 0.24±0.02 | - |
|
| |||
| Membranes two-site fit | |||
| Ki high (log M) | −7.73±0.04 | −7.18±0.03a | −8.38±0.05a |
| Ki low (log M) | −5.33±0.02 | −4.75±0.05a | −5.61±0.03a |
| Fraction high | 0.33±0.01 | 0.63±0.01a | 0.39±0.01a |
|
| |||
| Membranes+GTP two-site fit | |||
| Ki high (log M) | −5.89±0.06 | −5.02±0.16a | −5.86±0.05 |
| Ki low (log M) | −4.76±0.07 | −4.06±0.08a | −4.73±0.08 |
| Fraction high | 0.52±0.05 | 0.32±0.09b | 0.57±0.04 |
Ki values were calculated from [3H]-NMS displacement by carbachol shown in Fig. 1C, D and [3H]-NMS affinity indicated in Table 2.
P<0.01;
P<0.05; significantly different from corresponding controls by Anova and Tukey’s test. Preferred one or two site fits were determined by F-test (P<0.001).
Competition experiments in membranes prepared from control and treated cells (Fig. 1D, Table 3) exhibited two binding sites with significantly higher proportion of high-affinity sites in both cholesterol-depleted and to a lesser extent in cholesterol-enriched membranes (33±1% in control membranes; 63±1% and 39±1% in cholesterol-depleted and cholesterol-enriched membranes, respectively). The affinity of both high- and low-potency binding sites for carbachol was lowest in cholesterol-depleted and highest in cholesterol-enriched membranes (Ki of high affinity sites 66, 19, 4 nM, and low affinity sites 17.8, 4.7, 2.4 μM in cholesterol-depleted, control, and cholesterol-enriched membranes, respectively). Addition of 10 mM GTP expectedly caused a rightward shift of all displacement curves. Even under these conditions, however, obtained data better fitted to a two-site competition equation in all experimental groups (P<0.0001; F-test). The rightward shift of carbachol affinity ranged from 69 times to 331 times for high affinity sites and from 3.7 times to 7.6 times for low affinity sites in membranes prepared from control and cholesterol-enriched cells, respectively (Fig. 1D and Table 3).
Modification of cell membrane cholesterol content caused significant changes in M2 receptor activation of both preferential and non-preferential signaling pathways (Figs. 2, and 3AC, Table 4). As shown in Figs. 2A, 3A, and Table 4, changes of cholesterol concentrations influenced basal values of cAMP synthesis in control and pertussis toxin-treated cells as well as values of basal IP accumulation. For better clarity, concentration-response curves of carbachol effects on activation of individual signaling pathways were expressed as a net change of metabolite concentration (Figs. 2B and 3C, D).
Fig. 3.

Influence of changes in membrane cholesterol content on carbachol-induced inositol phosphates (IP) accumulation in intact cells expressing M2, M1, and M3 receptors. A and B: Shown are raw data (ordinate, percent of incorporated radioactivity; abscissa, log M concentration of carbachol) in cells expressing M2 receptor (A) or M1 and M3 receptors (B). C and D: For easier comparison, concentration-responses of carbachol-induced (abscissa, log M) IP accumulation are shown as a net increase above basal in percent of incorporated radioactivity (ordinate). The net stimulation of IP accumulation was calculated by subtracting resting values. A sigmoidal concentration-response equation was fitted to data. Statistical evaluation of the effect of treatments on resting values is shown in Table 4 and parameters of fits are shown in Table 5. Symbols are the same as in Figure 1. CHO-M1-M3, CHO cells expressing M1-M3 receptors individually.
Cholesterol depletion increased not only the efficacy (Emax) of carbachol in preferential inhibition of cAMP synthesis but also the Emax of nonpreferential stimulation of cAMP synthesis observed at higher carbachol concentrations. However, cholesterol depletion had no effect on the potency (EC50) of carbachol in activating either response (Fig. 2, Table 5). Although the data on cAMP synthesis stimulation (Fig. 2) apparently did not reach a plateau, fitting of the sigmoideal concentration-response curve is justified by the data on cAMP synthesis stimulation after pertussis toxin treatment that are devoid of the inhibitory influence of Gi/o G-proteins, and our previous observation of saturability with higher concentrations of carbachol in control cells (Michal et al., 2001). In concert, fitting a logistic bell-shaped concentration-response equation (Hornigold et al., 2003) to the data on cAMP synthesis obtained in native cells (Fig. 2A, full lines) provided virtually the same results as those obtained by fitting sigmoidal concentration-response curves to normalized data listed in Table 5 (i.e. Hill slope of unity for both stimulatory and inhibitory response; inhibitory pEC50 6.56±0.07, 6.49±0.05, and 6.70±0.06 for control, cholesterol-depleted, and cholesterol-enriched cells, respectively, with corresponding values of pEC50 3.78±0.09, 3.59±0.06, and 3.77±0.07 for the stimulatory component).
Table 5.
Influence of cholesterol content on carbachol-induced changes in cAMP synthesis and IP accumulation in intact CHO-M2 cells.
| Treatment | ||||
|---|---|---|---|---|
| Response | Parameter | Control | MBCD | Cholesterol-MBCD |
| CHO-M2 cells | ||||
| cAMP synthesis inhibition | Emax | −0.48±0.02 | −0.65/−0.04a | −0.37±0.02 |
| EC50 (log M) | −6.53±0.08 | −6.43±0.08 | −6.65±0.07 | |
| cAMP synthesis stimulation | Emax | 0.45/−0.04 | 0.76/−0.07a | 0.37±0.03 |
| EC50 (log M) | −3.76±0.11 | −3.51±0.11 | −3.65±1.11 | |
| cAMP synthesis stimulation after PTX | Emax | 0.57±0.01 | 1.51/−0.03a | 0.38±0.01a |
| EC50 (log M) | −4.88±0.05 | −4.59±0.04a | −5.20±0.06a | |
|
| ||||
| CHO-M2 cells | ||||
| IP accumulation | Emax | 7.88±0.21 | 5.15±0.21a | 7.80±0.26 |
| EC50 (log M) | −4.08±0.04 | −4.31±0.07 | −4.17±0.05 | |
|
| ||||
| CHO-M1 cells | ||||
| IP accumulation | Emax | 42.0±1.3 | 12.2±0.8a | 23.4±0.6a |
| EC50 (log M) | −5.45±0.05 | −5.19±0.09 | −5.53±0.04 | |
|
| ||||
| CHO-M3 cells | ||||
| IP accumulation | Emax | 38.9±2.1 | 10.9±0.8a | 35.4±1.4 |
| EC50 (log M) | −5.69±0.09 | −5.15±0.10a | −5.81±0.07 | |
Parameters are derived from concentration-response curves shown in Figs 2B and 3B. Results are mean±S.E.M. of values from two independent experiments done in hexaplicates (CHO-M2 cells) or three independent experiments done in triplicates (CHO-M1 and CHO-M3 cells). Emax, maximal change induced by carbachol in percent of incorporated radioactivity;
P<0.01, significantly different from corresponding controls by Anova and Tukey’s test.
As expected, inactivation of the preferential inhibitory Gi/o G-proteins by pertussis toxin treatment significantly increased the potency of carbachol in stimulating cAMP synthesis in all conditions (13, 12, and 37 fold in control, cholesterol-depleted, and cholesterol-enriched cells, respectively). In contrast, an appreciable significant increase in carbachol efficacy in stimulating cAMP synthesis compared to both pertussis toxin-treated control cells and corresponding native controls cells was apparent only in cholesterol-depleted cells (from 0.57% of tissue content in pertussis toxin-treated control cells and 0.76% in native cholesterol-depleted cells to 1.51% in pertussis toxin-treated cholesterol-depleted cells; Fig. 2B and Table 5).
Cholesterol enrichment in native cells had no major effect on carbachol potency and efficacy in inhibiting or stimulating cAMP synthesis (Fig. 2, Table 5). However, the stimulation of cAMP synthesis after pertussis toxin treatment demonstrated a significant decrease in carbachol efficacy that was accompanied with significant increase in potency, unlike in case of cAMP synthesis in native cells (Fig. 2, Table 5).
Since cAMP synthesis was determined by the conversion of [3H]ATP to [3H]cAMP in [3H]adenine-loaded cells, we tested the effects of treatment of cells with cholesterol-modifying agents on incorporation of [3H]adenine. The loading was 10944±93, 11688±73, and 10304±107 dpm/μg protein in control, MBCD-treated, and Ch-MBCD-treated cells that were not exposed to pertussis toxin, respectively (n = 144 individual samples in each group). The loading was only marginally higher in cholesterol-depleted compared to control and cholesterol-enriched cells (P<0.05 by ANOVA and Tukey’s test). The treatments had virtually no effect on [3H]adenine incorporation in pertussis toxin-treated cells, being 8793±118, 8484±113, and 8562±110 dpm/μg protein in control, MBCD-treated, and Ch-MBCD-treated cells, respectively (n=108 individual samples in each group).
In contrast to the increase of basal cAMP synthesis, cholesterol depletion decreased by 54% and cholesterol enrichment increased by 91% resting IP accumulation (from 1.79±0.16 to 0.83±0.15 and 3.42±0.21 percent of tissue content of radioactivity, respectively; Table 4). Maximal stimulation of IP accumulation by carbachol in cholesterol-depleted CHO-M2 cells was significantly smaller while cholesterol enrichment had no effect (Fig. 3A, C and Table 5). Cellular incorporation of [3H]-myo-inositol was not influenced by the subsequent treatments with cholesterol-modifying agents (1592±15, 1578±15, and 1552±14 dpm/μg protein in control, MBCD-treated, and Ch-MBCD-treated cells, respectively; n=84 individual samples in each group).
The treatments changed muscarinic receptor density, being 1.00±0.04, 1.67±0.10, and 0.76±0.07 pmol/mg protein in control, cholesterol-depleted, and cholesterol-enriched cells, respectively (mean±S.E.M. from four independent experiments in triplicates). The increase observed in cholesterol depleted cells was significant at P<0.01 (by ANOVA and Tukey’s test). Possible contribution of receptor internalization to the differential effects of cholesterol depletion on M2 receptor signaling pathways was addressed in experiments summarized in Fig. 4 and Table 6. Carbachol-induced receptor internalization was significantly attenuated in cholesterol-depleted cells, with a seven-fold increase in EC50 (from 4.7 to 34 μM) and reduction of maximal decrease in cell surface receptors from 73.7% to 54.5% (Fig. 4A and Table 6). In contrast, cholesterol enrichment had no effect. Similarly, studying the time-course of receptor internalization demonstrated significant changes in internalization kinetics in cholesterol-depleted but not in cholesterol-enriched cells (Fig. 4B, C and Table 6). Cell cholesterol depletion caused prolongation of the half-life of receptor internalization from 6.6 to 15 min at 10 μM carbachol and from 2.8 min to 6.8 min at 1 mM carbachol. Corresponding proportions of the receptors that were not internalized after stimulation with 10 μM and 1 mM carbachol (measured at the plateau) were increased from 32.5% to 80.3% and from 26.8% to 46.0%, respectively.
Table 6.
Internalization of M2 receptors in intact cells by carbachol
| Treatment | |||
|---|---|---|---|
| control | MBCD | Ch-MBCD | |
| Concentration-response | |||
| Emax (% of control binding) | 73.7±2.7 | 54.5±1.5a | 78.3±2.7 |
| EC50 (log M) | −5.32±0.09 | −4.46±0.06a | −5.38±0.09 |
|
| |||
| Internalization by 10 μM carbachol | |||
| Rate constant (1/min) | 0.104±0.008 | 0.046±0.017a | 0.104±0.009 |
| Plateau (% control) | 32.5±1.5 | 80.3±3.1a | 30.2±1.7 |
|
| |||
| Internalization by 1 mM carbachol | |||
| Rate constant (1/min) | 0.249±0.017 | 0.103±0.010a | 0.246±0.019 |
| Plateau (% control) | 26.8±1.1 | 46.0±1.5a | 27.2±1.2 |
Parameters of M2 receptor desensitization are mean±S.E.M. of two independent experiments in triplicates shown in Fig. 4.
P<0.01; significantly different from controls by Anova and Tukey’s test.
We performed several control experiments to exclude the possibility that the observed changes in M2 muscarinic receptor signaling were due to cell damage brought about by the treatments. As shown in Fig. 5, the treatments had no influence on total protein content or relative distribution of G-protein α-subunits in membranes. We also found no change in cell membrane permeability estimated as calcein retention or metabolic activity of cells determined as fluorescein oxidation.
Fig. 5.

Influence of treatments on protein content and cell integrity. A: Protein content in control cells and in cells treated with MBCD or Ch-MBCD. Results are expressed as percent (ordinate) of protein content in control cells in six independent experiments. B: Relative content in percent of controls (ordinate) of Gs, Gi/o, and Gq/11 G-protein α-subunits in membranes prepared from MBCD and Ch-MBCD treated cells. Data are shown as mean±S.E.M. of three independent experiments. Representative blots are shown above the columns. C: Rate of calcein leakage during one hour incubation in MBCD and cholesterol-MBCD treated cells expressed in percent (ordinate) of leakage in control cells. Columns represent mean±S.E.M. of two independent experiments in octaplicates. D: Oxidative activity of MBCD and cholesterol-MBCD treated cells is expressed in percent (ordinate) of that in control cells. Columns represent mean±S.E.M. of two independent experiments in octaplicates.
We also tested whether reduced Gq/11 G-protein activation in cholesterol-depleted CHO-M2 cells generalizes to the M1 and M3 receptors that are preferentially coupled to this signaling pathway. Results of these experiments are shown in Fig. 3B, D and summarized in Table 5. Cholesterol depletion significantly inhibited the efficacy of carbachol by 71% and 72% in CHO-M1 and CHO-M3 cells, respectively. This reduction was accompanied by a significant decrease of carbachol potency from 2.0 to 7.1 μM in CHO-M3 cells, with no effect in M1 cells. Cholesterol enrichment had no effect on carbachol potency at both receptor subtypes, but significantly reduced its efficacy by 44% only in CHO-M1 cells.
4. Discussion
Extracellular signal transduction to the cell interior through G-protein coupled receptors is effected by receptor interaction with preferred G-proteins (Baker and Hill, 2007; Kobilka, 2007; Ridge and Palczewski, 2007). However, we have recently shown that the muscarinic M2 receptors directly couples with all three major subclasses of heterotrimeric G-proteins, i.e. the preferential Gi/o G-proteins and the nonpreferential Gs and Gq/11 G-proteins, and activates their signaling pathways (Michal et al., 2007). Various muscarinic agonists induce receptor conformations differing in the extent of individual G-protein activation (Michal et al., 2001 and 2007; Jakubík et al., 2006). Lipid composition of the plasma membrane in which receptors, G-proteins, and more distal effector proteins are inserted also influences signal transduction (Escriba et al., 2007). Changes in cholesterol content alter membrane fluidity and thickness and can thus physically affect binding characteristics, functional properties, and metabolic fate of incorporated receptors and other proteins.
In the present work we demonstrated that altering membrane cholesterol content had significant influence on carbachol-evoked signal transduction at the M2 muscarinic receptor that differed for individual signaling pathways. Changes of membrane cholesterol content influenced either agonist potency or efficacy or both in activating different M2 receptor non-preferential signaling pathways (Figs 2, 3AC, and Table 5). However, there was no change in the M2 receptor selectivity in preferential activation of the Gi/o G-proteins. In case of preferred Gi/o G-protein signaling pathway there was an increase of maximal carbachol-induced inhibition of cAMP levels by 35% in cholesterol-depleted cells with no significant change in potency. In concert with our previous observations on cells in suspension (Michal et al., 2001 and 2007) we also observed a biphasic concentration-response relationship of carbachol effecst on forskolin-stimulated cAMP synthesis in attached cells. Our results differ from those of Mistry et al. (2005) who did not observe a stimulatory phase in attached cells. This discrepancy may be explained by the use of a phosphodiesterase inhibitor in our study. Concerning activation of the Gs signaling pathway by the M2 receptor, a decrease in cholesterol increased Emax in stimulating cAMP synthesis both in control and pertussis toxin-treated cells with significantly reduced potency only in pertussis toxin-treated CHO-M2 cells. In contrast, reduction of membrane cholesterol significantly reduced the efficacy of carbachol in inducing inositol phosphates accumulation with no change in carbachol potency. Generally, reduction of membrane cholesterol had more manifest effects than its augmentation on activation of the M2 receptor signal transduction. The increase of membrane cholesterol significantly decreased Emax and increased potency of carbachol in stimulating cAMP synthesis only in pertussis toxin-treated cells.
Because we measured second messenger responses that are distal to the initial step of agonist binding/G-protein/effector activation we performed several control experiments to exclude the possibility that cholesterol-modifying treatments impaired cell metabolism or induced redistribution of proteins involved in the signaling cascade. No change in total protein content or densities of α-subunits of Gi/o and Gq/11 G-proteins was detected in treated membranes. There was also no change in membrane permeability and oxidative activity of cells (Fig. 5). Finally, treatments had no appreciable influence on myo-inositol metabolic labeling or adenine incorporation into cells. These data thus support the view that the observed modulation of carbachol effects occurs at the agonist/receptor/G-protein/effector interaction level.
Density of membrane muscarinic receptors in intact cells was measured as specific binding of the membrane non-permeable antagonist [3H]NMS under conditions mimicking those used for determination of functional responses. Results showed that MBCD treatment caused a decrease in ligand affinity and a rather unexpected increase of its maximal binding. One possible interpretation of affinity change is the direct allosteric influence of cholesterol on the receptor (Colozo et al., 2007). Another is altered receptor conformation due to change of membrane biophysical properties. The observed increase in density of cell-surface muscarinic receptors by reducing membrane cholesterol content might be the result of effects on receptor recycling. Indeed, cholesterol depletion reduced the Emax, EC50, and rate constant of carbachol-induced receptor internalization (Fig. 4, Table 6) whereas cholesterol enrichment had no effect. In this context it is worth mentioning that the EC50 values of carbachol in inducing receptor internalization corresponded to the single affinity of carbachol binding in control and cholesterol-enriched cells, and to its low-affinity binding in cholesterol-depleted cells (Table 5). This suggests that the bulk of M2 receptors in control and cholesterol-enriched cells exist in the low-affinity conformation. However, factors other than reduced receptor internalization could also be implicated since an increase in membrane-bound receptors was also apparent when control membranes, instead of intact cells, were pretreated with MBCD. Neither reduced receptor internalization nor new receptor synthesis can take place under these conditions. The increase of the total cell receptor number in MBCD-treated cells was also observed in measurements with the membrane-permeable antagonist [3H]-quinuclidinylbenzilate that labels both surface and intracellular (but still intact) muscarinic receptors. This observation suggests that the increase in cell surface binding sites is not due to augmented incorporation of existing intracellular receptors to plasma membrane. Together, these findings are in favor of the notion that decreasing membrane cholesterol may make more membrane muscarinic binding sites accessible to the radioligands.
The increase in plasma membrane muscarinic receptor density in cholesterol-depleted cells offers plausible explanation for enhanced efficacy of carbachol in inhibiting cAMP synthesis via the Gi/o G-protein pathway and in increasing cAMP synthesis via activation of the Gs G-protein. It does not, however, fit with the decrease of carbachol efficacy in stimulating inositol phosphates accumulation via activation of the Gq/11 G-proteins pathway. Competition binding experiments in intact cells indicated that carbachol interacts with a single-affinity binding site in control and in cholesterol-enriched cells. In contrast, a two-site model with Ki high of 3.6 μM (24% binding sites) better fitted carbachol displacement data in cholesterol-depleted cells. The potency of carbachol at this high-affinity binding site approximates that of its single site in control and cholesterol-enriched cells (4.9 and 4.3 μM, respectively). Thus, cholesterol depletion might result in conversion of a substantial proportion of plasma membrane muscarinic receptors to a lower affinity agonist-binding conformation. Alternatively, the new muscarinic binding sites that become accessible as a result of cholesterol depletion might possess low agonist binding affinity.
In order to gain closer insight into changes of agonist binding properties of the orthosteric site we performed competition experiments on membranes prepared from control and treated cells. All competition curves exhibited high and low affinity binding sites. For both receptor conformations, the highest affinities were found in cholesterol-enriched and the lowest in cholesterol-depleted membranes (Fig. 1D, Table 3). However, membranes from cholesterol-depleted cells displayed a significantly larger proportion of high affinity binding sites (63% compared to 33% in control and 39% in cholesterol-enriched membranes). Addition of GTP to the incubation buffer of control membranes resulted in the expected robust rightward shift of the carbachol displacement curves that nevertheless still displayed two affinity binding sites. GTP caused large shifts of the high affinity carbachol binding site in all treatment groups (namely 69, 145, and 331 fold decrease in Ki high in control, cholesterol-depleted, and cholesterol-enriched membranes, respectively). The Ki high in the presence of GTP roughly corresponded to the Ki low obtained in its absence. In contrast, the shift of the low affinity binding site was only 3.7, 4.9, and 7.6 fold decrease in affinity in control, cholesterol-depleted, and cholesterol-enriched membranes, respectively. The presence of two binding sites with different low affinities for carbachol in the presence of GTP is most likely due to the presence of uncoupled receptors and receptors that interact with either „empty pocket (unliganded) Gαβγ“ or „Gα(GTP)Gβγ“ G-protein complexes (Abdulaev et al., 2006).
Changes in coupling of muscarinic receptors in cholesterol-depleted cells may be explained by altering the ratio of receptor monomers and oligomers. It has been recently shown that the monomeric form of the receptor is the receptor species that is capable of interaction with G-proteins and binding agonists with high affinity (Whorton et al., 2007). Changes in membrane fluidity by altering its cholesterol content could also modify diffusion of muscarinic receptors and therefore their interaction with G proteins. The latter phenomenon has been demonstrated in CHO-M2 and CHO-M3 cells (Azpiazu and Gautam, 2004). However, we observed opposite effects of cholesterol reduction on coupling of M2 receptors with respect to Gi/o and Gs signaling on the one hand and Gq/11 on the other hand.
Gq/11 signaling may have an important role in the pathogenesis of Alzheimer’s disease. It has been demonstrated that muscarinic receptor stimulation of the Gq/11 G-protein signaling pathway increases non-amyloidogenic cleavage of the amyloid precursor protein (Buxbaum et al., 1992; Nitsch et al., 1992). Furthermore, reduced activation of G-proteins by muscarinic receptors has been demonstrated in a transgenic mouse model of Alzheimer’s disease, a phenomenon expected to facilitate formation of the amyloid-beta protein (Machová et al., 2008). It was thus of interest to determine effects of membrane cholesterol manipulation at M1, M2, and M3 receptors that vary in their preference for coupling to this pathway. Cholesterol depletion decreased efficacy of carbachol in stimulating IP accumulation at all three receptor subtypes, but decreased agonist potency only at the M3 receptor. Cholesterol enrichment had no effect on carbachol stimulation of IP formation in CHO-M2 and CHO-M3 cells. However, the maximal response in CHO-M1 cells was significantly inhibited. Particularly interesting in this respect is the reduction of carbachol-induced IP accumulation by the M1 receptor in both cholesterol-depleted and cholesterol-enriched cells. The M1 receptor exhibits higher levels of expression in the brain in comparison to the other four muscarinic receptor subtypes (Flynn et al., 1995; Krejčí and Tuček, 2002) and plays an important role in learning and memory.
In summary, we demonstrated that depletion of plasma membrane cholesterol exerts significant influence on M2 muscarinic receptor function. This effect involves both attenuation of agonist-induced receptor internalization and changes in agonist potency and efficacy that are different for individual signaling pathways. The observed differences in the effects of altering cell cholesterol content between receptor subtypes eliminate the possibility of nonspecific effects distal to receptor/G protein/effector coupling. However, determination of the underlying molecular mechanisms that change receptor/G-protein/effector interactions will require further investigations. Regardless of the underlying mechanisms, demonstration of the attenuation of muscarinic receptor signaling through Gq/11 G-proteins consequent to changes in membrane cholesterol content may have importance in the pathogenesis of neurodegenerative disorders. Increased blood cholesterol level consequent to increased intake, aging, or hereditary malfunction of cholesterol metabolism are major risk factors in Alzheimer’s disease. However, brain membrane cholesterol level decreases during aging (Svennerholm et al., 1994) and this reduction is even more manifest in brains of a subset of Alzheimer’s patients (Ledesma et al., 2003), in line with the demonstrated physiological role of amyloid β1–40 fragments in inhibiting cholesterol synthesis (Grimm et al., 2005). A large body of evidence demonstrates that reduction of cell cholesterol levels inhibits amyloidogenic processing of the amyloid precursor protein (Hartmann et al., 2007) by inhibiting β- and γ-secretase activity (Grimm et al., 2008). On the other hand, an increase in the production of pathogenic β-amyloid fragments in membranes of cholesterol-depleted primary hippocampal neurons and CHO cells has been reported (Abad-Rodriguez et al., 2004). These effects may be related to attenuation of Gq/11 G-protein signaling and its associated non-amyloidogenic α-secretase cleavage of the amyloid precursor protein.
Acknowledgments
We appreciate the invaluable help of Dr. Jan Jakubík with mathematical processing of the data and beneficial comments on the manuscript and Ms. Danielle Johansson for language editing. This work was supported by Project AV0Z50110509 and by Grants GACR305/05/0452, IAA500110703, National Institutes of Health Grant NS25743, MSMT CR LC554, and EU project LipiDiDiet.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abad-Rodriguez J, Ledesma MD, Craessaerts K, Perga S, Medina M, Delacourte A, Dingwall C, De Strooper B, Dotti CG. Neuronal membrane cholesterol loss enhances amyloid peptide generation. J Cell Biol. 2004;167:953–960. doi: 10.1083/jcb.200404149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdulaev NG, Ngo T, Ramon E, Brabazon DM, Marino JP, Ridge KD. The receptor-bound „empty pocket“ state of the heterotrimeric G-protein alpha-subunit is conformationally dynamic. Biochemistry. 2006;45:12986–12997. doi: 10.1021/bi061088h. [DOI] [PubMed] [Google Scholar]
- Azpiazu I, Gautam N. A fluorescence resonance energy transfer-based sensor indicates that receptor access to a G protein is unrestricted in a living mammalian cell. J Biol Chem. 2004;279:27709–27718. doi: 10.1074/jbc.M403712200. [DOI] [PubMed] [Google Scholar]
- Baker JG, Hill SJ. Multiple GPCR conformations and signalling pathways: implications for antagonist affinity estimates. Trends Pharmacol Sci. 2007;28:374–381. doi: 10.1016/j.tips.2007.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonner TI. The molecular basis of muscarinic receptor diversity. Trends Neurosci. 1989;12:148–151. doi: 10.1016/0166-2236(89)90054-4. [DOI] [PubMed] [Google Scholar]
- Buxbaum JD, Oishi M, Chen HI, Pinkas-Kramarski R, Jaffe EA, Gandy SE, Greengard P. Cholinergic agonists and interleukin 1 regulate processing and secretion of the Alzheimer beta/A4 amyloid protein precursor. Proc Natl Acad Sci U S A. 1992;89:10075–10078. doi: 10.1073/pnas.89.21.10075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caulfield MP, Birdsall NJ. International Union of Pharmacology. XVII Classification of muscarinic acetylcholine receptors. Pharmacol Rev. 1998;50:279–290. [PubMed] [Google Scholar]
- Colozo AT, Park PS, Sum CS, Pisterzi LF, Wells JW. Cholesterol as a determinant of cooperativity in the M2 muscarinic cholinergic receptor. Biochem Pharmacol. 2007;74:236–255. doi: 10.1016/j.bcp.2007.04.009. [DOI] [PubMed] [Google Scholar]
- Doležal V, Tuček S. The effects of brucine and alcuronium on the inhibition of [3H]-acetylcholine release from rat striatum by muscarinic receptor agonists. Br J Pharmacol. 1998;124:1213–1218. doi: 10.1038/sj.bjp.0701966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escriba PV, Wedegaertner PB, Goni FM, Vogler O. Lipid-protein interactions in GPCR-associated signaling. Biochim Biophys Acta. 2007;1768:836–852. doi: 10.1016/j.bbamem.2006.09.001. [DOI] [PubMed] [Google Scholar]
- Flynn DD, Ferrari-DiLeo G, Mash DC, Levey AI. Differential regulation of molecular subtypes of muscarinic receptors in Alzheimer‘s disease. J Neurochem. 1995;64:1888–1891. doi: 10.1046/j.1471-4159.1995.64041888.x. [DOI] [PubMed] [Google Scholar]
- Fredriksson R, Lagerstrom MC, Lundin LG, Schioth HB. The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol Pharmacol. 2003;63:1256–1272. doi: 10.1124/mol.63.6.1256. [DOI] [PubMed] [Google Scholar]
- Grimm MO, Grimm HS, Patzold AJ, Zinser EG, Halonen R, Duering M, Tschape JA, De Strooper B, Muller U, Shen J, Hartmann T. Regulation of cholesterol and sphingomyelin metabolism by amyloid-beta and presenilin. Nat Cell Biol. 2005;7:1118–1123. doi: 10.1038/ncb1313. [DOI] [PubMed] [Google Scholar]
- Grimm MO, Grimm HS, Tomic I, Beyreuther K, Hartmann T, Bergmann C. Independent inhibition of Alzheimer‘s disease beta - and gamma-secretase cleavage by lowered cholesterol levels. J Biol Chem. 2008;283:11302–11311. doi: 10.1074/jbc.M801520200. [DOI] [PubMed] [Google Scholar]
- Haigh LS, Leatherman GF, O‘Hara DS, Smith TW, Galper JB. Effects of low density lipoproteins and mevinolin on cholesterol content and muscarinic cholinergic responsiveness in cultured chick atrial cells. Regulation of levels of muscarinic receptors and guanine nucleotide regulatory proteins. J Biol Chem. 1988;263:15608–15618. [PubMed] [Google Scholar]
- Hartmann T, Kuchenbecker J, Grimm MO. Alzheimer‘s disease: the lipid connection. J Neurochem. 2007;103(Suppl 1):159–170. doi: 10.1111/j.1471-4159.2007.04715.x. [DOI] [PubMed] [Google Scholar]
- Hornigold DC, Mistry R, Raymond PD, Blank JL, Challiss RA. Evidence for cross-talk between M2 and M3 muscarinic acetylcholine receptors in the regulation of second messenger and extracellular signal-regulated kinase signalling pathways in Chinese hamster ovary cells. Br J Pharmacol. 2003;138:1340–1350. doi: 10.1038/sj.bjp.0705178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulme EC, Birdsall NJ, Buckley NJ. Muscarinic receptor subtypes. Annu Rev Pharmacol Toxicol. 1990;30:633–673. doi: 10.1146/annurev.pa.30.040190.003221. [DOI] [PubMed] [Google Scholar]
- Jakubík J, Bacakova L, El-Fakahany EE, Tuček S. Positive cooperativity of acetylcholine and other agonists with allosteric ligands on muscarinic acetylcholine receptors. Mol Pharmacol. 1997;52:172–179. doi: 10.1124/mol.52.1.172. [DOI] [PubMed] [Google Scholar]
- Jakubík J, Bacakova L, el-Fakahany EE, Tuček S. Subtype selectivity of the positive allosteric action of alcuronium at cloned M1-M5 muscarinic acetylcholine receptors. J Pharmacol Exp Ther. 1995;274:1077–1083. [PubMed] [Google Scholar]
- Jakubík J, El-Fakahany EE, Doležal V. Differences in kinetics of xanomeline binding and selectivity of activation of g proteins at m1 and m2 muscarinic acetylcholine receptors. Mol Pharmacol. 2006;70:656–666. doi: 10.1124/mol.106.023762. [DOI] [PubMed] [Google Scholar]
- Jakubík J, Tuček S, El-Fakahany EE. Allosteric modulation by persistent binding of xanomeline of the interaction of competitive ligands with the M1 muscarinic acetylcholine receptor. J Pharmacol Exp Ther. 2002;301:1033–1041. doi: 10.1124/jpet.301.3.1033. [DOI] [PubMed] [Google Scholar]
- Kenakin T. Ligand-selective receptor conformations revisited: the promise and the problem. Trends Pharmacol Sci. 2003;24:346–354. doi: 10.1016/S0165-6147(03)00167-6. [DOI] [PubMed] [Google Scholar]
- Klein U, Gimpl G, Fahrenholz F. Alteration of the myometrial plasma membrane cholesterol content with beta-cyclodextrin modulates the binding affinity of the oxytocin receptor. Biochemistry. 1995;34:13784–13793. doi: 10.1021/bi00042a009. [DOI] [PubMed] [Google Scholar]
- Kobilka BK. G protein coupled receptor structure and activation. Biochim Biophys Acta. 2007;1768:794–807. doi: 10.1016/j.bbamem.2006.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krejčí A, Tuček S. Quantitation of mRNAs for M(1) to M(5) subtypes of muscarinic receptors in rat heart and brain cortex. Mol Pharmacol. 2002;61:1267–1272. doi: 10.1124/mol.61.6.1267. [DOI] [PubMed] [Google Scholar]
- Lazareno S, Birdsall NJ. Detection, quantitation, and verification of allosteric interactions of agents with labeled and unlabeled ligands at G protein-coupled receptors: interactions of strychnine and acetylcholine at muscarinic receptors. Mol Pharmacol. 1995;48:362–378. [PubMed] [Google Scholar]
- Lazareno S, Doležal V, Popham A, Birdsall NJ. Thiochrome enhances acetylcholine affinity at muscarinic M4 receptors: receptor subtype selectivity via cooperativity rather than affinity. Mol Pharmacol. 2004;65:257–266. doi: 10.1124/mol.65.1.257. [DOI] [PubMed] [Google Scholar]
- Ledesma MD, Abad-Rodriguez J, Galvan C, Biondi E, Navarro P, Delacourte A, Dingwall C, Dotti CG. Raft disorganization leads to reduced plasmin activity in Alzheimer‘s disease brains. EMBO Rep. 2003;4:1190–1196. doi: 10.1038/sj.embor.7400021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machová E, Jakubík J, Michal P, Oksman M, Iivonen H, Tanila H, Doležal V. Impairment of muscarinic transmission in transgenic APPswe/PS1dE9 mice. Neurobiol Aging. 2008;29:368–378. doi: 10.1016/j.neurobiolaging.2006.10.029. [DOI] [PubMed] [Google Scholar]
- Michal P, El-Fakahany EE, Doležal V. Muscarinic M2 receptors directly activate Gq/11 and Gs G-proteins. J Pharmacol Exp Ther. 2007;320:607–614. doi: 10.1124/jpet.106.114314. [DOI] [PubMed] [Google Scholar]
- Michal P, Lysíková M, Tuček S. Dual effects of muscarinic M(2) acetylcholine receptors on the synthesis of cyclic AMP in CHO cells: dependence on time, receptor density and receptor agonists. Br J Pharmacol. 2001;132:1217–1228. doi: 10.1038/sj.bjp.0703931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migeon JC, Nathanson NM. Differential regulation of cAMP-mediated gene transcription by m1 and m4 muscarinic acetylcholine receptors. Preferential coupling of m4 receptors to Gi alpha-2. J Biol Chem. 1994;269:9767–9773. [PubMed] [Google Scholar]
- Mistry R, Dowling MR, Challiss RA. An investigation of whether agonist-selective receptor conformations occur with respect to M2 and M4 muscarinic acetylcholine receptor signalling via Gi/o and Gs proteins. Br J Pharmacol. 2005;144:566–575. doi: 10.1038/sj.bjp.0706090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell DC, Straume M, Miller JL, Litman BJ. Modulation of metarhodopsin formation by cholesterol-induced ordering of bilayer lipids. Biochemistry. 1990;29:9143–9149. doi: 10.1021/bi00491a007. [DOI] [PubMed] [Google Scholar]
- Nitsch RM, Slack BE, Wurtman RJ, Growdon JH. Release of Alzheimer amyloid precursor derivatives stimulated by activation of muscarinic acetylcholine receptors. Science. 1992;258:304–307. doi: 10.1126/science.1411529. [DOI] [PubMed] [Google Scholar]
- Nováková J, Mikasová L, Machová E, Lisá V, Doležal V. Chronic treatment with amyloid beta(1–42) inhibits non-cholinergic high-affinity choline transport in NG108-15 cells through protein kinase C signaling. Brain Res. 2005;1062:101–110. doi: 10.1016/j.brainres.2005.09.021. [DOI] [PubMed] [Google Scholar]
- Peterson GL. A simplification of the protein assay method of Lowry et al. which is more generally applicable. Anal Biochem. 1977;83:346–356. doi: 10.1016/0003-2697(77)90043-4. [DOI] [PubMed] [Google Scholar]
- Pike LJ. Lipid rafts: bringing order to chaos. J Lipid Res. 2003;44:655–667. doi: 10.1194/jlr.R200021-JLR200. [DOI] [PubMed] [Google Scholar]
- Renaud JF, Scanu AM, Kazazoglou T, Lombet A, Romey G, Lazdunski M. Normal serum and lipoprotein-deficient serum give different expressions of excitability, corresponding to different stages of differentiation, in chicken cardiac cells in culture. Proc Natl Acad Sci U S A. 1982;79:7768–7772. doi: 10.1073/pnas.79.24.7768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridge KD, Palczewski K. Visual rhodopsin sees the light: structure and mechanism of G protein signaling. J Biol Chem. 2007;282:9297–9301. doi: 10.1074/jbc.R600032200. [DOI] [PubMed] [Google Scholar]
- Rudajev V, Novotný J, Hejnová L, Milligan G, Svoboda P. Dominant portion of thyrotropin-releasing hormone receptor is excluded from lipid domains. Detergent-resistant and detergent-sensitive pools of TRH receptor and Gqalpha/G11alpha protein. J Biochem. 2005;138:111–125. doi: 10.1093/jb/mvi114. [DOI] [PubMed] [Google Scholar]
- Simons K, Toomre D. Lipid rafts and signal transduction. Nat Rev Mol Cell Biol. 2000;1:31–39. doi: 10.1038/35036052. [DOI] [PubMed] [Google Scholar]
- Smith SM, Lei Y, Liu J, Cahill ME, Hagen GM, Barisas BG, Roess DA. Luteinizing hormone receptors translocate to plasma membrane microdomains after binding of human chorionic gonadotropin. Endocrinology. 2006;147:1789–1795. doi: 10.1210/en.2005-1046. [DOI] [PubMed] [Google Scholar]
- Svennerholm L, Bostrom K, Jungbjer B, Olsson L. Membrane lipids of adult human brain: lipid composition of frontal and temporal lobe in subjects of age 20 to 100 years. J Neurochem. 1994;63:1802–1811. doi: 10.1046/j.1471-4159.1994.63051802.x. [DOI] [PubMed] [Google Scholar]
- Tuček S, Musílková J, Nedoma J, Proška J, Shelkovnikov S, Vorlicek J. Positive cooperativity in the binding of alcuronium and N-methylscopolamine to muscarinic acetylcholine receptors. Mol Pharmacol. 1990;38:674–680. [PubMed] [Google Scholar]
- Vogel WK, Mosser VA, Bulseco DA, Schimerlik MI. Porcine m2 muscarinic acetylcholine receptor-effector coupling in Chinese hamster ovary cells. J Biol Chem. 1995;270:15485–15493. doi: 10.1074/jbc.270.26.15485. [DOI] [PubMed] [Google Scholar]
- Whorton MR, Bokoch MP, Rasmussen SG, Huang B, Zare RN, Kobilka B, Sunahara RK. A monomeric G protein-coupled receptor isolated in a high-density lipoprotein particle efficiently activates its G protein. Proc Natl Acad Sci U S A. 2007;104:7682–7687. doi: 10.1073/pnas.0611448104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson M, Siauw M, Horackova M. Modulation of cardiac M2 muscarinic receptor binding by progesterone-related steroids. J Mol Cell Cardiol. 1995;27:1831–1839. doi: 10.1016/0022-2828(95)90006-3. [DOI] [PubMed] [Google Scholar]