

Abstract
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
Organic anion transporting polypeptide 1B1 is an influx transporter expressed on the basolateral membrane of hepatocytes.
A common single nucleotide polymorphism, c.521T→C (p.Val174Ala), of the SLCO1B1 gene has been associated with increased plasma repaglinide concentrations in healthy volunteers.
Previous studies at low repaglinide doses have suggested that the effect of SLCO1B1 c.521T→C polymorphism on the pharmacokinetics of repaglinide could be dose-dependent.
WHAT THIS STUDY ADDS
Repaglinide peak plasma concentration and area under the plasma concentration–time curve increased linearly along with repaglinide dose ranging from 0.25 to 2 mg in both the predominant c.521TT and rare c.521CC genotype group.
The effect of SLCO1B1 c.521T→C polymorphism on repaglinide pharmacokinetics persists over a wide dose range.
AIMS
To establish whether the effect of SLCO1B1[encoding organic anion transporting polypeptide 1B1 (OATP1B1)] c.521T→C (p.Val174Ala) polymorphism on the pharmacokinetics of repaglinide is dose-dependent.
METHODS
Twelve healthy volunteers with the SLCO1B1 c.521TT genotype (controls) and eight with the c.521CC genotype ingested a single 0.25-, 0.5-, 1- or 2-mg dose of repaglinide in a dose-escalation study with a wash-out period of ≥1 week.
RESULTS
The mean area under the plasma concentration–time curve from time 0 to infinity (AUC0–∞) of 0.25, 0.5, 1 or 2 mg repaglinide was 82% (95% confidence interval 47, 125), 72% (24, 138), 56% (24, 95) or 108% (59, 171) (P ≤ 0.001) larger in participants with the SLCO1B1 c.521CC genotype than in those with the c.521TT genotype, respectively. Repaglinide peak plasma concentration and AUC0–∞ increased linearly along with repaglinide dose in both genotype groups (r > 0.88, P < 0.001). There was a tendency towards lower blood glucose concentrations after repaglinide administration in the participants with the c.521CC genotype than in those with the c.521TT genotype.
CONCLUSIONS
The effect of SLCO1B1 c.521T→C polymorphism on the pharmacokinetics of repaglinide persists throughout the clinically relevant dose range.
Keywords: OATP1B1, organic anion transporting polypeptide, pharmacogenetics, pharmacokinetics, repaglinide, SLCO1B1
Introduction
Repaglinide, a short-acting insulin secretagogue, is used in reducing postprandial hyperglycaemia in patients with Type 2 diabetes mellitus [1]. Repaglinide has an oral bioavailability of about 60% [1, 2] and is biotransformed via the cytochrome P450 (CYP) 3A4 and 2C8 enzymes to several inactive metabolites, including M1, M2 and M4 [3–7]. Hepatic uptake by organic anion transporting polypeptide 1B1 (OATP1B1) is an important step preceding the metabolism of repaglinide in the liver [8, 9]. Repaglinide is available as 0.5-, 1- and 2-mg tablets, and the maximum daily dose is 16 mg [10].
Large interindividual variability, partly genetically determined, exists in the pharmacokinetics of repaglinide [1]. The CYP2C8*3 allele has been associated with reduced plasma concentrations of repaglinide [11]. Moreover, subjects with the SLCO1B1 (encoding OATP1B1) c.521CC genotype have had a 188% or 72% larger area under the plasma repaglinide concentration–time curve (AUC) than those with the c.521TT genotype after ingestion of 0.25 or 0.5 mg repaglinide, respectively [8, 12]. These findings suggest that the effect of SLCO1B1 polymorphism on repaglinide pharmacokinetics could be less pronounced after higher repaglinide doses. One possible mechanism for these apparently dose-dependent findings is that the OATP1B1-mediated uptake of repaglinide is saturated already at low concentrations, and thus the effect of reduced OATP1B1 activity would be smaller at higher repaglinide doses. In this case, the effect of SLCO1B1 polymorphism would be of limited importance at the doses used in clinical practice.
To establish whether the effect of the SLCO1B1 c.521T→C (p.Val174Ala) single nucleotide polymorphism (SNP) on the pharmacokinetics of repaglinide is dose-dependent, we conducted a dose-escalation study in a panel of 20 healthy volunteers with different SLCO1B1 genotypes. To exclude the effect of CYP2C8*3 allele on the pharmacokinetics of repaglinide, only noncarriers of this allele were recruited.
Methods
Subjects
The study protocol was approved by the Coordinating Ethics Committee of the Helsinki and Uusimaa Hospital District and by the National Agency for Medicines. All subjects gave their written informed consent. Participants were selected on the basis of the SLCO1B1 c.521T→C SNP as well as the g.-11187G→A, g.-10499A→C and c.388A→G SNPs from a pool of genotyped healthy volunteers [13]. Haplotypes were assigned as described previously [13]. Only noncarriers of the CYP2C8*3 allele were included [11]. The c.521TT (control) group comprised seven women and five men (mean ± SD age 24 ± 4 years, height 171 ± 8 cm, weight 71 ± 12 kg) with the homozygous reference genotype at each position (SLCO1B1*1A/*1A). The c.521CC group (age 24 ± 5 years, height 172 ± 7 cm, weight 70 ± 6 kg) included five women and three men. One of them had the SLCO1B1*5/*17 genotype, three had the *15/*16 genotype and four had the *16/*17 genotype. Subjects were ascertained to be healthy by medical history, physical examination and laboratory tests before they were entered in the study. None was on any continuous medication and none was a tobacco smoker. Use of other drugs was prohibited for 1 week, use of grapefruit products for 3 days, and use of alcohol for 1 day before the day of repaglinide administration.
Study design
In a dose-escalation study with a wash-out period of ≥1 week, the subjects ingested, after an overnight fast, a single 0.25-, 0.5-, 1- or 2-mg dose of repaglinide (Novonorm; NovoNordisk, Bagsværd, Denmark) with 150 ml of water at 09.00 h. Repaglinide 0.25-mg tablets are not available and therefore repaglinide 0.5-mg tablets were halved and weighed by the investigators. Subjects remained seated for 3 h and received standardized meals: breakfast 15 min after administration of repaglinide, a snack after 1 and 2 h, a warm meal after 3 h, and a snack after 7 h. Additional carbohydrates were given if blood glucose concentration was low and/or the subject had symptoms of hypoglycaemia. Timed blood samples (5 or 10 ml each) were drawn prior to and 15, 30, 45, 60, 75, 90 and 105 min and 2, 2.5, 3, 4, 5 and 7 h after the administration of repaglinide. Blood glucose concentrations were measured immediately after each blood sampling by the glucose oxidase method with Precision G Blood Glucose Testing System (Medisense, Bedford, MA, USA). The between-day coefficient of variation (CV) of the method was 5.7% at 2.8 mmol l−1 and 4.4% at 18.6 mmol l−1 (n = 28). Plasma was separated within 30 min and stored at −70°C until analysis.
Determination of plasma drug concentrations
Plasma concentrations of repaglinide and its M1, M2 and M4 metabolites were quantified by use of an API 3000 liquid chromatography-tandem mass spectrometry (LC/MS/MS) system (Sciex Division of MDS Inc., Toronto, Ontario, Canada) as described previously [14]. The limit of quantification was 0.05 ng ml−1 for repaglinide, and the between-day CV was 9% at 0.1 ng ml−1 and 3% at 2.0 ng ml−1 and 20 ng ml−1 (n = 21). Because authentic metabolite standards were not available, plasma metabolite responses (relative concentrations) are given in arbitrary units relative to the ratio of the peak height of each metabolite to that of the internal standard, D5-repaglinide.
Pharmacokinetics and pharmacodynamics
The pharmacokinetics of repaglinide and its metabolites were characterized by the peak plasma concentration (Cmax), time to Cmax (tmax), elimination half-life (t1/2) and AUC0–∞. The elimination rate constant (ke) was determined by linear regression analysis of the log-linear part of the concentration–time curve. The t1/2 was calculated by the equation t1/2 = ln2/ke. AUC was calculated by a combination of the linear (for increasing concentrations) and log-linear (for decreasing concentrations) trapezoidal rules, with extrapolation to infinity, when appropriate, by division of the last measured concentration by ke. The blood glucose response to repaglinide was characterized by minimum blood glucose concentration, and the mean concentration from 0 to 3 h and from 0 to 7 h.
Statistical analysis
The data were analysed with the statistical program spss 13.0 (SPSS Inc., Chicago, IL, USA). Cmax and AUC data were logarithmically transformed before statistical analysis. The pharmacokinetic (except tmax) and pharmacodynamic variables of repaglinide between the two SLCO1B1 genotype groups were evaluated using repeated measures analysis of variance (anova) including dose as a within-subjects factor and genotype as a between-subjects factor. Post hoc testing between the genotypes was done with independent samples t-test. The tmax data were compared between genotypes during each treatment phase with the Mann–Whitney U-test. The geometric mean ratio (c.521CC/c.521TT) with 95% confidence interval was calculated for repaglinide Cmax and AUC0–∞ values. Dose linearity of repaglinide pharmacokinetics was investigated with linear regression analysis. A P-value <0.05 was considered to be statistically significant.
Results
Repaglinide pharmacokinetics
SLCO1B1 genotype was significantly associated with the pharmacokinetics of repaglinide after each dose (Table 1, Figure 1). The AUC0–∞ of repaglinide was 82, 72, 56 or 108% (P ≤ 0.001) larger in participants with the c.521CC genotype than in those with the c.521TT genotype after ingestion of 0.25, 0.5, 1 or 2 mg repaglinide, respectively. The Cmax of repaglinide was about 60–70% larger in the c.521CC participants than in the c.521TT participants (P < 0.01), except after the 1-mg dose with only a 32% larger (P = 0.068) Cmax. No significant differences were seen in the tmax or t1/2 of repaglinide between the genotype groups. The effect of SLCO1B1 genotype on the pharmacokinetic variables of repaglinide was independent of repaglinide dose (Table 1). The increase of repaglinide Cmax and AUC0–∞ was linear as a function of repaglinide dose in both genotype groups (Figure 2).
Table 1.
Pharmacokinetic variables of repaglinide and its M1, M2 and M4 metabolites in healthy subjects with the SLCO1B1 c.521TT (n = 12) or c.521CC (n = 8) genotype after a single oral 0.25-, 0.5-, 1- or 2-mg dose of repaglinide
| VariableSLCO1B1 genotype | Repaglinide0.25 mg | Repaglinide0.5 mg | Repaglinide1 mg | Repaglinide2 mg | anovaP-value for dose–group interaction |
|---|---|---|---|---|---|
| Repaglinide | |||||
| Cmax (ng ml−1) | |||||
| c.521TT | 3.7 ± 1.0 | 6.7 ± 2.4 | 16.5 ± 5.5 | 33.0 ± 8.5 | 0.485 |
| c.521CC | 6.2 ± 1.8** | 11.5 ± 4.0** | 21.6 ± 5.8 | 52.7 ± 11.7** | |
| Mean ratio (95% CI)† | 1.65 (1.25, 2.19) | 1.73 (1.19, 2.52) | 1.32 (0.98, 1.79) | 1.61 (1.26, 2.06) | |
| tmax (min) | |||||
| c.521TT | 30 (30–60) | 45 (30–120) | 38 (30–45) | 30 (30–45) | |
| c.521CC | 30 (30–45) | 30 (30–45) | 30 (30–45) | 45 (30–45) | |
| t1/2 (h) | |||||
| c.521TT | 1.5 ± 0.6 | 1.8 ± 0.5 | 1.7 ± 0.7 | 1.8 ± 0.6 | 0.680 |
| c.521CC | 1.7 ± 0.5 | 1.5 ± 0.3 | 1.7 ± 0.5 | 1.8 ± 0.4 | |
| AUC0–∞ (ng ml−1 h−1) | |||||
| c.521TT | 4.7 ± 1.1 | 10.3 ± 2.6 | 19.3 ± 4.2 | 37.2 ± 7.6 | 0.094 |
| c.521CC | 8.6 ± 1.8*** | 18.9 ± 9.6** | 30.4 ± 7.8** | 80.1 ± 27.4*** | |
| Mean ratio (95% CI)† | 1.82 (1.47, 2.25) | 1.72 (1.24, 2.38) | 1.56 (1.24, 1.95) | 2.08 (1.59, 2.71) | |
| M1 | |||||
| AUC0–∞ (U ml−1 h−1) | |||||
| c.521TT | 15.6 ± 14.1 | 35.8 ± 34.8 | 58.8 ± 38.1 | 106.5 ± 91.3 | 0.838 |
| c.521CC | 14.0 ± 6.2 | 27.7 ± 11.5 | 54.2 ± 27.2 | 96.5 ± 41.3 | |
| M1/Repaglinide AUC0–∞ ratio (U ng−1) | |||||
| c.521TT | 3.6 ± 3.8 | 3.9 ± 4.8 | 3.4 ± 2.9 | 3.0 ± 2.9 | 0.771 |
| c.521CC | 1.7 ± 0.9 | 1.7 ± 0.9 | 1.8 ± 0.7 | 1.3 ± 0.7 | |
| M2 | |||||
| AUC0–∞ (U ml−1 h−1) | |||||
| c.521TT | 20.9 ± 13.9 | 45.3 ± 34.2 | 93.2 ± 60.1 | 137.8 ± 95.7 | 0.883 |
| c.521CC | 49.7 ± 34.4** | 92.9 ± 57.1* | 193.0 ± 154.2* | 295.3 ± 162.6* | |
| M2/Repaglinide AUC0–∞ ratio (U ng−1) | |||||
| c.521TT | 4.6 ± 3.7 | 4.8 ± 5.0 | 5.3 ± 4.5 | 3.8 ± 3.1 | 0.384 |
| c.521CC | 6.0 ± 4.4 | 5.8 ± 3.8 | 6.0 ± 3.3 | 3.8 ± 2.0 | |
| M4 | |||||
| AUC0–∞ (U ml−1 h−1) | |||||
| c.521TT | 24.4 ± 19.8 | 47.1 ± 30.8 | 91.9 ± 60.2 | 169.6 ± 141.3 | 0.567 |
| c.521CC | 39.2 ± 15.6* | 83.0 ± 41.2* | 160.9 ± 103.4* | 308.3 ± 117.3** | |
| M4/Repaglinide AUC0–∞ ratio (U ng−1) | |||||
| c.521TT | 5.5 ± 5.4 | 4.9 ± 4.3 | 5.4 ± 5.1 | 4.8 ± 4.7 | 0.229 |
| c.521CC | 4.7 ± 2.0 | 5.0 ± 2.7 | 5.1 ± 2.1 | 4.0 ± 1.3 | |
Data are mean ± SD, tmax data are median (range). Cmax, peak plasma concentration; CI, confidence interval; tmax, time to Cmax; t1/2, elimination half-life; AUC0–∞, area under the plasma concentration–time curve from 0 h to infinity.
P < 0.05 vs. c.521TT group;
P < 0.01 vs. c.521TT group;
P < 0.001 vs. c.521TT group.
These data are geometric mean ratio (95% CI) c.521CC vs. c.521TT group.
Figure 1.
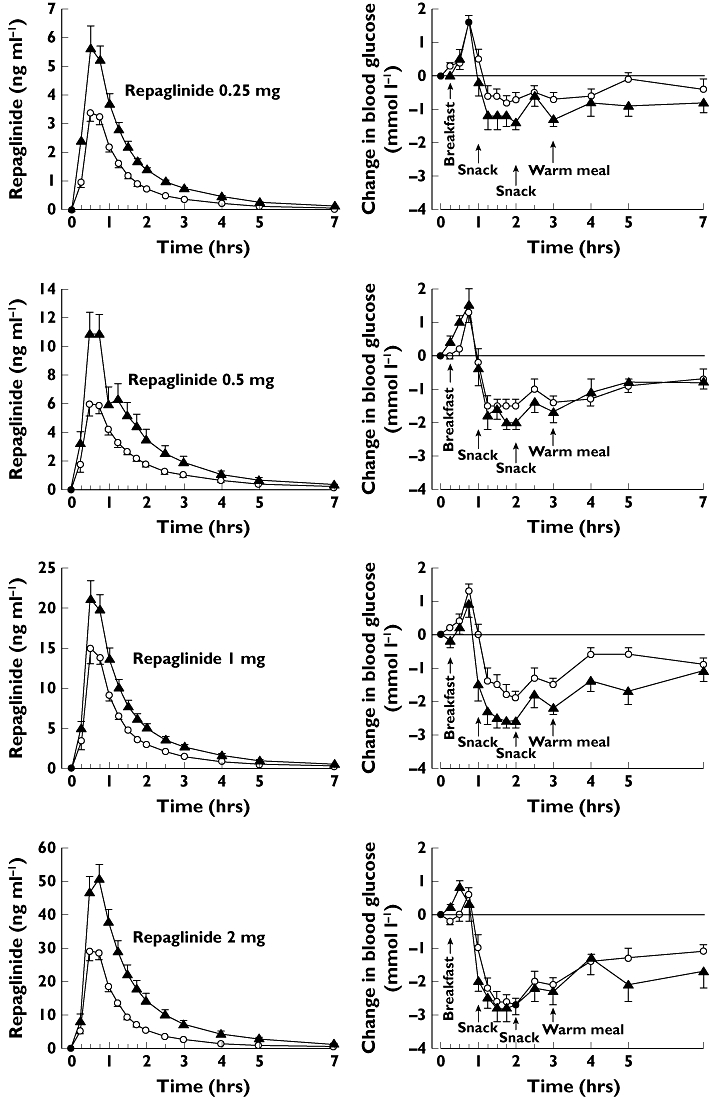
Mean ± SEM plasma concentrations of repaglinide and change in blood glucose concentrations after a single 0.25, 0.5, 1 or 2 mg oral dose of repaglinide in healthy subjects with the SLCO1B1 c.521TT (n = 12, open circles) or c.521CC (n = 8, solid triangles) genotype. Due to hypoglycaemia, additional carbohydrates were given after 2 mg repaglinide to four c.521CC participants and three c.521TT participants (14–123 g), after 1 mg repaglinide to one c.521TT participant (14 g), and after 0.5 mg repaglinide to one c.521CC participant (63 g). Some error bars have been omitted for clarity. c.521TT (○); c.521CC (▴)
Figure 2.
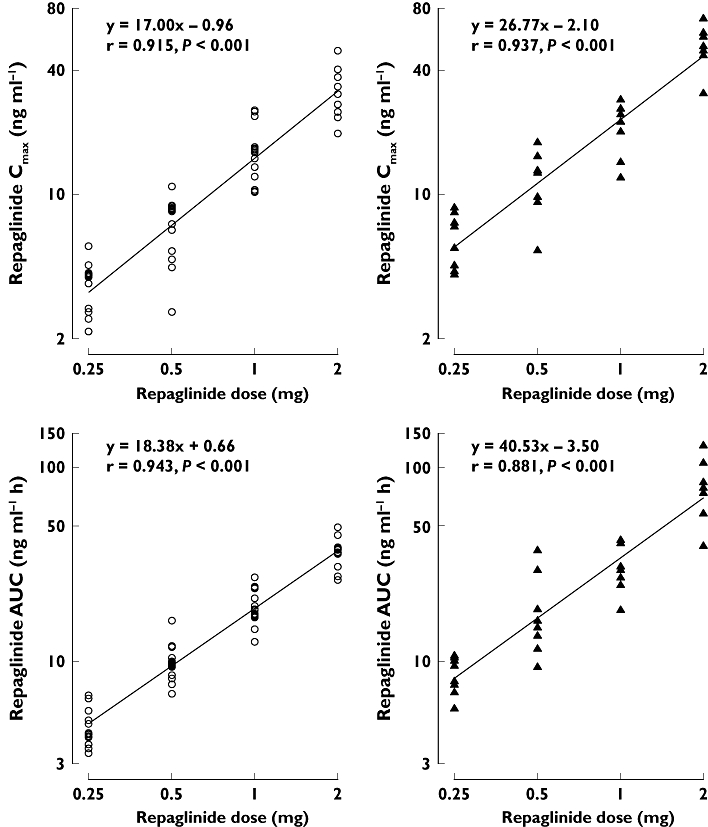
The individual repaglinide Cmax and AUC0–∞ values after a single 0.25-, 0.5-, 1- or 2-mg oral dose of repaglinide in healthy subjects with the SLCO1B1 c.521TT (n = 12, open circles) or c.521CC (n = 8, solid triangles) genotype. c.521TT (○); c.521CC (▴)
After each repaglinide dose, SLCO1B1 genotype was significantly associated with the AUC0–∞ of repaglinide M2 and M4 metabolites, but not with that of M1, and the effect was independent of repaglinide dose (Table 1). There were no differences in the metabolite/repaglinide AUC0–∞ ratios between the two SLCO1B1 genotype groups.
Repaglinide pharmacodynamics
There was a tendency towards lower blood glucose concentrations in the participants with the c.521CC genotype than in those with the c.521TT genotype during all phases (Figure 1), but no statistically significant differences were seen in repaglinide pharmacodynamic variables between the genotypes (Table 2). In addition to the standardized meals, four of the eight (50%) c.521CC participants and three of the 12 (25%) c.521TT participants received additional carbohydrates (14–123 g) after administration of 2 mg repaglinide to correct hypoglycaemia. After 1 mg repaglinide, additional carbohydrates were given to one c.521TT participant (14 g), and after 0.5 mg repaglinide to one c.521CC participant (63 g). Subjects reported only mild to moderate symptoms of hypoglycaemia, including reversible disturbance in attention, tremor, palpitations and headache.
Table 2.
Blood glucose variables of a single oral 0.25-, 0.5-, 1- or 2-mg dose of repaglinide in healthy subjects with the SLCO1B1 c.521TT (n = 12) or c.521CC (n = 8) genotype
| VariableSLCO1B1 genotype | Repaglinide0.25 mg | Repaglinide0.5 mg | Repaglinide1 mg | Repaglinide2 mg | anovaP-value for dose–group interaction |
|---|---|---|---|---|---|
| Minimum concentration (mmol l−1) | |||||
| c.521TT | 3.7 ± 0.6 | 3.2 ± 0.5 | 2.9 ± 0.8 | 2.3 ± 0.5 | 0.539 |
| c.521CC | 3.4 ± 0.6 | 3.1 ± 0.7 | 2.5 ± 0.6 | 2.1 ± 0.5 | |
| Mean concentration (mmol l−1) 0–3 h | |||||
| c.521TT | 4.9 ± 0.6 | 4.7 ± 0.6 | 4.3 ± 0.7 | 3.8 ± 0.5 | 0.610 |
| c.521CC | 4.9 ± 0.4 | 4.6 ± 0.7 | 4.1 ± 0.5 | 3.6 ± 0.3 | |
| Mean concentration (mmol l−1) 0–7 h | |||||
| c.521TT | 4.8 ± 0.4 | 4.5 ± 0.5 | 4.3 ± 0.5 | 3.9 ± 0.5 | 0.126 |
| c.521CC | 4.7 ± 0.5 | 4.5 ± 0.7 | 4.0 ± 0.6 | 3.5 ± 0.6 | |
Data are mean ± SD.
Discussion
To our knowledge, this is the first study to investigate the effects of SLCO1B1 polymorphism on drug pharmacokinetics at different doses. Repaglinide AUC0–∞ was 60–110% greater in participants with the c.521CC genotype than in those with the c.521TT genotype after ingestion of single repaglinide doses ranging from 0.25 to 2 mg. The effect of SLCO1B1 genotype on repaglinide was independent of repaglinide dose, and the increase in repaglinide Cmax and AUC0–∞ along with the dose was linear in both genotype groups. In agreement with the pharmacokinetic results, there was a tendency towards lower blood glucose concentrations after repaglinide administration in the participants with the c.521CC genotype than in those with the c.521TT genotype.
The SLCO1B1 c.521T→C polymorphism is associated with increased Cmax and AUC of repaglinide, with limited effects on its t1/2, suggesting increased oral bioavailability or reduced clearance in association with a reduced distribution volume, or both. However, little has been known about the consistency of this effect at different doses. In a retrospective study, the mean AUC of 0.25 mg repaglinide was 188% larger in subjects with the c.521CC genotype than in those with the c.521TT genotype [8]. However, in two prospective studies with 0.25 or 0.5 mg of repaglinide, the mean AUC of repaglinide was only about 70% larger in the c.521CC group than in the c.521TT group [12, 15]. In any case, the results of the present study demonstrate that the effect of SLCO1B1 polymorphism on repaglinide pharmacokinetics is independent of repaglinide dose. The apparent discrepancies in the previous studies could have been caused by random sampling variation, or by the low-activity SLCO1B1*1B (c.388G-c.521T) allele [16, 17], the carriers of which were not excluded from the retrospective study [8].
The difference in the AUC of repaglinide between the two SLCO1B1 genotype groups was larger, although not statistically significantly, after ingesting 2 mg repaglinide than after the lower repaglinide doses. Therefore, the possibility that the effect of SLCO1B1 genotype on repaglinide pharmacokinetics could increase after administering ≥2 mg repaglinide cannot be excluded. In this study, higher repaglinide doses were not administered due to the risk of profound hypoglycaemia in healthy volunteers.
The present study has confirmed the relevance of the SLCO1B1 c.521T→C polymorphism to the pharmacokinetics of repaglinide over the dose range used in clinical practice. Moreover, the pharmacokinetics of repaglinide proved to be linear in both the predominant c.521TT and the rare c.521CC (about 2–5% of Whites and 1–2% of Asians) [13, 18–21] genotype group, similar to what has been seen in subjects not genotyped for SLCO1B1 polymorphism [1].
It should be recognized that the pharmacodynamics of repaglinide cannot be directly extrapolated from healthy subjects to patients with Type 2 diabetes mellitus. The pharmacokinetics of repaglinide, however, is similar in both healthy subjects and patients, and repaglinide exhibits a clear dose–response [1]. Therefore, patients homozygous for the SLCO1B1 c.521C allele are probably more susceptible to the blood glucose-lowering effect of repaglinide than those with other genotypes. Based on the pharmacokinetic results of the present study, the optimal starting dose of repaglinide could be about 50% lower in patients with the c.521CC genotype than in those with the c.521TT genotype. Obviously, the dose of repaglinide should be adjusted according to the actual blood glucose-lowering response. Selecting the starting dose according to genotype might reduce the time needed to reach the correct maintenance dose, potentially with a smaller risk of hypoglycaemia.
In addition to repaglinide, the SLCO1B1 c.521T→C polymorphism has been associated with the pharmacokinetics of several other drugs, such as fexofenadine, pravastatin, simvastatin, atorvastatin and rosuvastatin [22–27], but different doses have not been addressed in these studies. For example, statins generally have a large dose range of up to eightfold [28], and data on the effect of the SLCO1B1 genotype in relation to dose could be useful in adjusting statin dosage. Such information might also be valuable in tailoring treatment of drugs with a narrow therapeutic range, such as SN-38, the active metabolite of the antineoplastic agent irinotecan, the concentrations of which have been higher in patients carrying the SLCO1B1 c.521C allele than in noncarriers [29].
In conclusion, the effect of SLCO1B1 c.521T→C polymorphism on the pharmacokinetics of repaglinide persists throughout the clinically relevant dose range. Further studies are warranted to establish the relevance of SLCO1B1 polymorphism on the blood glucose-lowering effect of repaglinide in patients with Type 2 diabetes mellitus.
Acknowledgments
The authors thank Ms Kerttu Mårtensson, Ms Eija Mäkinen-Pulli, Ms Lisbet Partanen, Ms Anna-Riitta Pasanen and Mr Jouko Laitila for skilful assistance, and Mr Harri Paakkulainen for statistical advice. This study was supported by grants from the Helsinki University Central Hospital Research Fund (Helsinki, Finland) and the Sigrid Jusélius Foundation (Helsinki).
REFERENCES
- 1.Hatorp V. Clinical pharmacokinetics and pharmacodynamics of repaglinide. Clin Pharmacokinet. 2002;41:471–83. doi: 10.2165/00003088-200241070-00002. [DOI] [PubMed] [Google Scholar]
- 2.Dornhorst A. Insulinotropic meglitinide analogues. Lancet. 2001;358:1709–16. doi: 10.1016/S0140-6736(01)06715-0. [DOI] [PubMed] [Google Scholar]
- 3.Bidstrup TB, Bjørnsdottir I, Sidelmann UG, Thomsen MS, Hansen KT. CYP2C8 and CYP3A4 are the principal enzymes involved in the human in vitro biotransformation of the insulin secretagogue repaglinide. Br J Clin Pharmacol. 2003;56:305–14. doi: 10.1046/j.0306-5251.2003.01862.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Niemi M, Backman JT, Neuvonen M, Neuvonen PJ. Effects of gemfibrozil, itraconazole, and their combination on the pharmacokinetics and pharmacodynamics of repaglinide: potentially hazardous interaction between gemfibrozil and repaglinide. Diabetologia. 2003;46:347–51. doi: 10.1007/s00125-003-1034-7. [DOI] [PubMed] [Google Scholar]
- 5.Niemi M, Kajosaari LI, Neuvonen M, Backman JT, Neuvonen PJ. The CYP2C8 inhibitor trimethoprim increases the plasma concentrations of repaglinide in healthy subjects. Br J Clin Pharmacol. 2004;57:441–7. doi: 10.1046/j.1365-2125.2003.02027.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kajosaari LI, Laitila J, Neuvonen PJ, Backman JT. Metabolism of repaglinide by CYP2C8 and CYP3A4 in vitro: effect of fibrates and rifampicin. Basic Clin Pharmacol Toxicol. 2005;97:249–56. doi: 10.1111/j.1742-7843.2005.pto_157.x. [DOI] [PubMed] [Google Scholar]
- 7.Kajosaari LI, Niemi M, Backman JT, Neuvonen PJ. Telithromycin, but not montelukast, increases the plasma concentrations and effects of the cytochrome P450 3A4 and 2C8 substrate repaglinide. Clin Pharmacol Ther. 2006;79:231–42. doi: 10.1016/j.clpt.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 8.Niemi M, Backman JT, Kajosaari LI, Leathart JB, Neuvonen M, Daly AK, Eichelbaum M, Kivistö KT, Neuvonen PJ. Polymorphic organic anion transporting polypeptide 1B1 is a major determinant of repaglinide pharmacokinetics. Clin Pharmacol Ther. 2005;77:468–78. doi: 10.1016/j.clpt.2005.01.018. [DOI] [PubMed] [Google Scholar]
- 9.Bachmakov I, Gläser H, Fromm MF, König J. Interaction of oral antidiabetic drugs with hepatic uptake transporters: focus on OATPs and OCT1. Diabetes. 2008;57:1463–9. doi: 10.2337/db07-1515. [DOI] [PubMed] [Google Scholar]
- 10.Prandin (Repaglinide) Prescribing Information. [accessed 28 March 2008]. Available at http://www.prandin.com.
- 11.Niemi M, Leathart JB, Neuvonen M, Backman JT, Daly AK, Neuvonen PJ. Polymorphism in CYP2C8 is associated with reduced plasma concentrations of repaglinide. Clin Pharmacol Ther. 2003;74:380–7. doi: 10.1016/S0009-9236(03)00228-5. [DOI] [PubMed] [Google Scholar]
- 12.Kalliokoski A, Neuvonen M, Neuvonen PJ, Niemi M. Different effects of SLCO1B1 polymorphism on the pharmacokinetics and pharmacodynamics of repaglinide and nateglinide. J Clin Pharmacol. 2008;48:311–21. doi: 10.1177/0091270007311569. [DOI] [PubMed] [Google Scholar]
- 13.Pasanen MK, Backman JT, Neuvonen PJ, Niemi M. Frequencies of single nucleotide polymorphisms and haplotypes of organic anion transporting polypeptide 1B1 SLCO1B1 gene in a Finnish population. Eur J Clin Pharmacol. 2006;62:409–15. doi: 10.1007/s00228-006-0123-1. [DOI] [PubMed] [Google Scholar]
- 14.Tornio A, Niemi M, Neuvonen M, Laitila J, Kalliokoski A, Neuvonen PJ, Backman JT. The effect of gemfibrozil on repaglinide pharmacokinetics persists for at least 12 h postdose: evidence for mechanism-based inhibition of CYP2C8 in vivo. Clin Pharmacol Ther. 2008;84:403–11. doi: 10.1038/clpt.2008.34. [DOI] [PubMed] [Google Scholar]
- 15.Kalliokoski A, Backman JT, Kurkinen KJ, Neuvonen PJ, Niemi M. Effects of gemfibrozil and atorvastatin on the pharmacokinetics of repaglinide in relation to SLCO1B1 polymorphism. Clin Pharmacol Ther. 2008 doi: 10.1038/clpt.2008.74. Apr 16 [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 16.Mwinyi J, Johne A, Bauer S, Roots I, Gerloff T. Evidence for inverse effects of OATP-C (SLC21A6) 5 and 1b haplotypes on pravastatin kinetics. Clin Pharmacol Ther. 2004;75:415–21. doi: 10.1016/j.clpt.2003.12.016. [DOI] [PubMed] [Google Scholar]
- 17.Maeda K, Ieiri I, Yasuda K, Fujino A, Fujiwara H, Otsubo K, Hirano M, Watanabe T, Kitamura Y, Kusuhara H, Sugiyama Y. Effects of organic anion transporting polypeptide 1B1 haplotype on pharmacokinetics of pravastatin, valsartan, and temocapril. Clin Pharmacol Ther. 2006;79:427–39. doi: 10.1016/j.clpt.2006.01.011. [DOI] [PubMed] [Google Scholar]
- 18.Nozawa T, Nakajima M, Tamai I, Noda K, Nezu J, Sai Y, Tsuji A, Yokoi T. Genetic polymorphisms of human organic anion transporters OATP-C (SLC21A6) and OATP-B (SLC21A9): allele frequencies in the Japanese population and functional analysis. J Pharmacol Exp Ther. 2002;302:804–13. doi: 10.1124/jpet.302.2.804. [DOI] [PubMed] [Google Scholar]
- 19.König J, Seithel A, Gradhand U, Fromm MF. Pharmacogenomics of human OATP transporters. Naunyn Schmiedebergs Arch Pharmacol. 2006;372:432–43. doi: 10.1007/s00210-006-0040-y. [DOI] [PubMed] [Google Scholar]
- 20.Niemi M. Role of OATP transporters in the disposition of drugs. Pharmacogenomics. 2007;8:787–802. doi: 10.2217/14622416.8.7.787. [DOI] [PubMed] [Google Scholar]
- 21.Pasanen MK, Neuvonen PJ, Niemi M. Global analysis of genetic variation in SLCO1B1. Pharmacogenomics. 2008;9:19–33. doi: 10.2217/14622416.9.1.19. [DOI] [PubMed] [Google Scholar]
- 22.Nishizato Y, Ieiri I, Suzuki H, Kimura M, Kawabata K, Hirota T, Takane H, Irie S, Kusuhara H, Urasaki Y, Urae A, Higuchi S, Otsubo K, Sugiyama Y. Polymorphisms of OATP-C (SLC21A6) and OAT3 (SLC22A8) genes: consequences for pravastatin pharmacokinetics. Clin Pharmacol Ther. 2003;73:554–65. doi: 10.1016/S0009-9236(03)00060-2. [DOI] [PubMed] [Google Scholar]
- 23.Niemi M, Schaeffeler E, Lang T, Fromm MF, Neuvonen M, Kyrklund C, Backman JT, Kerb R, Schwab M, Neuvonen PJ, Eichelbaum M, Kivistö KT. High plasma pravastatin concentrations are associated with single nucleotide polymorphisms and haplotypes of organic anion transporting polypeptide-C (OATP-C, SLCO1B1) Pharmacogenetics. 2004;14:429–40. doi: 10.1097/01.fpc.0000114750.08559.32. [DOI] [PubMed] [Google Scholar]
- 24.Niemi M, Kivistö KT, Hofmann U, Schwab M, Eichelbaum M, Fromm MF. Fexofenadine pharmacokinetics are associated with a polymorphism of the SLCO1B1 gene (encoding OATP1B1) Br J Clin Pharmacol. 2005;59:602–4. doi: 10.1111/j.1365-2125.2005.02354.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Niemi M, Pasanen MK, Neuvonen PJ. SLCO1B1 polymorphism and sex affect the pharmacokinetics of pravastatin but not fluvastatin. Clin Pharmacol Ther. 2006;80:356–66. doi: 10.1016/j.clpt.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 26.Pasanen MK, Neuvonen M, Neuvonen PJ, Niemi M. SLCO1B1 polymorphism markedly affects the pharmacokinetics of simvastatin acid. Pharmacogenet Genomics. 2006;16:873–9. doi: 10.1097/01.fpc.0000230416.82349.90. [DOI] [PubMed] [Google Scholar]
- 27.Pasanen MK, Fredrikson H, Neuvonen PJ, Niemi M. Different effects of SLCO1B1 polymorphism on the pharmacokinetics of atorvastatin and rosuvastatin. Clin Pharmacol Ther. 2007;82:726–33. doi: 10.1038/sj.clpt.6100220. [DOI] [PubMed] [Google Scholar]
- 28.Grundy SM, Cleeman JI, Merz CN, Brewer HB, Jr, Clark LT, Hunninghake DB, Pasternak RC, Smith SC, Jr, Stone NJ. Implications of recent clinical trials for the National Cholesterol Education Program Adult Treatment Panel III guidelines. Circulation. 2004;110:227–39. doi: 10.1161/01.CIR.0000133317.49796.0E. [DOI] [PubMed] [Google Scholar]
- 29.Xiang X, Jada SR, Li HH, Fan L, Tham LS, Wong CI, Lee SC, Lim R, Zhou QY, Goh BC, Tan EH, Chowbay B. Pharmacogenetics of SLCO1B1 gene and the impact of *1b and *15 haplotypes on irinotecan disposition in Asian cancer patients. Pharmacogenet Genomics. 2006;16:683–91. doi: 10.1097/01.fpc.0000230420.05221.71. [DOI] [PubMed] [Google Scholar]