

Abstract
Angiotensin-converting enzyme 2 (ACE2), a homologue of angiotensin-converting enzyme (ACE), converts angiotensin (Ang) I to Ang(1−9) and Ang II to Ang(1−7), but does not directly process Ang I to Ang II. Cardiac function is compromised in ACE2 null mice; however, the importance of ACE2 in the processing of angiotensin peptides within the murine heart is not known. We determined the metabolism of angiotensins in wild-type (WT), ACE (ACE−/−) and ACE2 null mice (ACE2−/−). Angiotensin II was converted almost exclusively to Ang(1−7) in the cardiac membranes of WT and ACE−/− strains, although generation of Ang(1−7) was greater in the ACE−/− mice (27.4 ± 4.1 versus 17.5 ± 3.2 nmol−1 mg h−1 for WT). The ACE2 inhibitor MLN4760 significantly attenuated Ang II metabolism and the subsequent formation of Ang(1−7) in both strains. In the ACE2−/− hearts, Ang II metabolism and the generation of Ang(1−7) were significantly attenuated; however, the ACE2 inhibitor reduced the residual Ang(1−7)-forming activity in this strain. Angiotensin I was primarily converted to Ang(1−9) (WT, 28.9 ± 3.1 nmol−1 mg h−1; ACE−/−, 49.8 ± 5.3 nmol−1 mg h−1; and ACE2−/−, 35.9 ± 5.4 nmol−1 mg h−1) and to smaller quantities of Ang(1−7) and Ang II. Although the ACE2 inhibitor had no effect on Ang(1−9) formation, the carboxypeptidase A inhibitor benzylsuccinate essentially abolished the formation of Ang(1−9) and increased the levels of Ang I in cardiac membranes. In conclusion, our studies in the murine heart suggest that ACE2 is the primary pathway for the metabolism of Ang II and the subsequent formation of Ang(1−7), a peptide that, in contrast to Ang II, exhibits both antifibrotic and antiproliferative actions.
Angiotensin-converting enzyme 2 (ACE2) is a homologue of angiotensin-converting enzyme (ACE) and potentially a new member of the renin–angiotensin system (RAS). Unlike ACE, ACE2 functions as a carboxypeptidase within the RAS that converts angiotensin I (Ang I) to des-Leu10-Ang I [Ang(1−9)] and Ang II to Ang(1−7), but does not directly process Ang I to Ang II (Tipnis et al. 2000; Vickers et al. 2002). Angiotensin-converting enzyme 2 is predominantly found in the heart, testis and kidney, and the expression of the enzyme is markedly reduced in several hypertensive models, as well as in the kidneys of streptozotocin-induced diabetic rats (Crackower et al. 2002; Tikellis et al. 2003). Moreover, ACE2 is not attenuated by conventional ACE inhibitors, and our data suggest that ACE2 expression is upregulated by chronic ACE or AngII type 1 (AT1) receptor blockade (Chappell et al. 2002). Indeed, the increased expression of ACE2 may contribute to the elevated levels of Ang(1−7) following either ACE or AT1 receptor inhibition (Iyer et al. 1998; Nakamura et al. 2003). However, at present, there are few studies that have assessed the functional significance of ACE2 or determined the role of this enzyme in the local or tissue RAS. Crackower et al. (2002) reported a severe attenuation in cardiac contractility that was associated with increased cardiac and circulating levels of Ang II in an ACE2 knockout model. An ACE/ACE2 double knockout model rescued the deleterious phenotype of the ACE2 deletion (Crackower et al. 2002). Moreover, Yammamoto et al. (1992) found that ACE2 knockout mice developed greater severity of cardiac damage than wild-type mice following pressure overload, which was associated with higher tissue levels of Ang II.
Although these studies suggest that ACE2 may play a key role in the regulation of the cardiac RAS, the contribution of ACE2 to the tissue processing of angiotensin peptides within the murine heart is not known. In this regard, we tested the hypothesis that ACE2 and ACE are key enzymes that contribute to both the formation and the metabolism of angiotensin peptides within the mouse heart. For this study, we determined the metabolism of angiotensins in wild-type mice, ACE2 knockout (ACE2−/−) and ACE knockout (ACE−/−) mice. In addition to the use of the endogenous substrates (Ang I and Ang II) and a specific non-peptide inhibitor to define ACE2 activity (Dales et al. 2002), we compared the extent to which angiotensin processing is altered in mice that do not express either ACE or ACE2.
Methods
The study, including the protocols for the care and use of animals, was approved by the Institutional Animal Care and Use Committee (University of Texas Southwestern Medical Center and the Institute for Molecular Biotechnology of the Austrian Academy of Sciences). Male homozygous ACE−/− and ACE2−/− mice were generated using targeted homologous recombination as previously described (Crackower et al. 2002; Modrall et al. 2004) with the wild-type C57Black/6 used for control tissues. The genotype of the mice was confirmed using polymerase chain reaction analysis. Animals were killed at 12−14 weeks old, tissues rapidly removed, snap frozen in liquid nitrogen and stored at −80°C.
Membranes were prepared from frozen mouse heart by homogenizing the tissue in an Ultra-Turrax T25 (IKA, Staufen, Germany) in 10 mm Hepes sodium salt, 125 mm NaCl, 0.01 mm ZnCl2, pH 7.4, followed by centrifugation at 20 000g at 4°C for 20 min (Allred et al. 2000). The pellets were washed with the buffer, homogenized, and centrifuged as before. The resultant pellet was diluted to a volume of 1 ml with buffer and homogenized to yield a protein concentration of approximately 1 mg ml−1.
Angiotensin peptides (100 μmol l−1 final concentration, Bachem, Torrance, CA, USA) were incubated in 0.5 ml of buffer containing 50 μg of heart membranes at 37°C for 60−240 min in the absence or presence of various peptidase inhibitors. The reaction was terminated by dilution in 1.0% phosphoric acid, membranes centrifuged at 15 000 g for 15 min in a Sorvall Biofuge (Germany) and filtered (0.2 μm) prior to peptide analysis by reverse phase-high performance liquid chromatography (RP-HPLC) as previously described (Allred et al. 2000). In brief, peptide separation was achieved on a Waters NovaPak C18 microbore column (2 × 150 mm, Milford, MA, USA) using a mobile phase of 0.1% phosphoric acid and acetonitrile. Heart membrane activities were expressed as nanomoles of product formed per hour per milligram of protein. Protein was determined using a Bradford protein assay kit (BioRad, Hercules, CA, USA) with bovine immunoglobulin G protein as a standard. The selective ACE2 inhibitor MLN4760 (final concentration of 10 μm) was pre-incubated with cardiac membranes for 5 min at 37°C prior to addition of the substrates and was obtained from Millennium Pharmaceuticals (Baltimore, MD, USA). Chymostatin and benzylsuccinate (both from Sigma, St Louis, MI, USA) were also used at a final concentration of 10 μm.
For the immunocytochemical distribution of Ang(1−7) and ACE2, hearts were rapidly excised from the animal and immersed in 4% formalin acetate for 24 h and then transferred to a solution of 70% ethanol (Averill et al. 2003). Tissues were blocked in paraffin, sections of 5 μm thickness cut and deparaffinized by washing in xylene, reduced concentrations of ethanol (100, 95 and 75%) and distilled water. Sections were blocked with 3% H202, rinsed in phosphate-buffered saline (PBS, pH 7.4), dried and blocked with 5% normal goat serum for 60 min. Slides were then rinsed in PBS and incubated in the primary antibody to either ACE2 [1:100 dilution in 1% bovine serum albumin (BSA)/PBS, v/v] or Ang(1−7) (1:50 dilution) overnight at 4°C in a humidified chamber. Slides were rinsed in PBS, incubated with biotinylated goat anti-rabbit immunoglobulin G (1:400 dilution, v/v in 1% BSA/PBS) for 3 h at 4°C and visualized with avidin–biotin (Vector Laboratories, Burlingame, CA, USA) and alkaline phosphatase I (Sigma) in 50 mm Tris-PBS, pH 7.6. Specificity of staining was determined by the absence of the primary antibody or pre-incubation with either 10 μm Ang(1−7) or recombinant ACE2 (10 ng). Sections were counterstained with Haematoxylin and Eosin, dehydrated in ethanol and coverslipped with Histomount (National Diagnostics, Atlanta, GA, USA). Tissue sections were examined under a light microscope and photographed using a Zeiss AxioCam digital camera and AxioVision software (Zeiss; München-Hallbergmoos, Germany).
Data are expressed as the means ± s.e.m. Comparisons between the wild-type and knockout mice were evaluated using multiple regression analysis and Dunnett's post hoc analysis (GraphPad Stat Mate), while the inhibitor results were assessed by Student's paired t test. All figures were constructed with GraphPad Prism plotting and statistical software (San Diego, CA, USA) A value of P < 0.05 was required for statistical significance.
Results
We initially assessed the pattern of Ang I metabolism in cardiac membranes from the wild-type, ACE−/− and ACE2−/− mice. As shown in the chromatographs of Fig. 1, the major peak of Ang I processing was identified as the nonapeptide Ang(1−9), with smaller peaks corresponding to Ang(1−7) and Ang II (Fig. 1A). A similar pattern for Ang I processing was also observed in membranes isolated from the ACE null mice; however, we identified an additional peak corresponding Ang(1−4) (Fig. 1B). Inclusion of the chymase inhibitor chymostatin abolished the Ang(1−4) peak and increased the peak of Ang(1−9) (Fig. 1C). Although ACE2 was initially reported to generate Ang(1−9) from Ang I in cardiomyocytes and proximal tubules (Donoghue et al. 2000; Li et al. 2004), addition of the ACE2 inhibitor MLN4760 did not attenuate the peak corresponding to Ang(1−9) (Fig. 1D). However, addition of the carboxypeptidase A inhibitor benzylsuccinate inhibited the peak of Ang(1−9) and increased the Ang I peak (Fig. 1E). The Ang(1−7) and Ang II peaks also appeared to diminish to some extent with the carboxypeptidase inhibitor. Angiotensin(1−9) was also the predominant peak arising from the metabolism of Ang I in ventricular membranes of the ACE2−/− mouse (Fig. 1F). Figure 2 summarizes the metabolism studies of Ang I (Fig. 2A) and the formation of Ang(1−9) (Fig. 2B). These data again reveal that Ang(1−9) was the major product formed from Ang I and that inhibition of a carboxypeptidase A-like activity protected Ang I from enzymatic metabolism in the heart membranes. Moreover, a chymostatin-sensitive activity was significantly increased in the cardiac membranes of the ACE knockout mice, which contributed to the metabolism of Ang(1−9) to Ang(1−4) (Fig. 2B).
Figure 1. Metabolism of Ang I by cardiac membranes.

Angiotensin I was incubated with cardiac membranes (50 μg) for 120 min at 37°C and the metabolites separated by HPLC. A, Ang I was hydrolysed primarily to Ang(1−9) in the heart membranes from wild-type mice. B, increased metabolism of Ang I results in a peak of Ang(1−4) in the heart membranes of ACE−/− mice. C, addition of the chymase inhibitor chymostatin (10 μm) reduced the peak of Ang(1−4) and increased the peak of Ang(1−9) in ACE−/− mice. D, addition of the ACE2 inhibitor MLN4760 (10 μm) did not attenuate the formation of Ang(1−9). E, the carboxypeptidase A inhibitor benzylsuccinate reduced the peak of Ang(1−9) and increased the level of Ang I. F, Ang I was hydrolysed primarily to Ang(1−9) in the heart membranes from ACE2−/− mice. The HPLC metabolism results are representative of four separate experiments.
Figure 2. The quantification of the Ang I metabolism (A) and Ang(1−9) formation (B) from cardiac membranes of wild-type, ACE−/− and ACE2−/− mice.

Angiotensin I metabolism was significantly enhanced in hearts of ACE−/− mice versus the wild-type (WT). Addition of the chymase inhibitor chymostatin (CHM, 10 μm) did not alter Ang I metabolism (A), but increased Ang(1−9) (B) in ACE−/− mice. The ACE2 inhibitor MLN4760 in the presence of chymostatin (CHM-A2I, 10 μm) did not alter Ang I or Ang(1−9) metabolism compared with CHM alone. Addition of the carboxypeptidase A benzylsuccinate (BSC, 10 μm) significantly reduced the metabolism of Ang I and subsequent formation of Ang(1−9). Data are means + s.e.m. *P < 0.05 versus control (CON); #P < 0.05 versus chymostatin (CHM); n = 4.
We then evaluated the metabolism of Ang II in the mouse heart membranes. The chromatographs in Fig. 3 reveal that the major peak for Ang II processing was identified as Ang(1−7) (Fig. 3A). An identical pattern for Ang II metabolism was evident in membranes isolated from the ACE null mice; however, the Ang(1−7)-forming activity was greater than that of the wild-type mice (Fig. 3B). We also observed a peak corresponding to Ang(1−4) which may result from the metabolism of either Ang(1−7) or Ang II (Fig. 3B). Addition of the ACE2 inhibitor substantially reduced the peak of Ang(1−7) and increased the Ang II peak (Fig. 3C). Inhibition of ACE2 was also associated with the appearance of peaks that corresponded to Ang(2−8) and Ang(3−8)/(4−8), suggesting a minor role for aminopeptidase activities at this time point. The role for ACE2 in the processing of Ang II was confirmed in the ACE2 null mice, in which only minor conversion to Ang(1−7) was evident (Fig. 3D). Figure 4 summarizes the metabolism data for Ang II and the formation of the Ang(1−7) in the cardiac membranes. The overall metabolism of Ang II was significantly reduced by the ACE2 inhibitor or in the ACE2 null mice (Fig. 4A). The ACE2 inhibitor also tended to reduce Ang II metabolism in cardiac membranes from the ACE2−/− mice, suggesting some residual level of ACE2 activity. Angiotensin-converting enzyme 2 was the major activity contributing to Ang(1−7) formation in mouse cardiac membranes in wild-type and ACE−/− mice (Fig. 4B). In this case, the ACE2 inhibitor significantly reduced the residual Ang(1−7) forming activity in the ACE2 null mice (Fig. 4B). The data also demonstrate that ACE2 activity was increased in the ACE null mice (Fig. 4B).
Figure 3. Metabolism of Ang II by cardiac membranes.

Angiotensin II was incubated with cardiac membranes (50 μg) for 120 min at 37°C and the metabolites separated by HPLC. A, Ang II was hydrolysed primarily to Ang(1−7) in the heart membranes from wild-type mice. B, increased metabolism of Ang II to Ang(1−7) in the heart membranes of ACE−/− mice. C, addition of the ACE2 inhibitor MLN4760 (10 μm) reduced the peak of Ang(1−7) and increased the peak of Ang II in ACE−/− mice. D, attenuated metabolism of Ang II in the in the heart membranes of ACE2−/− mice. The HPLC metabolism results are representative of four separate experiments.
Figure 4. The quantification of the Ang II metabolism (A) and Ang(1−7) formation (B) from the cardiac membranes of the wild-type, ACE−/− and ACE2−/− mice.

Angiotensin II metabolism was significantly enhanced in hearts of ACE−/− mice versus the wild-type (WT). The ACE2 inhibitor MLN4760 (A2I, 10 μm) reduced the metabolism of Ang II and the subsequent formation of Ang(1−7) in ACE−/− mice. Angiotensin II metabolism and Ang(1−7) generation were significantly reduced in the ACE2−/− mice. The ACE2 inhibitor (A2I) further attenuated the formation of Ang(1−7) in ACE2−/− mice. Data are the means ± s.e.m. *P < 0.05 versus control wild-type (CON); #P < 0.05 versus ACE−/− control; ϕP < 0.05 versus ACE2−/− control; n = 4.
In the rat heart, we demonstrated intense immunocytochemical (ICC) staining for Ang(1−7), predominantly within the cardiomyocytes (Averill et al. 2003). Therefore, we assessed the ICC distribution of Ang(1−7) and ACE2 in the wild-type murine heart. The ICC studies in cardiac tissue revealed widespread staining throughout the left ventricle for both components that is likely to reflect the contribution of cardiomyocytes; however, the ICC staining for ACE2 was more evident in the coronary vessels, which is consistent with the distribution of the enzyme in the human heart (Fig. 5).
Figure 5. Immunocytochemical staining for Ang(1−7) and ACE2 in the left ventricle of the mouse heart.
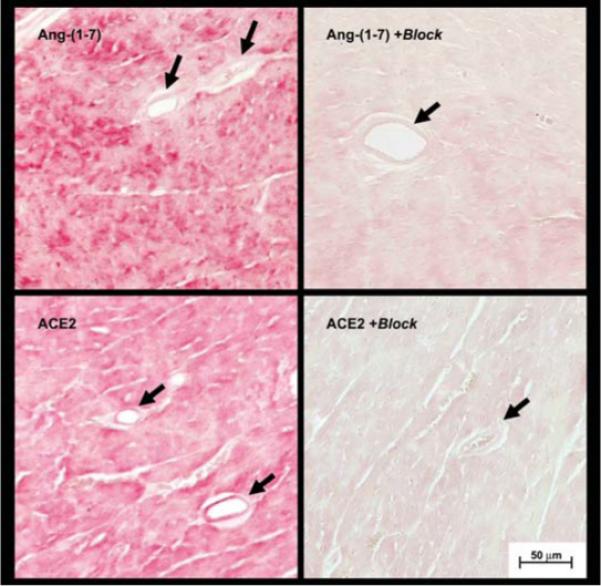
Sections of the left ventricle (5 μm) from the heart of wild-type mice were reacted with polyclonal antibodies against Ang(1−7) and ACE2. In the top left panel, Ang (1−7) staining is evident throughout the field, which is primarily composed of cardiomyocytes. In the bottom left panel, ACE2 staining was evident throughout the field and in coronary vessels. Pre-incubation of primary antibodies with Ang(1−7) (top right panel) or recombinant ACE2 (bottom right panel) essentially abolished the immunocytochemical signals. Data are representative of tissues from three animals. Arrows indicate coronary vessels and the scale bar represents 50 μm.
Discussion
In the present study, we characterized the angiotensin processing pathways in the membrane fraction isolated from the hearts of wild-type, ACE−/− and ACE2−/− mice. In the wild-type, ACE−/− and ACE2−/− strains, Ang I was converted primarily to Ang(1−9) in cardiac membranes through a carboxypeptidase A-like enzyme. Angiotensin(1−7) and Ang II peaks were also identified, although these were minor products in comparison to Ang(1−9). We also showed that Ang(1−7) was the primary peptide formed from Ang II in the heart. In contrast to the carboxypeptidase A-dependent formation of Ang(1−9), the activity responsible for the processing of Ang II to Ang(1−7) was identified as ACE2. In comparison to the wild-type and ACE−/− mice, the processing of Ang II to Ang(1−7) was significantly attenuated (>80%) in the hearts of ACE2 null mice. Moreover, addition of the ACE2 inhibitor ML4760 essentially abolished the conversion of Ang II to Ang(1−7) in the wild-type and ACE−/− strains, emphasizing the predominance of the enzyme in the heart membranes. Finally, in support of the biochemical data, we demonstrated the immunocytochemical presence of Ang(1−7) and ACE2, primarily within the cardiomyocytes of the murine heart.
The present findings reveal several novel aspects of the intracardiac angiotensin system regarding the processing of Ang I and Ang II. Our studies are the first to demonstrate a direct role for ACE2 in the metabolism of Ang II in cardiac membranes of the mouse, consistent with earlier studies by Zimsan and colleagues in the human heart (Zimsan et al. 2003). In addition, we show that Ang II is the preferred substrate over Ang I for ACE2 in the murine heart, which supports previous kinetic studies that revealed a 500-fold greater catalytic constant to the Michalis-Menten constant (kcat/Km) ratio for Ang II versus Ang I (Vickers et al. 2002). Indeed, we did not observe ACE2-dependent formation of Ang(1−9) from Ang I in the presence of carboxypeptidase A inhibition, but simply the accumulation of Ang I. These results also support the observed phenotype of the ACE2−/− model that exhibits an impaired cardiac contractility and is associated with increased levels of Ang II in the heart, kidney and circulation (Crackower et al. 2002). In this regard, the attenuated expression of ACE2 would favour an enhanced accumulation of tissue or interstitial levels of Ang II, particularly under conditions of an activated RAS. Indeed, Yammamoto et al. (2006) reported that cardiac Ang II levels were higher in their ACE2−/− model than in the wild-type mice following pressure overload. The importance of ACE2 may not solely reflect its ability to metabolize Ang II, but also its capacity to generate Ang(1−7), a biologically active product of the RAS that primarily opposes the actions of Ang II. The potential impact of ACE2 in the formation of Ang(1−7) is revealed in several recent studies. Loot et al. (2002) demonstrated that Ang(1−7) improved cardiac function and coronary perfusion and reversed endothelial dysfunction in an experimental model heart failure. Ferreira et al. (2001) find that Ang(1−7) exhibited protective effects following ischaemia–reperfusion that were abrogated by the selective antagonist [d-Ala7]-Ang(1−7). Indeed, the anti-inflammatory actions of Ang(1−7) may underlie the ability of the peptide to reverse cardiac fibrosis in the deoxycorticosterone acetate (DOCA)-salt rat model, since Katovitch and colleagues (Grobe et al. 2006) demonstrate that the antifibrotic actions of Ang(1−7) are independent of changes blood pressure. In isolated myocytes, Ang(1−7) reduces collagen formation and the expression of transforming growth factor β (Iwata et al. 2005). Mas-deficient mice also exhibit impaired cardiac function that is associated with increased collagen and fibronectin (Santos et al. 2006). Moreover, infusion of Ang(1−7) or its nonpeptide agonist AVE0991 provides comparable cardioprotection from induced ischaemia in l-NAME-treated or streptozotocin-treated rats (Benter et al. 2006). Although the functional role of Ang(1−7) in the normal or failing heart is far from defined, our studies provide biochemical evidence for these alternative components of the RAS in the murine heart. Moreover, our data clearly confirm a recent study by Averill et al. (2003) demonstrating increased staining for Ang(1−7) in a rat model of heart failure and that of Wei et al. (2002a), who found that interstitial Ang(1−7) was increased with Ang II in the microdialysis perfusate of the canine heart.
In the present study, we found no evidence for the predominant processing of Ang I to Ang II by ACE or chymase. Indeed, the conversion of Ang I to Ang(1−9) was the major metabolic pathway in cardiac membranes from wild-type and both transgenic strains. Our results in the murine heart are similar to those of Kokkonen et al. (1997), who reported that Ang(1−9) was the predominant product formed from Ang I by a human heart carboxypeptidase A. Jackman et al. (2002) also reported significant conversion of Ang I to Ang(1−9) via cathepsin A, as well as the formation of Ang II and Ang(1−7) in human atrial tissues, although neither the precursor nor the Ang(1−7)-forming enzyme(s) was identified. The present findings contrast with those of Wei et al. (2002b), who reported increased chymase activity that formed Ang II in a different transgenic ACE−/− model. In this regard, we did not apply a cocktail of peptidase inhibitors to the heart membranes or chelating agents such as phenanthroline or EDTA, which would probably inhibit carboxypeptidase A and ACE2, since both are metallopeptidases. Moreover, the comparison of angiotensin metabolism in the latter and current studies is mitigated by the differences in the methods of tissue preparation: soluble or membrane fractions; use of detergents; and the potential presence of endogenous inhibitors. We did find that addition of the chymase inhibitor chymostatin increased the recovery of Ang(1−9) in the heart membranes from the ACE−/− mice. There are multiple isoforms of chymase in various species, and these isotypes can either form or degrade Ang II and Ang I, although we are not aware of data regarding whether chymase can directly metabolize Ang(1−9). Chymase and carboxypeptidase A are both soluble enzymes synthesized by mast cells within the heart (Kokkonen et al. 1997). Upon degranulation, the released enzymes form proteoglycan complexes bound to the extracellular matrix within the interstitial space of the heart. Thus, we do not know the extent of extracellular carboxypeptidase A activity versus that released from mast cells upon freezing of the heart tissue and preparation of the membrane fraction. In contrast, ACE2 is an ectocellular enzyme, primarily expressed in cardiomyocytes and coronary endothelial cells, although the enzyme may undergo shedding to release a soluble and active form (Donoghue et al. 2000).
Blockade of the RAS by ACE inhibitors and/or AT1 antagonists are effective treatments in experimental models of heart failure and in the clinical setting. There is substantial evidence for an intracardiac RAS and its activation in heart failure; however, recent studies have questioned the influence of local Ang II in the heart (De Mello & Danser, 2000; Reudelhuber et al. 2007). Reudelhuber and colleagues (van Kats et al. 2001) used a novel viral expression system to increase myocyte production and secretion of Ang II within the murine heart. Despite a 40-fold increase in the interstial levels of Ang II, no changes in cardiac function or histology were evident. In this model, alterations in cardiac function were present only when Ang II expression increased to the point whereby circulating Ang II resulted in higher blood pressure (van Kats et al. 2001). Baker et al. (2004), however, demonstrated that increased intracellular expression of Ang II within cardiomyocytes markedly stimulated cardiac hypertrophy without alterations in blood pressure. Overexpression of ACE2 in the heart of the spontaneously hypertensive rat reduced the extent of cardiac fibrosis, although in these studies the lentiviral ACE2 construct also significantly lowered blood pressure (Díez-Freire et al. 2006). Tissue levels of Ang II or Ang(1−7) were not determined in this study; however, the overexpression of cardiac ACE2 may have increased the circulating levels of the enzyme and reduced the Ang II to Ang(1−7) ratio in plasma. In this regard, ACE2 may function to dampen Ang II within the heart, and active shedding of ACE2 may influence interstitial or circulating levels of Ang II as well. Indeed, our studies in the perfused rat heart revealed significant ACE2 activity and protein in the perfusate that converted Ang II to Ang(1−7) (Trask et al. 2007).
In conclusion, the present study demonstrates that normal heart tissue expresses an effective enzymatic pathway in ACE2 that may efficiently regulate the intracardiac levels of Ang II. In addition to the specificity of the enzyme, the removal of a single phenylalanine moiety from Ang II not only yields a peptide with little or no affinity for the typical Ang II receptor, but creates an alternative hormone with distinct biological actions that may functionally antagonize Ang II (Ferrario, 2006; Chappell, 2007).
Acknowledgements
The authors gratefully acknowledge the technical expertise of Nancy T. Pirro and Brian M. Westwood for their contribution to these studies. This work is supported in part by grants from the National Heart Lung and Blood Institute, NIH (HL-51952 and HL-56973). An unrestricted grant from the Unifi Corporation (Greensboro, NC, USA) and the Farley-Hudson Foundation (Jacksonville, NC, USA) is also acknowledged. Portions of these studies were previously presented at the 2003 Council for High Blood Pressure meeting in Washington, DC, USA.
References
- Allred AJ, Diz DI, Ferrario CM, Chappell MC. Pathways for angiotensin-(1−7) metabolism in pulmonary and renal tissues. Am J Physiol Renal Physiol. 2000;279:F841–F850. doi: 10.1152/ajprenal.2000.279.5.F841. [DOI] [PubMed] [Google Scholar]
- Averill DB, Ishiyama Y, Chappell MC, Ferrario CM. Cardiac angiotensin-(1−7) in ischemic cardiomyopathy. Circulation. 2003;108:2141–2143. doi: 10.1161/01.CIR.0000092888.63239.54. [DOI] [PubMed] [Google Scholar]
- Baker KM, Chernin MI, Schreiber T, Sangh S, Haiderazidi S, Booz GW, Dostal DE, Kumar R. Evidence of a novel intracrine mechanism in angiotensin II-induced cardiac hypertrophy. Regul Pept. 2004;120:5–13. doi: 10.1016/j.regpep.2004.04.004. [DOI] [PubMed] [Google Scholar]
- Benter IF, Yousif MHM, Cojocel C, Al-Maghrebi M, Diz DI. Angiotensin-(1−7) prevents diabetes-induced cardiovascular dysfunction. Am J Physiol Heart Circ Physiol. 2006;292:H666–H672. doi: 10.1152/ajpheart.00372.2006. [DOI] [PubMed] [Google Scholar]
- Chappell MC. Emerging evidence for a functional angiotensin-converting enzyme 2–angiotensin-(1−7)–MAS receptor axis; more than regulation of blood pressure? Hypertension. 2007;50:596–599. doi: 10.1161/HYPERTENSIONAHA.106.076216. [DOI] [PubMed] [Google Scholar]
- Chappell MC, Jung FF, Gallagher PE, Averill DB, Crackower MA, Penninger JM, Ferrario CM. Omapatrilat treatment is associated with increased ACE-2 and angiotensin-(1−7) in spontaneously hypertensive rats. Hypertension. 2002;40:409. [Google Scholar]
- Crackower MA, Sarao R, Oudit GY, Yagil C, Kozieradski I, Scanga SE, Oliveira-Dos-Santos AJ, Da Costa J, Zhang L, Pei Y, Scholely J, Bray MR, Ferrario CM, Backxx PH, Manoukian AS, Chappell MC, Yagil Y, Penninger JM. Angiotensin-converting enzyme 2 is an essential regulator of heart function. Nature. 2002;417:822–828. doi: 10.1038/nature00786. [DOI] [PubMed] [Google Scholar]
- Dales NA, Gould AE, Brown JA, Calderwood EF, Guan B, Minor CA, Gavin JM, Hales P, Kaushik VK, Stewart M, Tummino PJ, Vickers CS, Ocain TD, Patane MA. Substrate-based design of the first class of angiotensin-converting enzyme-related carboxypeptidase (ACE2) inhibitors. J Am Chem Soc. 2002;124:11852–11853. doi: 10.1021/ja0277226. [DOI] [PubMed] [Google Scholar]
- De Mello WC, Danser AH. Angiotensin II and the heart: on the intracrine renin-angiotensin system. Hypertension. 2000;35:1183–1188. doi: 10.1161/01.hyp.35.6.1183. [DOI] [PubMed] [Google Scholar]
- Díez-Freire C, Vásques J, Correa De Adjounian MF, Ferrari MFR, Yuan L, Silver X, Torres R, Raizada MK. ACE2 gene transfer attenuates hypertension-linked pathophysiological changes in the SHR. Physiol Genomics. 2006;27:12–19. doi: 10.1152/physiolgenomics.00312.2005. [DOI] [PubMed] [Google Scholar]
- Donoghue M, Hsieh F, Baronas E, Godbout K, Gosselin M, Stagliano N, Dononvan ANM, Woolf B, Robinson K, Jeyaseelan R, Breitbart RE, Acton S. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1−9. Circ Res. 2000;87:E1–E9. doi: 10.1161/01.res.87.5.e1. [DOI] [PubMed] [Google Scholar]
- Ferrario CM. Angiotensin-converting enzyme 2 and angiotensin-(1−7): an evolving story in cardiovascular regulation. Hypertension. 2006;47:515–621. doi: 10.1161/01.HYP.0000196268.08909.fb. [DOI] [PubMed] [Google Scholar]
- Ferreira AJ, Santos RA, Almeida AP. Angiotensin-(1−7): cardioprotective effect in myocardial ischemia/reperfusion. Hypertension. 2001;38:665–668. doi: 10.1161/01.hyp.38.3.665. [DOI] [PubMed] [Google Scholar]
- Grobe JL, Mecca AP, Mao H, Katovich MJ. Chronic angiotensin-(1−7) prevents cardiac fibrosis in DOCA-salt model of hypertension. Am J Physiol Heart Circ Physiol. 2006;290:H2417–H2423. doi: 10.1152/ajpheart.01170.2005. [DOI] [PubMed] [Google Scholar]
- Iwata M, Cowling RT, Gurantz D, Moore C, Zhang S, Yuan JXJ, Greenberg BH. Angiotensin-(1−7) binds to specific receptors on cardiac fibroblasts to initiate antifibrotic and antitrophic effects. Am J Physiol Heart Circ Physiol. 2005;289:H2356–H2363. doi: 10.1152/ajpheart.00317.2005. [DOI] [PubMed] [Google Scholar]
- Iyer SN, Chappell MC, Averill DB, Diz DI, Ferrario CM. Vasodepressor actions of angiotensin-(1−7) unmasked during combined treatment with lisinopril and losartan. Hypertension. 1998;31:699–705. doi: 10.1161/01.hyp.31.2.699. [DOI] [PubMed] [Google Scholar]
- Jackman HL, Massad MG, Sekosan M, Tan NF, Brovokovych V, Marcic BM, Eerdos EG. Angiotensin 1−9 and 1−7 release in human heart: role of cathepsin A. Hypertension. 2002;39:976–981. doi: 10.1161/01.hyp.0000017283.67962.02. [DOI] [PubMed] [Google Scholar]
- Kokkonen JO, Saarinen J, Kovanen PT. Regulation of local angiotensin II formation in the human heart in the presence of interstitial fluid: inhibition of chymase by protease inhibitors of interstitial fluid and of angiotensin-converting enzyme by Ang(1−9) formed by heart carboxypeptidase A-like activity. Circ Res. 1997;80:1455–1463. doi: 10.1161/01.cir.95.6.1455. [DOI] [PubMed] [Google Scholar]
- Li N, Zimpelmann J, Cheng K, Wilkins JA, Burns KD. The role of angiotensin converting enzyme 2 in the generation of angiotensin 1−7 by rat proximal tubules. Am J Physiol Renal Physiol. 2004;288:F353–F362. doi: 10.1152/ajprenal.00144.2004. [DOI] [PubMed] [Google Scholar]
- Loot AE, Roks AJM, Henning RH, Tio RA, Suurmeijier AHH, Boomsma F, Vangilst WH. Angiotensin-(1−7) attenuates the development of heart failure after myocardial infarction in rats. Circulation. 2002;105:1548–1550. doi: 10.1161/01.cir.0000013847.07035.b9. [DOI] [PubMed] [Google Scholar]
- Modrall JG, Sadjadi J, Brosnihan KB, Gallagher PE, Ya C-H, Kremaer GL, Bernstein KE, Chappell MC. Depletion of tissue ace differentially influences the intrarenal and urinary expression of angiotensins. Hypertension. 2004;43:849–853. doi: 10.1161/01.HYP.0000121462.27393.f6. [DOI] [PubMed] [Google Scholar]
- Nakamura S, Averill DB, Chappell MC, Diz DI, Brosnihan KB, Ferrario CM. Angiotensin receptors contribute to blood pressure homeostasis in salt-depleted SHR. Am J Physiol Regul Integr Comp Physiol. 2003;284:R164–R173. doi: 10.1152/ajpregu.00210.2002. [DOI] [PubMed] [Google Scholar]
- Reudelhuber TL, Bernstein KE, Delfontaine P. Is angiotensin II a direct mediator of left ventricular hypertrophy? Hypertension. 2007;49:1196–1201. doi: 10.1161/HYPERTENSIONAHA.106.075085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos RAS, Castro CH, Gava E, Pineheiro SVB, Almeida AP, De Paula RD, Cruz JS, Ramos AS, Rosa KT, Irigoyen MC, Bader M, Alenina N, Kitten GT, Ferreira AJ. Impairment of in vitro and in vivo heart function in angiotensin-(1−7) receptor mas knockout mice. Hypertension. 2006;47:996–1002. doi: 10.1161/01.HYP.0000215289.51180.5c. [DOI] [PubMed] [Google Scholar]
- Tikellis C, Johnston CI, Forbers JM, Burns WC, Burrell LM, Risvantis J, Cooper ME. Characterization of renal angiotensin-converting enzyme 2 in diabetic nephropathy. Hypertension. 2003;41:392–397. doi: 10.1161/01.HYP.0000060689.38912.CB. [DOI] [PubMed] [Google Scholar]
- Tipnis SR, Hooper NM, Hyde R, Karran E, Christie G, Turner AJ. A human homolog of angiotensin-converting enzyme. Cloning and functional expression of a captopril-insensitive carboxypeptidase. J Biol Chem. 2000;275:33238–33243. doi: 10.1074/jbc.M002615200. [DOI] [PubMed] [Google Scholar]
- Trask AJ, Averill DB, Ganten D, Chappell MC, Ferrario CM. Primary role of angiotensin converting enzyme 2 in cardiac production of angiotensin-(1−7) in transgenic Ren-2 hypertensive rats. Am J Physiol Heart Circ Physiol. 2007;292:H3019–H3024. doi: 10.1152/ajpheart.01198.2006. [DOI] [PubMed] [Google Scholar]
- van Kats JP, Methot D, Paraids P, Silversides DW, Reudelhuber TL. Use of a biological peptide pump to study chronic peptide hormone action in transgenic mice. Direct and indirect effects of angiotensin II on the heart. J Biol Chem. 2001;276:44012–44017. doi: 10.1074/jbc.M106132200. [DOI] [PubMed] [Google Scholar]
- Vickers C, Hales P, Kaushik V, Dick L, Gavin J, Tang J, Godbout K, Parsons T, Baronas E, Hsieh F, Acton S, Patane M, Nichols A, Tummino P. Hydrolysis of biological peptides by human angiotensin-converting enzyme-related carboxypeptidase. J Biol Chem. 2002;277:14838–14843. doi: 10.1074/jbc.M200581200. [DOI] [PubMed] [Google Scholar]
- Wei CC, Ferrario CM, Brosnihan KB, Farrell DM, Bradley WE, Jaffa AA, Dell'Italia LJ. Angiotensin peptides modulate bradykinin levels in the interstitium of the dog heart in vivo. J Pharmacol Exp Ther. 2002a;300:324–329. doi: 10.1124/jpet.300.1.324. [DOI] [PubMed] [Google Scholar]
- Wei CC, Tian B, Perry G, Meng QC, Chen Y-F, Oparil S, Dell'Italia LJ. Differential ANG II generation in plasma and tissue of mice with decreased expression of the ACE gene. Am J Physiol Heart Circ Physiol. 2002b;282:H2254–H2258. doi: 10.1152/ajpheart.00191.2001. [DOI] [PubMed] [Google Scholar]
- Yammamoto K, Chappell MC, Brosnihan KB, Ferrario CM. In vivo metabolism of angiotensin I by neutral endopeptidase (EC 3.4.24.11) in spontaneously hypertensive rats. Hypertension. 1992;19:692–696. doi: 10.1161/01.hyp.19.6.692. [DOI] [PubMed] [Google Scholar]
- Yammamoto K, Ohishi M, Katsuya T, Ito N, Ikushima M, Kaibe M, Tatara Y, Shiota A, Sugano S, Takeda S, Rouge H, Gadara T. Deletion of angiotensin converting enzyme 2 accelerates pressure overload-induced cardiac dysfunction by increasing local angiotensin II. Hypertension. 2006;47:718–726. doi: 10.1161/01.HYP.0000205833.89478.5b. [DOI] [PubMed] [Google Scholar]
- Zimsan LS, Keller RS, Weaver B, Lin Q, Speth R, Bristow MR, Canver CC. Increased angiotensin-(1−7)-forming activity in failing human heart ventricles: evidence for upregulation of the angiotensin-converting enzyme homologue ACE2. Circulation. 2003;108:1707–1712. doi: 10.1161/01.CIR.0000094734.67990.99. [DOI] [PubMed] [Google Scholar]