

Abstract
Although cisplatin plays a vital role in the treatment of several types of human cancer, its wide use is limited by the development of drug resistance and associated toxic side effects. Gold and gold complexes have been used to treat a wide range of ailments for many centuries. In recent years, the use of gold(III) complexes as an alternative to cisplatin treatment was proposed due to the similarities of gold and platinum. Gold(III) is isoelectronic with platinum(II) and gold(III) complexes have the same square-planar geometries as platinum(II) complexes, such as cisplatin. Although it was originally thought that gold(III) complexes might have the same molecular target as cisplatin, several lines of data indicated that proteins, rather than DNA, are targeted by gold complexes. We have recently evaluated cytotoxic and anti-cancer effects of several gold(III) dithiocarbamates against human breast cancer cells in vitro and in vivo. We have identified the tumor proteasome as an important target for gold(III) complexes and have shown that proteasome inhibition by gold(III) complexes is associated with apoptosis induction in breast cancer cells in vitro and in vivo. Furthermore, treatment of human breast tumor-bearing nude mice with a gold(III) dithiocarbamate complex was associated with tumor growth inhibition, supporting the significance of its potential development for breast cancer treatment.
Keywords: gold(III) complexes, gold coordination chemistry, proteasome inhibitors, breast cancer, anti-cancer drugs
1. History: Gold application in medicine
Medical properties of gold have been explored throughout the history of civilization. Even ancient cultures in India and Egypt exploited gold and used it for various medicinal preparations. The earliest therapeutic application of gold can be tracked back to 2500 BC in China where gold was used to treat smallpox, skin ulcers, and measles [1]. Moreover, at that time, people thought of gold as being immortal and everlasting, and associated gold with longevity. Some cultures still believe in the healing properties of gold. As an illustration, Japanese tradition suggests that thin gold-foils placed into tea, sake and food could be beneficial to human health.
Despite the therapeutics effects attributed to gold, several toxic side effects were also observed. To avoid toxicity, alchemists sought to solve the problem by making elixirs of “drinkable gold”. The elixirs were prepared by mixing gold with various plants, including Chinese herbs traditionally used for medicine. Although the ancient alchemists did not know much about chemistry, they were still able to coax gold into solution, mostly due to gold's interactions with cyanide present in the plants [2]. As a result, “medicinal gold” (golden alloys, mosaic gold and other man-made gold preparations) and “potable gold” (neutralized gold solutions) were developed as elixirs [3].
Although gold and gold complexes have historically been used for the treatment of a wide range of ailments, the rational use of gold in medicine began in the early 1920s when gold was clinically tested for its in vitro bacteriostatic effect. The first gold complex used for that purpose was gold cyanide, employed by bacteriologist Robert Koch to kill the mycobacteria, the causative agent of tuberculosis [4]. After its initial use for pulmonary tuberculosis, toxicity associated with that treatment was observed, and the treatment was switched to the less toxic gold(I) thiolate complexes. In the early 1930s, the French physician Jacque Forestier introduced the same thiolate complexes for the treatment of rheumatoid arthritis, a condition which he believed was related to tuberculosis [2].
Rheumatoid arthritis is an autoimmune inflammatory disease with the characteristic progressive erosion of the joints leading to their deformities, immobility and pain. While several new strategies for the treatment of this disease are currently under development, there is still no complete cure. The first gold(I) thiolates used to treat rheumatoid arthritis were aurothiomalate (ATM) and aurothioglucose (ATG) (Figure 1), administered by an intramuscular injection [5]. Although these gold complexes were rapidly cleared from plasma, much of the drug was still distributed to organs throughout the body. The highest concentration of gold accumulates in kidneys, resulting in nephrotoxicity [1]. For many decades, gold(I) thiolate complexes were considered the drug of choice for rheumatoid arthritis treatment. However, the toxic side effects observed with these complexes prompted the search for new, less toxic, therapeutic gold complexes.
Figure 1.

Chemical structures of gold(I) compounds for rheumatoid arthritis treatment.
The main requirement for a new gold-based drug, in addition to improved toxicological and pharmacological profile, is its capacity for oral administration in low doses on a daily basis [6]. This treatment modality would give stable serum levels of the active drug, an improved therapeutic response, and reduced toxicity. As a result of intense work on synthesis and evaluation of many new gold complexes, triethylphosphine complexes have optimal pharmacological activity in models of rheumatoid arthritis and one of these complexes, named auranofin, [tetra-O-acetyl-β-D- (glucopyranosyl)thio](triethylphosphine)gold(I) (Figure 1), was developed as a new drug for the rheumatoid arthritis treatment. Although having the advantage of being orally administered on a daily basis [7], the disadvantage of auranofin is its reduced effectiveness compared to either ATM or ATG.
Unfortunately, not all the patients benefit from gold treatment and for those who do respond most suffer from deleterious side-effects to varying extents. These side effects include dermatitis, diarrhea and problems with internal organs such as kidney and the lungs. For those reasons, gold(I) drugs are currently used more as a last-resort treatment for severe cases of rheumatoid arthritis [8]. Since the introduction of auranofin in 1985, there has been no new clinically approved gold drug for either rheumatoid arthritis or any other disease.
Meanwhile, a possible connection between rheumatoid arthritis and infection, together with the known therapeutic benefit of gold complexes, has prompted an exploration of their activities against other diseases. Since gold possesses a high degree of resistance to bacterial colonization, it became a material of choice for implants that are at risk of infection, such as the inner ear. Gold has a long tradition of use in this application and is considered to be a valuable metal in microsurgery of the ear.
In the last few decades, the properties of some gold compounds, such as ATM and ATG, have been evaluated against human immunodeficiency virus (HIV) agents for the treatment of AIDS [9,10]. Gold complexes have also been explored for effectiveness against acute forms of asthma (e.g. chronic corticosteroid-dependent asthma), pemphigus (an autoimmune disease of the skin), and arthritis. Additionally, current research has described promising results using gold complexes to treat malaria [11], Chagas disease [12], and cancer [13].
2. Gold complexes as potential anti-cancer drugs
One of the major public health problems worldwide is cancer. Annually, an estimated 10 million people worldwide are diagnosed with cancer, while approximately 6.2 million die from this disease [14,15]. Although the initiative to develop suitable chemotherapeutic agents is a focus of medical research, most of the currently available chemotherapeutic agents are lacking ability to selectively target cancer cells. Since gold is one of the oldest metals used in medicine and gold-containing or coordinated complexes are the most extensively investigated metal complexes [2], it is not surprising that interests for gold as a potential anti-cancer drug have dramatically increased.
2.1 Lesson learned from platinum (Pt) complexes
One of the first metal complexes used for cancer treatment is cisplatin (Figure 2) with its anti-cancer activity discovered in the 1960s [16]. Cisplatin is responsible for the cure of more than 90% of testicular cancer cases and plays a vital role in the treatment of other cancers such as ovarian, head and neck, bladder, cervical cancer, melanoma, and lymphomas [17].
Figure 2.

Chemical structures of platinum-based drugs, cisplatin, carboplatin, and oxaliplatin.
Cisplatin exerts its anti-cancer effect by interacting with DNA and forming adducts that interfere with transcription and replication, thereby triggering programmed cell death (apoptosis) [18]. The interactions of cisplatin with DNA have been extensively studied and it is now well-known that a Pt-GG intrastrand cross-link is the critical lesion that leads to cisplatin toxicity [19].
However, the effectiveness of cisplatin is hindered by the phenomenon of tumor resistance and toxic side effects. To reduce toxic side effects, an alternative approach to design a second-generation drug was undertaken. Since then, thousands of platinum complexes with various ligands have been synthesized and tested for their anti-tumor activities. These results showed that toxic side effects on tumor cells were directly related to the rate of ligand loss [19], indicating that beneficial effect of the drug is a result of platinum-ligand coordination. Even though over 28 new platinum(II) compounds have entered human clinical trials, only few of them, carboplatin, oxaliplatin (Figure 2), nedaplatin and lobaplatin, received approval and achieved routine clinical use [20]. However, all these platinum(II) drugs are active in the same range of tumors and are still administered intravenously [17]. As a result, their impact as anti-cancer drugs is still reduced by the significant side effects, such as gastrointestinal and hematological toxicity, and drug-resistance phenomena [20].
Regardless of the observed side effects, the successes achieved with cisplatin in cancer chemotherapy stimulated search for other coordinated metal complexes as potential anti-cancer agents. At that time, it was believed that metal complexes that bore chemically and structurally similarities to cisplatin, or with similar chemical coordination to that of cisplatin, would posses a mechanism of action resembling that of cisplatin [21]. However, metal centers are very important for the biological activity of various metal-containing proteins and enzymes, and metals in a defined coordinating position are often responsible for the activity of organic drugs. The classic example is again cisplatin, which exerts its anti-tumor activity by interacting with DNA [22], forming a unique lesion that has not been mimicked by any other organic drugs. Therefore, it is clear that a general metal-biomolecule interaction is critical for the activity of a metal-based drug as is the case for the platinum-DNA interaction observed with cisplatin.
On the other hand, the activity of metal complexes is not determined solely by the presence of the metal itself, especially when the metals exist in different oxidation states, thereby having rich coordination chemistry [23]. In that case, even subtle changes in the charge of a metal could result in a great change in the coordination structure of the metal complex, which would lead to dramatic alterations of the physicochemical and thus biological properties of this drug. Therefore, the activity of a metal complex depends not only on the metal itself, but also on its oxidation state, number and types of ligands bound, and the coordination geometry of the metal complex. Since many metal-based drugs act as pro-drugs that undergo ligand substitution and redox reactions before they reach their targets, it is important to recognize these processes and learn how to control them.
If the precise molecular structure of many metal-based drugs is not completely established, then the difficulty in understanding their mechanisms of action is further increased. However, even for some well-characterized compounds, it is still difficult to predict their biological activities and molecular targets. For example, gold(III) complexes were synthesized in a way to reproduce the main features of cisplatin, and were later found to have completely different molecular targets (as discussed below).
2.2 Gold(I) complexes
The known immunosuppressive and anti-inflammatory actions of anti-cancer drugs, such as 6-mercaptopurine and cyclophosphamide, have established at least in principle, a connection between anti-arthritic and anti-cancer therapies [24]. Importantly, one long-term study showed that risk of cancer for rheumatoid arthritis patients was not increased and even suggested that rheumatoid arthritis patients treated with gold-based drugs have lower malignancy rate [25].
The most common coordination for gold(I) complexes is linear two-coordinate, although trigonal or tetrahedral coordinations are also possible. Using auranofin as an example (Figure 1), the Au(I) atom is coordinated with the S atom on the glucopyranose ring and the P atom on a PEt3 group, and the S–Au–P angle is nearly linear. After the demonstration that auranofin presented an in vitro activity similar to or even greater than cisplatin [26,27], a series of gold(I) coordination complexes, including auranofin analogs, were evaluated for in vitro cytotoxic potency and in vivo anti-tumor activity [28]. Auranofin and a number of its analogs showed potent cytotoxic activity against melanoma and leukemia cell lines in vitro and anti-tumor activity against leukemia in vivo. However, these complexes were completely inactive against solid tumors [29]. The main observations from these experiments were that (i) lack of potency in vitro correlates well with lack of anti-tumor activity; (ii) potent cytotoxicity in vitro does not necessarily translate into anti-tumor activity in vivo; and (iii) in vivo anti-tumor activity is generally optimized by ligation of Au(I) with a substituted phosphine.
Based on that, a series of digold phosphine complexes, such as gold(I) 1,2-bis(diphenylphosphine)ethane (DPPE), was synthesized and found to confer in vitro cytotoxic activity especially in some cisplatin-resistant cell lines [30]. Surprisingly, mechanistic studies suggested that, in contrast to cisplatin, DNA was not the primary target for these gold(I) complexes and that their cytotoxicity was mediated by their ability to alter mitochondrial function and inhibit protein synthesis by interfering with DNA-protein interactions [2]. This could be explained by higher affinity of gold(I) toward so-called soft ligands with sulfur (e.g. thiolates) and phosphorus (e.g. phosphines), and the lower affinity for nitrogen and oxygen-containing ligands. Although these gold(I) complexes had marked cytotoxic and anti-tumor activity against P388 leukemia, they had limited activity against solid tumor models. These complexes never entered clinical trials, due to problems associated with cardiotoxicity highlighted during pre-clinical toxicology studies [31].
2.3 Gold(III) complexes
Since gold(III) complexes are highly reactive, they have not been as comprehensively investigated as gold(I) complexes. Considering their high redox potential and relatively poor stability, the use of gold(III) complexes as anti-cancer drugs under physiological conditions was questioned [32]. Under the reducing conditions characteristic for mammalian environment, gold(III) complexes could be easily reduced to gold(I) and metallic gold in vivo, which makes them less effective as drugs [33]. However, higher stability of gold(III) complexes could be achieved by using appropriate ligands, predominantly multidentate ligands such as polyamines, cyclams, terpyridines, phenanthrolines and damp (N-benzyl-N,N-dimethylamine). In recent years, this approach renewed the interest for gold(III) complexes as judged from the increased number of new gold(III) complexes synthesized by using better ligands that usually have nitrogen atoms as donor groups [34]. For example, the chelating effect and great electron-donating ability of dithiocarbamates induces a large stabilization of gold center in the 3+ oxidation state. Therefore, gold coordination by dithiocarbamates provides stability for these complexes in different solvents, which is important for their use in biological systems [33].
The interests for gold(III) complexes were further stimulated with the promising results of platinum(II) complexes against selected types of cancer. Being isoelectronic (d8) with platinum(II) and having tetracoordinate complexes with the same square-planar geometries as cisplatin [33], gold(III) and its complexes became the subject of increased anti-cancer research.
A series of gold(III) complexes with 2-[(dimethylamino)methyl]phenyl] (damp) ligand, synthesized in such a way to stabilize gold in its 3+ oxidation state, showed some promising results. Their cytotoxic effects against several human cancer cell lines were comparable to, or greater than cisplatin. More importantly, these complexes retained cytotoxic activity even against cisplatin-resistant cell lines [31].
Another class of gold(III) complexes, cycloaurated N,N-dimethylbenzylamine complexes, was synthesized by Dinger and Henderson using either diphenylurea [35] or salicylate and thiosalicylate [36] as the dianionic ligands. All these complexes demonstrated cytotoxic activity in vitro, with the methoxy substituted N,N-dimethylbenzylamine cyclometallated gold(III) thiosalicylate complex being the most potent. However, their effect was evaluated only against the P388 leukemia cell line, and their in vivo anti-tumor potential needs further investigation.
Recently, in vitro activities of a series of gold(III) complexes, [Au(en)2]Cl3, [Au(dien)Cl]Cl2, [Au(cyclam)](ClO4)Cl2, [Au(terpy)Cl]Cl2, and [Au(phen)Cl2]Cl, against the A2780 ovarian cancer cell line and a cisplatin-resistant variant were described [37,38]. The relative order of cytotoxicity was: Au(terpy) ≫ Au(phen) > Au(en), Au(dien) ≫ Au(cyclam). Although the coordination mode of gold is the same in all the complexes, the geometry of the used ligands is quite different since only the terpy and phen ligands have planar structures. Interestingly, the three most active complexes retained activity against the cisplatin-resistant cell line [37,38].
Some of the other gold(III) complexes were synthesized and evaluated by numerous groups including those led by Calamai and Bruni. Calamai et al tested a group of square planar gold(III) complexes containing at least two gold-chlorine bonds in cis-position [21], while Bruni et al synthesized four gold(III) complexes: trichloro(2-pyridylmethanol) gold(III) [AuCl3(Hpm)], dichloro(N-ethylsalicylaldiminato) gold(III) [AuCl2(esal)], trichlorodiethyl-endiamine gold(III) [AuCl(dien)] Cl2, and trichlorobisethylendiamine gold(III) [Au(en)2] Cl3 [39]. All these complexes showed significant cytotoxic effects against the A2780 human ovarian cancer cell line, comparable to or even greater than cisplatin, and were able to overcome resistance to cisplatin to a large extent [39].
A number of other gold(III) complexes was synthesized by different groups and their anti-proliferative and pro-apoptotic effects were demonstrated in vitro against leukemia cells including those resistant to cisplatin [21,40], cisplatin-resistant ovarian cancer cells [21,37], and cervical cancer HeLa cells [41,42]. Buckley et al synthesized a series of five gold(III) complexes with damp ligand and tested them against a panel of cancer cell lines in vitro and in vivo. Importantly, with some of these complexes, non-cross-resistance to cisplatin was observed in an acquired cisplatin-resistant ovarian cancer cell line in vitro, and significant tumor growth inhibition was found in mice with either human bladder or ovarian cancer xenografts [43].
Results from cellular and molecular studies with gold(III) (damp) complexes further supported the previously proposed concept of different anti-cancer mechanisms utilized by gold complexes compared to cisplatin. These complexes were equally potent against cisplatin-sensitive and cisplatin-resistant ovarian cancer cells and showed only minimal activity against ADJ/PC6 murine tumor that is highly sensitive to cisplatin [31]. Taken together, all these studies imply that different mechanisms are employed by platinum(II) and gold(III) coordinated complexes in exerting cytotoxic effects against cancer cells in vitro and in vivo.
2.4 Search for molecular targets of gold complexes in cancer: DNA or proteins
The big successes of cisplatin in chemotherapy on one side, and toxic side effects related to cisplatin treatment on the other side, each in its own way stimulated the search for new and more selective metal-based anti-cancer drugs. Since gold(III) is isoelectronic (d8) with platinum(II) and tetracoordinate gold(III) complexes have the same square-planar geometries as cisplatin [33], the potential anti-cancer activity of gold(III) complexes was investigated.
Unlike cisplatin, the main biological targets for gold complexes and mechanisms responsible for gold–induced cytotoxicity are still mainly unknown. Considering that some gold drugs have already been in use for decades and that many gold complexes have been investigated for their potential use in medicine, it is surprising that their mechanisms of action are still not well understood. Gold(I) drugs are in fact pro-drugs that are rapidly metabolized in the patients generating the pharmacologically active species coordinated to a biologically relevant thiol [8]. In the case of auranofin, the metabolic pathway most likely involves Au-S bond cleavage and coordination of the albumin from the blood [8]. Subsequent metabolism probably involves reduction of the original albumin, the coordination of another thiol (e.g., from albumin or glutathione), and oxidation of the phosphine with Au-P bond cleavage. This results in the gold being embedded in a ball of protein that is delivered to the site of inflammation [8].
A number of mechanistic studies with auranofin identified several potential biological targets important for its anti-inflammatory and anti-cancer activities. One of the first studies suggested that auranofin inhibits DNA synthesis, RNA synthesis, and protein synthesis [44], while later studies identified the ability of auranofin to generate reactive oxygen species (ROS) in leukemia and ovarian cancer cells [45,46]. Auranofin also inhibits mitochondrial thioredoxin reductase (TrxR) [47] and cathepsin B [48]. Recently, Rigobello et al demonstrated that auranofin, in the presence of calcium ions, induces mitochondrial swelling, decreases mitochondrial membrane potential, and stimulates respiration, a process dependent on the permeability transition of mitochondrial membrane [49]. Several other groups have also suggested that mitochondria are primary target for gold(I) phosphine complexes [13,50-53] and Rackham et al even demonstrated that bis-chelated gold(I) phosphine complexes, by targeting mitochondria, selectively induced apoptosis in breast cancer but not normal cells [54].
In addition to studies on mitochondria, the most recent observatios of Kim et al suggested that the anti-inflammatory effect of auranofin is associated with inhibition of Janus family of tyrosine kinase/signal transducer and activator of transcription ((JAK1/STAT3) signalling pathway [55]. By blocking this important pathway, auranofin interferes with interleukin-6 (IL-6) signalling that regulates inflammation, proliferation, and differentiation in various cell types [56]. Constitutively activated STAT signalling, one of significant regulators of apoptosis, cell cycle progression, and angiogenesis, was found in a number of leukemias and solid tumors, directly contributing to cancer development and progression [55].
Since gold(III) complexes were synthesized in such a way to closely resemble the structure of cisplatin, they were expected to have the same molecular target - DNA. Therefore, the probable binding mode of gold(III) to DNA has been modeled by thorough crystallographic and spectroscopic investigations of gold(III) complexes with nucleosides and nucleotides [57]. In addition, a number of studies based on different physicochemical techniques were performed and suggested that probable binding sites for gold(III) are N(1)/N(7) atoms of adenosine, N(7) or C(6)O of guanosine, N(3) of cytidine, and N(3) of thymidine, which are analogous to the possible binding sites for the isoelectronic platinum(II) ion [58]. However, later studies showed that the in vitro interactions of some gold(III) complexes with calf thymus DNA are weak, whereas significant binding to model proteins takes place. This implies that their mechanism of action is substantially different from that of the clinically established Pt(II) compounds and from Pt(II) complexes with the same ligands [59].
Although one recent study showed that DNA binding affinity of four new gold(III)-terpyridine complexes correlated to their cytotoxicity in a variety of cancer cell lines [60] suggesting that intracellular DNA is their primary target in vitro, a number of other studies support the concept that proteins, rather than DNA, are mainly targeted by gold complexes.
One such a study was conducted by Fricker et al who demonstrated a clear preference of a gold(III)-damp complex for S-donor ligands such as glutathione and cysteine, with only limited reactivity against nucleosides and their bases [29]. This group proposed a new mechanism by which proteins containing exposed cysteine residues might be proper targets for that class of gold(III) complexes. Gold(III) complexes cleave the disulfide bond of cystine [61-64], and oxidize methionine [65-67] and glycine [68], suggesting the possibility that amino terminus of peptides and proteins could be deaminated by gold(III). Gold(III) complexes also interact with bovine serum albumin [69], making very stable adducts that, once formed, were destroyed only by the addition of strong ligands for gold(III) such as cyanide [70]. Based on these observations, it has been proposed that selective modification of surface protein residues by gold(III) complexes in a defined coordination geometry could be the molecular basis for their biological effects. This has prompted a new search for gold-protein interactions to identify possible targets responsible for the biological effects of gold complexes.
Similarly to gold(I) complexes, thioredoxin reductase (TrxR) is also a target for various gold(III) complexes with none, one, two or three coordinated carbon-gold bonds [71]. Importantly, TrxR inhibition by these complexes did not correlate with their potency to kill MCF-7 breast cancer cells in vitro. Moreover, although one of these complexes was able to inhibit colony formation by MCF-7 cells, it was completely inactive against MCF-7 breast cancer and HT-29 colon cancer xenografts in SCID mice [71].
Recently, Wang et al demonstrated the role of mitochondria in apoptotic cell death induced by gold(III) porphyrin 1a in human nasopharyngeal carcinoma cell lines [72]. The treatment was followed by depletion of mitochondrial membrane potential, alterations of Bcl-2 family proteins, release of cytochrome c and apoptosis-inducing factor (AIF), ROS production and apoptosis induction. By conducting functional proteomic studies, the same group also identified several clusters of altered proteins, following treatment with gold(III) porphyrin 1a [73]. Some of these proteins include cellular structure and stress-related chaperones, proteins involved in ROS, translation factors, and proteins that mediate cell proliferation and differentiation. Taken together, it is likely that multiple gold targets are involved in drug cytotoxicity that eventually leads to apoptotic cell death.
Most recently, Saggioro et al have confirmed that gold(III) dithiocarbamates exert their cytotoxic effects, at least partly, by stimulating production of ROS [74]. These gold(III) dithiocarbamates also modify some mitochondrial functions, inactivate both cytosolic and mitochondrial TrxR, and interfere with extracellular signal-regulated kinases (ERK) pathway, inducing cancer cell death through both apoptotic and non-apoptotic mechanisms [74]. Additionally, our recent study suggests that the tumor proteasome is one of the important targets of these gold(III) complexes in human breast cancer cells in vitro and in vivo (see below).
3. Targeting the ubiquitin-proteasome pathway by novel gold(III) complexes for breast cancer treatment
3.1 Breast cancer - overview
Breast cancer is a major health problem worldwide. By affecting one in 8 to 12 women, breast cancer is the most frequent form of cancer in women [75] and according to recent statistics, only in 2002 an estimated 1.15 million new cases were diagnosed [76]. Breast cancer is also the primary cause of cancer death among women globally, and was responsible for about 375,000 deaths in the year of 2000 [77].
Although the prognosis from breast cancer is generally good, it still remains the leading cause of cancer mortality in women (and ranks as the fifth cause of death from cancer overall). It has been estimated that about 1.5% of the US female population are breast cancer survivors [76] and that the higher survival rate of breast cancer is partly due to early detection, and advanced screening programs and techniques.
After a suspicious lesion is identified by mammography or examination, the patient's complete history and physical condition are assessed. When breast cancer is confirmed, the standard protocol is surgery (mastectomy or lumpectomy) that is usually followed by postoperative adjuvant therapy. Mastectomy removes all the breast tissue, while lumpectomy removes the tumor and some tissue surrounding the tumor and is followed by postoperative radiation therapy. Radiation therapy is used to “sterilize” the remaining breast by destroying cells through production and release of free radicals.
Frequently, adjuvant chemotherapy is also applied and it provides a 20-30% improvement in long term survival. The type of adjuvant chemotherapy is also determined based on the status of clinical markers in removed tumors. These markers include the status of estrogen and progesterone receptors, ploidy, HER/2neu, erbB-2, and MIB status. The most common regiments for breast cancer chemotherapy include CMF (cyclophosphamide, methotrexate and 5-fluorouracil), CAF (cyclophosphamide, doxorubicin, and fluorouracil), and AC (doxorubicin, cyclophosphamide).
Although adjuvant chemotherapy therapy increases survival rate, it is commonly associated with toxic side effects including pain, diarrhea, constipation, mouth sores, hair loss, nausea and vomiting, as well as blood-related side effects. Therefore, there is still a need for new and improved chemotherapeutics with reduced or no toxic side-effects. One of the promising aproaches to treat cancer is focused on targeting the ubiquitin-proteasome pathway. Since aberant proteasome-dependent proteolysis is associated with the development and progression of some types of cancer, the use of proteasome inhibitors might prove useful as an anti-cancer tool.
3.2 The ubiquitin-proteasome pathway
The ubiquitin-proteasome pathway (Figure 3) is the predominant means of proteolysis in eukaryotic cells. It plays a major role in the degradation of proteins that regulate cell cycle progression, proliferation, and apoptosis and abnormal proteins that result from oxidative damage and mutations. By having these important functions, the ubiquitin-proteasome pathway is essential for controlling intracellular levels of myriad proteins and maintaining normal cellular function.
Figure 3.
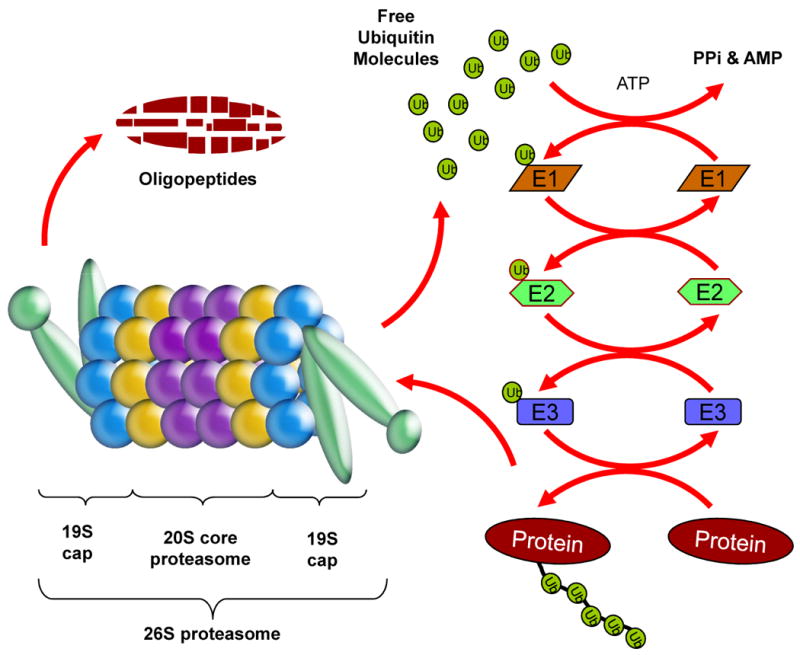
The ubiquitin-proteasome pathway. A target protein degraded by the ubiquitin-proteasome pathway is first covalently modified by multiple ubiquitin (Ub) molecules in a three-stepped, highly regulated enzymatic process involving an Ub-activating (E1), Ub-conjugating (E2), and the Ub-ligating (E3) enzymes. The ubiquitinated protein is then escorted to the 26S proteasome, recognized by the 19S cap, de-ubiquitinated and then degraded by the catalytic 20S core into oligopeptides. The ubiquitin molecules are released and recycled.
Two distinct steps of the ubiquitin-proteasome pathway are: (i) the covalent attachment of multiple ubiquitin molecules to a protein substrate and (ii) degradation of the polyubiquitylated protein by the 26S proteasome. With few exceptions, only proteins containing polyubiquitin chains on sequential lysine residues are recognized and degraded by the proteasome. The best known ubiquitin-independent proteasome substrate is the enzyme ornithine decarboxylase [78]. Although subject to ubiquitin-dependent degradation, cell cycle regulators p53 and p21 are also degraded via ubiquitin-independent mechanism [79]. Finally, under conditions of cellular stress, many structurally abnormal, misfolded, or highly oxidized proteins could also be degraded by the ubiquitin-independent proteasomal degradation [80].
Ubiquitin is a highly conserved 76-amino acid protein that gets covalently attached to a target protein by a sequential activity of Ub-activating (E1), Ub-conjugating (E2), and the Ub-ligating (E3) enzymes. The attachment of an ubiquitin molecule is a three-step process that starts with ubiquitin activation by the E1 enzyme in an ATP-dependent reaction that generates a high-energy thiol ester intermediate, E1-S∼ubiquitin. Activated ubiquitin is then transferred from E1, by one of several ubiquitin-conjugating enzymes, E2, via an additional high-energy thiol ester intermediate, E2-S∼ubiquitin. From E2- to the E3-bound substrate, the activated ubiquitin can be then transferred directly or via a third high-energy thiol ester intermediate, E3-S∼ubiquitin [81]. Once multiple ubiquitin molecules are attached to the target protein, it is escorted to the proteasome for degradation (Figure 3).
The proteasome is a large multi-subunit protease complex localized in the nucleus and cytosol where it selectively degrades intracellular proteins. The typical and most studied proteasome is the 26S proteasome that contains at least 44 polypeptides and possesses a molecular weight of about 2,400 kDa [82]. It is composed of the 20S catalytic core and two 19S regulatory particles, located on each side of the catalytic core (Figure 3) [83-86]. The role of 19S particles is to recognize ubiquitinated proteins, and regulate protein entry to the 20S core. In some immunoproteasomes, an 11S particle with a heteroheptameric ring structure can bind to each end of the 20S core, acting as a potent activator of the proteasome [87-89].
The eukaryotic 20S proteasome is a barrel-shaped structure with the primary sites of proteolytic activities located on the inside of the barrel. It consists of 28 different individual subunits, 14 alpha subunits and 14 beta subunits [90-92] arranged in four heptameric, tightly stacked rings (7α, 7β, 7β, 7α) to form a cylindrical structure (Figure 3) [93]. Two outer rings are composed of the α-subunits while two inner rings contain the β-subunits (Figure 3). The α-rings guard the entrance to the active site of the complex by allowing access only to unfolded and extended substrate polypeptides. The proteolytic activities are confined to the β-rings that confer the unique and distinguishing proteasome feature of multiple peptidase activities. Some of these activities include chymotrypsin-like (cleavage after hydrophobic residues, mediated by the β5 subunit), peptidylglutamyl peptide hydrolyzing-like or PGPH-like (cleavage after acidic residues, mediated by the β1 subunit), and trypsin-like (cleavage after basic residues, mediated by the β2 subunit) activities [93].
A common component of most proteases, including the proteasome, is the presence of a catalytic triad that contains three interacting amino acids, one of which is responsible for cleaving the substrate. The primary residue in the proteasome responsible for substrate peptide cleavage is the amino (N)-terminal threonine [90]. The oxygen on the side chain of this N-terminal threonine is nucleophilic by nature, and its nucleophilicity seems to be enhanced by surrounding amino acids of the catalytic triad. This threonine, present in all three catalytically active beta subunits, is responsible for catalysis and does so through nucleophilic attack [90]. Several binding pockets, identified near N-terminal threonine, recognize the side chains of peptides giving each catalytic site its specific activity. Based on specificity of residues for these binding pockets, several proteasome inhibitors have been designed [82-83,94] and played an essential role in studying the functions of the proteasome [85,95-98].
Proteasome inhibitors can mechanistically work in several ways [82]. One of the most common mechanisms requires binding to the proteasomal binding pockets, which then prevents substrate binding. Another frequently observed mechanism involves inactivation of the primary catalytic amino acid residue, the N-terminal threonine. Many of the most potent proteasome inhibitors combine both of these aspects of inhibition, by binding the proteasome active site tightly and modifying the N-terminal threonine residue. Some of those proteasome inhibitors include Ac-LLnL, with an acetyl group on its N-terminus that can form a hemi(thio)acetal with the proteasomal N-terminal threonine [82] and ester bond-containing proteasome inhibitors, such as lactacystin [99] (Figure 4) and green tea polyphenols [100] (epigallocatechin gallate or EGCG, Figure 4), which can both inhibit the proteasome via covalent interaction with the N-terminal threonine.
Figure 4.

The chemical structures of naturally occurring and synthetic proteasome inhibitors are depicted. Lactacystin is a natural product derived from a microbial metabolite, EGCG is green tea polyphenol, and bortezomib is a synthetic dipeptide boronic acid analogue.
After the observations that aberrant proteasome-dependent proteolysis might be associated with the pathophysiology of some malignancies, proteasome inhibitors were suggested as a novel class of anti-cancer drugs. Increased proliferation of cancer cells is associated with higher rate of synthesis and degradation of many regulatory proteins. Since the key mechanism for proteolytic degradation in the cells is the ubiquitin-proteasome pathway, the increased activity of this pathway in cancer was anticipated. Indeed, higher proteasome activity is associated with more malignant and aggressive carcinomas, including colorectal carcinoma [101], leukemia [102] and prostate cancer [103]. As a result, cancer cells were expected to be more dependent upon proteasomal activity and more sensitive to its inhibition.
In recent years, this higher sensitivity of cancer cells, compared to their normal counterparts was undoubtedly confirmed by many groups [96,104-105]. A number of studies have demonstrated that inhibition of the ubiquitin-proteasome pathway in cancer cells results in accumulation of tumor suppressor and pro-apoptotic proteins, followed by cell cycle arrest and apoptosis [106]. Since activation of cellular apoptosis is a current strategy for the treatment of human cancer, the use of proteasome inhibitors that could selectively induce apoptosis in cancer but not normal cells was proposed. More importantly, proteasome inhibitors have better apoptosis-inducing potency compared to current anti-cancer drugs, when tested in various human cancer cell lines [96-98]. Some studies even indicate that proteasome inhibitors are able not only to overcome cancer cell resistance to cytotoxic therapies, but also to selectively target transformed and cancer, but not normal, human cells [107-110].
The possibility of targeting the ubiquitin-proteasome pathway therapeutically was questioned mainly because this pathway plays an important role in normal cellular homeostasis, as well. However, many groups have shown that this pathway is more active in cancer cells, causing them to be more dependent upon proteasomal activity and more sensitive to proteasome inhibitors [96,104-105]. These observations provide a strong rationale for using proteasome inhibitors in anti-cancer treatment and suggest that the possibility of targeting the ubiquitin-proteasome pathway in cancer therapy is a viable option.
The first naturally occurring proteasome inhibitor described to effectively induce apoptosis [111] was lactacystin, derived from a microbial metabolite [112]. However, lack of specificity or potency led to the development of synthetic proteasome inhibitors. A number of studies have shown that newly synthesized proteasome inhibitors are active in tumor cells inducing cell growth arrest and/or apoptosis [96-98]. As a logical expansion of these studies, some of these inhibitors were tested in in vivo human xenograft animal models [113-117]. These studies showed that proteasome inhibitors could reach the proteasome in vivo and be sustained at therapeutic levels in mice. It should be noted that tumor growth regression induced by proteasome inhibitors was accomplished without measurable toxicity to the host. Based on these promising data, some proteasome inhibitors were considered for clinical trials in human patients and one of them, bortezomib (Figure 4), received regular approval from the U.S. Food and Drug Administration (FDA) for multiple myeloma treatment.
3.3 Targeting ubiquitin-proteasome pathway in breast cancer
Although proteasome inhibitors were initially synthesized to study function of the proteasome, after the pro-apoptotic effect of these inhibitors was demonstrated in a number of cancer cell lines, they were proposed to be developed as a new class of anti-cancer drugs. Results from preclinical studies suggest that proteasome inhibitors might have potential for efficient breast cancer treatment. By targeting several major pathways of importance for breast cancer growth and progression, proteasome inhibitors could help overcome breast cancer resistance to chemotherapy. One of these pathways, the one mediated by transcription factor NF-κB and regulated by the ubiquitin-proteasome pathway is frequently elevated in breast cancer. By blocking this pathway, through accumulation of NF-κB inhibitor - IκB, proteasome inhibitors would be able to stop cancer cell proliferation and stimulate the apoptotic signaling pathway, leading to cancer cell death, which is the ultimate goal of anti-cancer therapy.
Proteasome inhibitors might also be used in combination with other chemotherapeutic drugs and help overcome resistance of cancer cells to chemotherapeutic treatment. For example, proteasome has a critical role in maturation of P-glycoprotein, a membrane pump that, among others, promotes the efflux of chemotherapeutic drugs [118]. By inhibiting the proteasome, P-glycoprotein levels in cancer cells membranes will decrease, allowing for accumulation of the used chemotherapeutic and consequently increasing the drug's effectiveness [118].
Targeting the ubiquitin-proteasome pathway might have already played an important role in breast cancer treatment with anthracyclines. One drug from this group, doxorubicin (Adriamycin) (Table 1), binds the 20S proteasome, which then translocates to the nucleus. 20S proteasome is thereby acting as a carrier for doxorubicin and helping doxorubicin exerting its cytotoxic effects [119]. Several other drugs (Table 1), although originally aimed at different targets, also have a potential to affect either ubiquitination or proteasomal degradation in a direct or indirect manner. One drug that has an indirect effect on the ubiquitin-proteasome pathway is arsenic trioxide. By preventing IκB phosphorylation and its subsequent degradation, arsenic indirectly inhibits NF-κB activation [120], having an anti-proliferative and pro-apoptotic effect (Table 1). Since arsenic trioxide also inhibits expression and signaling through the estrogen receptor pathway [121], its possible application in breast cancer treatment alone or in combination with other agents has been evaluated in several clinical trials [118].
Table 1. Various drugs with the effect on the ubiquitin-proteasome pathway.
| Drug name | Effect on the ubiquitin-proteasome pathway | Side effects | Major application |
|---|---|---|---|
| Doxorubicin | Translocation of 20S proteasome to the nucleus | Cardiotoxicity, neutropenia, alopecia | Hematological malignancies, carcinomas, sarcomas |
| Arsenic trioxide | Inhibition of ubiquitination by affecting IκB kinase | Overall toxicity | Leukemia, autoimmune diseases |
| Cyclosporine A | Uncompetitive inhibition of proteasomal CT-like activity | Nephropathy | Solid organ and bone marrow transplantation |
| Rapamycin | Inhibition of proteasome activator PA28 | Impaired wound healing | Kidney transplantation |
| Anti-retroviral agents | Inhibition of proteasomal CT-like and T-like activities | Various, mild to serious | Viral infections, (including HIV) |
| Lovastatin | Mechanism unknown, but structurally related to lactacysin | Myopathy, rhabdomyolysis | Hypercholesterolemia treatment |
| Vinca alkaloids | Inhibition of proteasomal CT-like, T-like and PGPH-like activities | Neurotoxicity, myelosuppression | Various cancers |
| Bortezomib | Inhibition of proteasomal CT-like and T-like activities | Nausea, diarrhea, peripheral neuropathy | Multuple myeloma |
Some immunosuppressive agents (Table 1) also inhibit the ubiquitin-proteasome pathway and were evaluated for breast cancer treatment. One of them, cyclosporine A is an uncompetitive proteasome inhibitor which is used in the breast cancer treatment mostly to block drug resistance [118]. Another immunosuppressive agent, rapamycin, inhibits proteasomal function indirectly by inhibiting expression of the proteasome activator PA28 [122] (Table 1). Given that rapamycin also prevents estrogen-mediated G1 to S phase progression of breast cancer cells [123], its potential use in breast cancer treatment should be further investigated.
Other chemotherapeutic agents that were not originally discovered as proteasome inhibitors but have an effect on the proteasomal activity include tannic acid [124], anti-retroviral agents [125], lipid-lowering dug lovastatin [126], vinblastine and vincristine (Table 1) [125]. Possible application of these agents against breast cancer has not been evaluated, yet. However, vinca alkaloid vinorelbine (Naelbine®) has been successfully used for breast cancer treatment for over a decade [127] (Table 1). It is still not clear how much vinorelbine's anti-cancer effect is attributed to its proteasome-inhibitory activity.
3.3.1 Bortezomib: promises and limitations
Bortezomib (Velcade/PS-341; Figure 4, Table 1) is the most popular and best known proteasome inhibitor. It is a dipeptide boronic acid analogue with cell death-inducing activity found in several cancer cell lines and animal models, including breast [128], prostate [113], ovarian [129], lung [130], and colorectal cancer [131], and non-Hodgkins lymphoma [132]. Bortezomib works through inhibition of the β5 (chymotrypsin-like) and β1 (PGPH-like) subunits with the β5 subunit as the predominant cell-death inducing target [133]. Treatment with bortezomib effectively blocks NFκB activation, which is mostly responsible for clinically observed chemoresistance [134]. However, several lines of evidence indicate that, although important, the down-regulation of NFκB activity is not the only mechanism responsible for the activity of proteasome inhibitors. Since the proteasome degrades almost all endogenous proteins, proteasome inhibitors induce up-regulation of different proteins, including transcription factors that stimulate proliferation. When the cell is exposed to these contradictory signals, it is believed that the conflicting situation is resolved by initiation of apoptotic pathways [135].
With the demonstration that it was well-tolerated and had activity in models of human malignancies in vivo [116], the proteasome inhibitor bortezomib was introduced into Phase I safety trials [113]. The data from the bortezomib trials showed acceptable toxicity with significant clinical benefit [136] and as a result, in the year of 2003, bortezomib (Velcade®, of Millennium Pharmaceuticals, Inc., Takeda Oncology Company, and Johnson and Johnson Pharmaceutical Research and Development) became the first proteasome inhibitor to receive a regulatory approval from the U.S. FDA for the treatment of more refractory multiple myeloma [137]. Two years later, bortezomib was also approved for the treatment of patients with multiple myeloma who had received at least one prior therapy. Most recently, on June 20, 2008, bortezomib received the U.S. FDA approval for injection for the treatment of patients with multiple myeloma. This approval results from a clinical trial using bortezomib as an initial treatment for patients with multiple myeloma.
Although bortezomib appears to exhibit the most substantial potency against multiple myeloma and non-Hodgkin lymphoma [138], other hematological aberrations and solid tumors are being investigated as potential candidates for proteasome inhibition [139-140]. In a phase II study with twelve metastatic breast cancer patients treated with bortezomib, no objective responses were observed, since the disease remained stable only in one patient and progressed in the remaining eleven [141]. Similar results were reported from another phase II study conducted on the same number of patients with metastatic breast cancer [142]. Since all of the twelve patients experienced disease progression while treated with bortezomib, the study was ended after the first stage. Therefore, it was suggested that for breast cancer treatment in future, bortezomib should be combined with other conventional chemotherapeutics [143].
Indeed, an additive effect has been observed in cancer cells treated with combination of bortezomib and traditional chemotherapeutics and, in some cases, drug resistance is overcome [110,131,144]. When used in combination with trastuzumab, in different regimen combinations, bortezomib induced apoptosis and necrosis in various breast cancer cell lines and has been currently evaluated in phase I trials [145]. Most recently, a phase I/II clinical trial with 35 metastatic breast cancer patients was conducted to evaluate the combination of bortezomib and capecitabine in anthracycline-pretreated and/or taxane-pretreated patients. Although this combination treatment was well tolerated, it had only moderate anti-tumor activity since the overall response rate was 15% while 27% patients had stable disease [146]. Several Phase I clinical trials with bortezomib and various conventional agents showed promising results for breast cancer treatment. Some of these trials include studies with bortezomib and Doxil® (liposomal doxorubicin) conducted at the University of North Carolina at Chapel Hill [118]; bortezomib and doxorubicin at the University of Wisconsin [147]; bortezomib with 5-fluorouracil [148]; bortezomib with irinotecan [149], and bortezomib with gemcitabine [150]. Therefore, the anti-tumor activity of bortezomib has been examined not only as a mono-therapy but also as a sensitizer in combination with various cytotoxic agents in multiple clinical trials.
Unfortunately, bortezomib treatment is sometimes associated with toxic side effects, the most frequent of which include nausea, fatigue, and diarrhea [151-152]. More adverse events include thrombocytopenia, peripheral neuropathy, neutropenia, lymphopenia, and hyponatremia. Thrombocytopenia was observed to dissipate during the rest phase of treatments. However, individuals that started bortezomib treatment with low baseline platelet counts were at the greatest risk of developing clinically significant thrombocytopenia [151]. Peripheral neuropathy led to the highest proportion of discontinuations in phase II trials [151-152]. While baseline symptoms of peripheral neuropathy were often present and were attributed to prior treatment with neurotoxic agents, neuropathy was improved or was resolved in the majority of patients upon discontinuation of bortezomib [151]. Therefore, there is still a need for new and improved proteasome inhibitors with less or no toxic side-effects.
3.3.2 Gold(III) complexes inhibit the proteasome in breast cancer cells in vitro and in vivo
To obtain gold (III) complexes with superior chemotherapeutic index in terms of increased bioavailability, higher cytotoxicity, and lower side effects than cisplatin, Ronconi et al synthesized new gold(III) dithiocarbamate derivatives with defined coordination geometry [32]. The choice of dithiocarbamate ligands was not accidental; they were being evaluated for their efficacy as inhibitors of cisplatin-induced nephrotoxicity without decreasing its anti-tumor activity [153-155].
The synthesized gold complexes were tested for their in vitro cytotoxic activity toward a panel of human tumor cell lines. Remarkably, most of them, in particular gold(III) derivatives of N,N-dimethyldithiocarbamate and ethylsarcosinedithiocarbamate, (Compounds 1 – 4; Figure 5), were shown to be 1- to 4-fold more cytotoxic than cisplatin and to overcome to a large extent both intrinsic and acquired resistance to cisplatin.
Figure 5.

Chemical structures of gold(III) dithiocarbamate complexes, Compounds 1-4.
Coordination of all of these derivatives takes place in a near square-planar geometry through the sulfur-donating atoms, the –NCSS group coordinating the metal center in a bidentate symmetrical mode and lying in the same plane [33]. Moreover, the remaining coordination positions are occupied by two cis-gold(III)-halogen bonds that may undergo easy hydrolysis [33]. These observations further supported the idea that the coordinated gold(III) dithiocarbamates have different molecular targets than cisplatin. The key proteins that are modified by gold(III) complexes and responsible for triggering apoptosis remained still unknown.
In our previous work, we investigated the molecular mechanism responsible for gold(III) dithiocarbamate-mediated anti-cancer activity [156]. We first tested the effect of selected gold(III) dithiocarbamate complex, compound 2 (Figure 5) or Au(DMDT)Br2, on proliferation of different breast epithelial cell lines, including pre-malignant MCF10K.cl2 cells and malignant MCF10dcis.com, estrogen receptor α-positive MCF7, and estrogen receptor α-negative MDA-MB-231 cancer cell lines. We found that 24 hour treatment with compound 2 inhibited proliferation of all four tested cell lines in a dose-dependent manner. Also, the effect of compound 2 was much stronger than the effect of cisplatin under the same experimental conditions. Compound 2 at 5 μM inhibited 85% of MDA-MB-231 cell proliferation, compared to less than 20% inhibition by 5 μM of cisplatin. Even when 50 μM of cisplatin was used, only ∼40% inhibition was observed. Moreover, compound 2 at 5 μM for 2 h induced apoptotic morphological changes in MDA-MB-231 cells, while cisplatin at 50 μM for 48 h did not induce such changes [156]. We selected highly malignant, metastatic and invasive breast cancer MDA-MB-231 cell line for further investigation. It is important to note that anti-proliferative effect was attributed strictly to the gold(III) coordinated complex, compound 2, while neither ligand DMDT nor gold salt KAuBr4 alone had any effect on proliferation of MDA-MB-231 cells.
Our data are consistent with previous implications that the mechanism of action utilized by gold complexes is substantially different from that of the cisplatin. Moreover, it has been proposed that selective modification of surface protein residues by gold(III) complexes could be the molecular basis for their biological effects. Since we have previously shown that some copper complexes were capable of irreversible inhibition of the proteasome in time- and concentration-dependent manner under in vitro conditions [157-158], we hypothesized that gold and copper complexes might use the same mechanism against cancer cells. We found that four tested gold(III) dithiocarbamates inhibited the proteasomal chymotrypsin-like activity in MDA-MB-231 whole cell extract in a concentration-dependent manner. To provide direct evidence for proteasome inhibition by gold compounds, we performed a cell-free proteasome activity assay using purified 20S proteasome and compound 2. We discovered that compound 2 significantly inhibited all three activities of the purified 20S proteasome, especially its chymotrypsin-like activity [156]. This observation is particularly important since inhibition of the proteasomal chymotrypsin-like activity is associated with growth arrest and/or apoptosis induction in cancer cells [159,107].
After we demonstrated that compound 2 could inhibit the purified proteasomal chymotrypsin-like activity, we then tested its effect in intact MDA-MB-231 cells and found similar inhibitory effects. Proteasomal inhibition by compound 2 was confirmed by decreased proteasomal activity and increased levels of ubiquitinated proteins and the proteasome target protein p27. Most importantly, inhibition of the proteasome activity and accumulation of p27 were also found in MDA-MB-231 xenografts treated with compound 2 [156]. All together, these observations clearly indicate that compound 2 can directly target the tumor proteasome in vivo.
It seems that coordination mode of gold is an important factor for proteasome-inhibitory effect of gold complexes since gold(III) dithiocarbamate compound 3 had much stronger inhibitory effect on the purified and tumor proteasome compared to its gold(I) analog [(ESDT)Au]2 (our unpublished data). Gold(III) dithiocarbamate compound 3 has a near-square-planar geometry through the sulfur-donating atoms; the NCSS group coordinates the gold center in a bidentate symmetrical mode, while the two remaining coordination positions are occupied by two cis-gold(III)-chlorine bonds. However, in gold(I) derivative [(ESDT)Au]2, each ligand binds the two gold centers in a bidentate symmetrical mode, leading to formation of binuclear cyclic complex with a characteristic S-Au-S linear structure [32]. This different coordination mode of gold apparently has a huge impact on the biological effects of gold(III) versus gold(I) complexes.
3.3.3 Gold(III) complexes induce apoptosis in human breast cancer cells in vitro and in vivo
Various proteasome inhibitors potently induce tumor cell apoptosis [96,104-105,107,159]. Therefore, we investigated if gold(III) compound 2 behaved similarly in vitro and in vivo. Indeed, we found that inhibition of the proteasomal chymotrypsin-like activity by compound 2 induced apoptosis in cultured MDA-MB-231 breast cancer cells and in the tumors developed from the same breast cancer cell line grown in nude mice. Induction of apoptosis by compound 2 in vitro and in vivo was confirmed by multiple assays that measure characteristic cellular and biochemical hallmarkers. For example, apoptotic morphological changes, the presence of apoptotic nuclei, and apoptosis-specific poly(ADP-ribose) polymerase (PARP) cleavage were observed in cultured MDA-MB-231 cells treated with compound 2. In the treated tumors, apoptosis induction was confirmed by PARP cleavage, Terminal nucleotidyl transferase-mediated nick end labeling (TUNEL) and Hematoxylin and Eosin (H & E staining) assays [156].
Our data strongly suggest that the proteasome is one of the primary targets for gold(III) dithiocarbamates and that inhibition of the proteasomal activity by gold(III) dithiocarbamates is associated with apoptosis in cancer cells. We found that the effect of compound 2 could be completely blocked by two different S-donor ligands, 1,4-Dithio-DL-threitol (DTT), or N-Acetyl-L-Cysteine (NAC). DTT at 1 mM entirely reversed inhibition of the purified 20S proteasome by compound 2, and NAC at 200 μM completely blocked proteasome inhibition by compound 2 in intact MDA-MB-231 cells at both early (4 h) and later (24 h) time points. Consistently, apoptotic morphological changes, PARP cleavage, and accumulation of p36/Bax were also completely prevented in the cells co-treated with compound 2 and NAC [156]. It should be noted that NAC at lower concentrations was unable to inhibit copper-mediated proteasome inhibition in vitro and in tumor cells [157]. We are currently investigating whether NAC at higher concentrations could reverse organic copper-induced events.
There are several possible mechanisms that might be responsible for the reversal of compound 2-mediated proteasomal inhibition by NAC and DTT. First, some gold(III) complexes could coordinatively bind S-donor ligands, such as glutathione and cysteine, and cleave their disulfide bond(s) [68], an event which might be responsible for the biological effects of gold(III) complexes [70]. Therefore, it is possible that gold(III) dithiocarbamates could bind sulfhydryl groups found in NAC or DTT. NAC or DTT at high concentrations could react with all the compound 2 molecules, thereby preventing binding to and inhibition of the proteasome. Second, NAC or DTT could reduce gold(III) to gold(I), changing the coordination mode of gold to an ionic state that does not have the affinity to bind the proteasome and inhibit its activity. Third, gold(III) porphyrin 1a induces intracellular oxidation, altering glutathione (GSH) levels in the cell [72]. GSH is the main antioxidant system in the cell, and its depletion might facilitate accumulation of reactive oxygen species (ROS) in cells treated with anti-cancer drugs, which in turn increases the drug lethality [160]. Compound 2 might stimulate production of ROS, which then oxidize and inactivate the proteasome. This argument is supported by a report that the proteasome is susceptible to oxidative modification and inactivation upon exposure to free radical generating systems [161]. Moreover, we observed that the effect of NAC is much stronger in intact cells compared to cell-free conditions. When intact cells were treated with compound 2, NAC at much lower concentrations could reverse proteasomal inhibition induced by compound 2, arguing that NAC can increase the cellular pool of ROS scavengers.
These gold(III) dithiocarbamates stimulate production of ROS [74]. Moreover, they modify some mitochondrial functions, inactivate both cytosolic and mitochondrial thioredoxin reductase (TrxR), and interfere with ERK pathway, inducing cancer cell death through both apoptotic and non-apoptotic mechanisms [74]. It is important to emphasize that under physiological conditions, these gold(III) dithiocarbamates undergo hydroxylation process delivering two moles of halide and two moles of hydrogen ions per mole of starting complex. While DMDT derivatives (Compounds 1 and 2; Figure 5) still remain gold center in 3+ oxidation state, ESDT derivatives (Compounds 3 and 4; Figure 5), upon hydrolysis undergo a subsequent reduction process within 24 hours, forming binuclear gold(I) complex [(ESDT)Au]2. However, this reduction process should not have any effect on cytotoxic properties of ESDT derivatives since their cytotoxic effect is exerted mainly within first twelve hours at nanomolar concentrations [33] and within 4 hours at micromolar concentrations (our unpublished data). Also, binuclear gold(I) complex [(ESDT)Au]2 is less toxic than gold(III) analogues (Compounds 3 and 4; Figure 5) against a panel of human tumor cells ([32] and our unpublished data). Taken together, these observations strongly suggest that coordination mode of gold is very important for biological effect of gold complexes, including their cytotoxic and anti-cancer effects.
3.4 Perspectives of using gold complexes for human breast cancer therapy
During the past decades, breast cancer treatment has been rapidly evolved leading to substantial reduction in breast cancer mortality. Although sometimes compounds with encouraging preclinical data give disappointingly low responses in clinical settings, the next generation of potential drugs is synthesized and evaluated. One class of drugs that is getting more attention and is showing promising results in the area of anti-cancer drug discovery is a class of gold-based complexes. Considerable interest in this area is encouraged by a variety of new gold complexes that have already been synthesized or are still waiting to be synthesized and evaluated.
While developing new gold-based anti-cancer drugs, it is essential to design a drug that would target a specific biological site, resulting in reduced or no toxic side effects. For that reason, it is of paramount importance to better understand the coordination chemistry and molecular and biochemical mechanisms of gold complexes, which will also provide the new impetus for development of gold-based drugs. By following the interactions between gold complexes and various biological targets, much progress will be made in understanding the mode of action of gold complexes. Therefore, in vitro screening should be mandatory and should be followed by relevant in vivo studies, which remain the most important evaluation of drug effectiveness in preclinical settings.
One of the molecular targets of gold(III) dithiocarbamates recently identified by our group is the proteasome. One of the gold(III) dithiocarbamates, compound 2, showed promising results in animal studies and has a potential for further development in breast cancer treatment. Currently, there is mainly one proteasome inhibitor approved for multiple myeloma treatment – bortezomib. While very effective against some other hematological malignancies as well, bortezomib did not show promising results as a single treatment in breast cancer and other solid tumors. However, when combined with other conventional chemotherapeutics, more promising results were observed.
Although bortezomib is mainly focused on the proteasome itself, it seems that potentially promising strategy would also be in targeting other components of the ubiquitin-proteasome pathway. For example, by targeting specific E3 ubiquitin ligases involved in the process of ubiquitination, a more restricted set of proteins could be affected, resulting in more specific and better tolerated treatment. Future studies focused on this approach remain to be conducted.
4. Conclusion
The therapeutic and beneficial effect of gold on human health have been known for centuries. Despite the considerable effort to develop new, efficient, and safe gold-based drugs, very few advances have been made. A significant amount of effort in medicinal gold chemistry involves the development of novel anti-cancer drugs and identification of their novel molecular targets. The tumor proteasome is one of the identified in vivo molecular targets of gold(III) complexes. Proteasome inhibition by gold(III) complexes is associated with apoptosis induction in breast cancer cultures and tumors, supporting the conclusion that inhibition of tumor proteasome activity contributes to the anti-tumor activity of gold(III) complexes. By further investigating gold interactions with other potential biological targets in cancer cells and identifying them, more potent and more specific gold-based drugs could be designed, synthesized and, hopefully in near future, developed for treatment of human breast cancer and other cancers.
Acknowledgments
We thank Drs. Kenyon Daniel, Wayne Guida and Guoqing Shi for critical reading and valuable suggestions. This work is supported by Karmanos Cancer Institute of Wayne State University, Department of Defense Breast Cancer Research Program awards W81XWH-04-1-0688 and DAMD17-03-1-0175, and National Cancer Institute grant CA112625 (Q. P. Dou) and the National Cancer Institute/NIH Cancer Center Support grant (Karmanos Cancer Institute).
List of abbreviations
- AC
doxorubicin, cyclophosphamide
- Ac-LLnL
N-Acetyl-Leu-Leu-Norleu
- AIDS
Acquired Immune Deficiency Syndrome
- AIF
apoptosis-inducing factor
- ATG
aurothioglucose
- ATM
aurothiomalate
- CAF
cyclophosphamide, doxorubicin, and fluorouracil
- CMF
cyclophosphamide, methotrexate and 5-fluorouracil
- damp
[(dimethylamino)methyl]phenyl
- DMDT
N,N-Dimethyldithiocarbamate
- DNA
Deoxyribonucleic acid
- DPPE
diphenylphosphine
- DTT
1,4-Dithio-DL-threitol
- EGCG
epigallocatechin-3-gallate
- ERK
extracellular signal-regulated kinases
- ESDT
Ethylsarcosinedithiocarbamate
- FDA
U.S. Food and Drug Administration
- GSH
glutathione
- H & E
Hematoxylin and Eosin
- HIV
human immunodeficiency virus
- IL-6
interleukin-6
- JAK
Janus family of tyrosine kinase
- NAC
N-Acetyl-L-Cysteine
- NF-κB
nuclear factor kB
- PARP
poly (ADP-ribose) polymerase
- PGPH-like
peptidylglutamyl peptide hydrolyzing-like
- Pt
platinum
- RNA
Ribonucleic acid
- ROS
reactive oxygen species
- STAT3
Signal transducer and activator of transcription3
- TrxR
thioredoxin reductase
- TUNEL
Terminal deoxynecleotydyl transferase-mediated dUPT-biotin nick end-labeling
- Ub
Ubiquitin
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Fricker SP. Gold bulletin. 1996;29:53. [Google Scholar]
- 2.Kostova I. Anticancer Agents Med Chem. 2006;6:19. doi: 10.2174/187152006774755500. [DOI] [PubMed] [Google Scholar]
- 3.Huanzi Z, Yuantao N. Gold bulletin. 2001;34:24. [Google Scholar]
- 4.Orvig C, Abrams MJ. Chem Rev. 1999;99:2201. doi: 10.1021/cr980419w. [DOI] [PubMed] [Google Scholar]
- 5.Champion GD, Graham GG, Ziegler JB. Baillieres Clin Rheumatol. 1990;4:491. doi: 10.1016/s0950-3579(05)80005-6. [DOI] [PubMed] [Google Scholar]
- 6.Gottlieb NL. J Rheumatol. 1982;Suppl 8:99. [PubMed] [Google Scholar]
- 7.Shaw CF., III . The biochemistry of gold. In: Schmidbaur H, editor. Gold: Progress in chemistry, biochemistry and technology. John Wiley & Sons; New York: 1999. [Google Scholar]
- 8.Tiekink ERT. Bioinorg Chem Appl. 2003;53:1. doi: 10.1155/S1565363303000050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Okada T, Patterson BK, Ye SQ, Gurney ME. Virology. 1993;192:631. doi: 10.1006/viro.1993.1079. [DOI] [PubMed] [Google Scholar]
- 10.Yamaguchi K, Ushijima H, Hisano M, Inoue Y, Shimamura T, Hirano T, Müller WE. Microbiol Immunol. 2001;45:549. doi: 10.1111/j.1348-0421.2001.tb02657.x. [DOI] [PubMed] [Google Scholar]
- 11.Navarro M, Perez H, Sanchez-Delgado RA. J Med Chem. 1997;40:1937. doi: 10.1021/jm9607358. [DOI] [PubMed] [Google Scholar]
- 12.Navarro M, Cisneros-Fajardo EJ, Lehmann T, Sánchez-Delgado RA, Atencio R, Silva P, Lira R, Urbina JA. Inorg Chem. 2001;40:6879. doi: 10.1021/ic0103087. [DOI] [PubMed] [Google Scholar]
- 13.McKeage MJ, Maharaj L, Berners-Price SJ. Coord Chem Rev. 2002;232:127. [Google Scholar]
- 14.Parkin DM, Pisani P, Ferlay J. CA Cancer J Clin. 1999;49:33. doi: 10.3322/canjclin.49.1.33. [DOI] [PubMed] [Google Scholar]
- 15.Parkin DM, Bray F, Ferlay J, Pisani P. Int J Cancer. 2001;94:153. doi: 10.1002/ijc.1440. [DOI] [PubMed] [Google Scholar]
- 16.Alderden RA, Matthew DH, Trevor WH. J Chem Edu. 2006;83:728. [Google Scholar]
- 17.Wong E, Giandomenico CM. Chem Rev. 1999;99:2451. doi: 10.1021/cr980420v. [DOI] [PubMed] [Google Scholar]
- 18.Eckhardt S. Curr Med Chem Anti-Cancer Agents. 2002;2:419. doi: 10.2174/1568011024606389. [DOI] [PubMed] [Google Scholar]
- 19.Abrams MJ, Murrer BA. Science. 1993;261:725. doi: 10.1126/science.8102010. [DOI] [PubMed] [Google Scholar]
- 20.Ott I, Gust R. Arch Pharm Chem Life Sci. 2007;340:117. doi: 10.1002/ardp.200600151. [DOI] [PubMed] [Google Scholar]
- 21.Calamai P, Carotti S, Guerri A, Mazzei T, Messori L, Mini E, Orioli P, Speroni GP. Anticancer Drug Des. 1998;13:67. [PubMed] [Google Scholar]
- 22.Zhu C, Raber J, Eriksson LA. J Phys Chem B. 2005;109:12195. doi: 10.1021/jp0518916. [DOI] [PubMed] [Google Scholar]
- 23.Sadler PJ. 1982 J Rheumatol. 1982;Suppl 8:71. [PubMed] [Google Scholar]
- 24.Ward JR. Am J Med. 1988;85:39. doi: 10.1016/0002-9343(88)90361-0. [DOI] [PubMed] [Google Scholar]
- 25.Fries JF, Bloch D, Spitz P, Mitchell DM. Am J Med. 1985;78:39. doi: 10.1016/0002-9343(85)90247-5. [DOI] [PubMed] [Google Scholar]
- 26.Simon TM, Kunishima DH, Vibert GJ, Lorber A. J Rheumatol. 1979;Suppl 5:91. [PubMed] [Google Scholar]
- 27.Ni Dhubhghail OM, Sadler PJ. Metal complexes in cancer chemotherapy. Weinheim: VHC; 1993. [Google Scholar]
- 28.Mirabelli CK, Johnson RK, Hill DT, Faucette LF, Girard GR, Kuo GY, Sung CM, Crooke ST. J Med Chem. 1986;29:218. doi: 10.1021/jm00152a009. [DOI] [PubMed] [Google Scholar]
- 29.Fricker SP, Skerjl R, Cameron BR, Mosi R, Zhu Y. GOLD. AnorMED Inc; Langley BC, V2Y 1N5, Canada: 2003. Recent developments in gold drugs 2003. [Google Scholar]
- 30.Sadler PJ, Sue RE. Metal Based Drugs. 1994;1:107. doi: 10.1155/MBD.1994.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fricker SP. Metal Based Drugs. 1999;6:291. doi: 10.1155/MBD.1999.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ronconi L, Giovagnini L, Marzano C, Bettio F, Graziani R, Pilloni G, Fregona D. Inorg Chem. 2005;44:1867. doi: 10.1021/ic048260v. [DOI] [PubMed] [Google Scholar]
- 33.Ronconi L, Marzano C, Zanello P, Corsini M, Miolo G, Macca C, Trevisan A, Fregona D. J Med Chem. 2006;49:1648. doi: 10.1021/jm0509288. [DOI] [PubMed] [Google Scholar]
- 34.Messori L, Marcon G, Orioli P. Bioinorg Chem Appl. 2003;1:177. doi: 10.1155/S1565363303000141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dinger MB, Henderson W. J Organomet Chem. 1998;557:231. [Google Scholar]
- 36.Dinger MB, Henderson W. J Organomet Chem. 1998;560:233. [Google Scholar]
- 37.Messori L, Abbate F, Marcon G, Orioli P, Fontani M, Mini E, Mazzei T, Carotti S, O'Connell T, Zanello P. J Med Chem. 2000;43:3541. doi: 10.1021/jm990492u. [DOI] [PubMed] [Google Scholar]
- 38.Marcon G, Carotti S, Coronnello M, Messori L, Mini E, Orioli P, Mazzei T, Cinellu MA, Minghetti G. J Med Chem. 2002;45:1672. doi: 10.1021/jm010997w. [DOI] [PubMed] [Google Scholar]
- 39.Bruni B, Guerri A, Marcon G, Messori L, Orioli P. Croatica Chemica Acta. 1999;72:221. [Google Scholar]
- 40.Aldinucci D, Lorenzon D, Stefani L, Giovagnini L, Colombatti A, Fregona D. Anticancer Drugs. 2007;18:323. doi: 10.1097/CAD.0b013e328011ae98. [DOI] [PubMed] [Google Scholar]
- 41.Giovagnini L, Ronconi L, Aldinucci D, Lorenzon D, Sitran S, Fregona D. J Med Chem. 2005;48:1588. doi: 10.1021/jm049191x. [DOI] [PubMed] [Google Scholar]
- 42.Casas JS, Castaño MV, Cifuentes MC, García-Monteagudo JC, Sánchez A, Sordo J, Abram U. J Inorg Biochem. 2004;98:1009. doi: 10.1016/j.jinorgbio.2004.02.019. [DOI] [PubMed] [Google Scholar]
- 43.Buckley RG, Elsome AM, Fricker SP, Henderson GR, Theobald BR, Parish RV, Howe BP, Kelland LR. J Med Chem. 1996;39:5208. doi: 10.1021/jm9601563. [DOI] [PubMed] [Google Scholar]
- 44.Mirabelli CK, Johnson RK, Sung CM, Faucette L, Muirhead K, Crooke ST. Cancer Res. 1985;45:32. [PubMed] [Google Scholar]
- 45.Kim IS, Jin JY, Lee IH, Park SJ. Br J Pharmacol. 2004;142:749. doi: 10.1038/sj.bjp.0705708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Marzano C, Gandin V, Folda A, Scutari G, Bindoli A, Rigobello M. Free Radic Biol Med. 2007;42:872. doi: 10.1016/j.freeradbiomed.2006.12.021. [DOI] [PubMed] [Google Scholar]
- 47.Gromer S, Arscott LD, Williams CH, Jr, Schirmer RH, Becker K. J Biol Chem. 1998;273:20096. doi: 10.1074/jbc.273.32.20096. [DOI] [PubMed] [Google Scholar]
- 48.Gunatilleke SS, Barrios AM. J Med Chem. 2006;49:3933. doi: 10.1021/jm060158f. [DOI] [PubMed] [Google Scholar]
- 49.Rigobello MP, Scutari G, Folda A, Bindoli A. Biochem Pharmacol. 2004;67:689. doi: 10.1016/j.bcp.2003.09.038. [DOI] [PubMed] [Google Scholar]
- 50.Humphreys AS, Filipovska A, Berners-Price SJ, Koutsantonis GA, Skelton BW, White AH. Dalton Trans. 2007:4943. doi: 10.1039/b705008a. [DOI] [PubMed] [Google Scholar]
- 51.Jellicoe MM, Nichols SJ, Callus BA, Baker MV, Barnard PJ, Berners-Price SJ, Whelan J, Yeoh GC, Filipovska A. Carcinogenesis. 2008;29:1124. doi: 10.1093/carcin/bgn093. [DOI] [PubMed] [Google Scholar]
- 52.Barnard PJ, Baker MV, Ho AYY, Day DA, Berners-Price SJ. GOLD. 2003 abstract. [Google Scholar]
- 53.McKeage MJ, Papathanasiou P, Salem G, Sjaarda A, Swiegers GF, Waring P, Wild SB. Met Based Drugs. 1998;5:217. doi: 10.1155/MBD.1998.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rackham O, Nichols SJ, Leedman PJ, Berners-Price SJ, Filipovska A. Biochem Pharmacol. 2007;74:992. doi: 10.1016/j.bcp.2007.07.022. [DOI] [PubMed] [Google Scholar]
- 55.Kim NH, Lee MY, Park SJ, Choi JS, Oh MK, Kim IS. Immunology. 2007;122:607. doi: 10.1111/j.1365-2567.2007.02679.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hirano T. Int Rev Immunol. 1998;16:249. doi: 10.3109/08830189809042997. [DOI] [PubMed] [Google Scholar]
- 57.Novelli F, Recine M, Sparatore F, Juliano C. Farmaco. 1999;54:232. doi: 10.1016/s0014-827x(99)00019-1. [DOI] [PubMed] [Google Scholar]
- 58.Crooke ST, Mirabelli CK. Am J Med. 1983;75:109. doi: 10.1016/0002-9343(83)90482-5. [DOI] [PubMed] [Google Scholar]
- 59.Marzano C, Bettio F, Baccichetti F, Trevisan A, Giovagnini L, Fregona D. Chem Biol Interact. 2004;148:37. doi: 10.1016/j.cbi.2004.04.002. [DOI] [PubMed] [Google Scholar]
- 60.Shi P, Jiang Q, Zhao Y, Zhang Y, Lin J, Lin L, Ding J, Guo Z. J Biol Inorg Chem. 2006;11:745. doi: 10.1007/s00775-006-0120-y. [DOI] [PubMed] [Google Scholar]
- 61.Shaw CF, III, Cancro MP, Witkiewicz PL, Eldridge JE. Inorg Chem. 1980;19:3198. [Google Scholar]
- 62.Witkiewicz PL, Shaw CF., III J Chem Soc, Chem Commun. 1981:1111. [Google Scholar]
- 63.Brown DH, Smith WE. Am Chem Soc Symp Ser. 1983;209:401. [Google Scholar]
- 64.Smith WE, Reglinski J. Perspect Bioinorg Chem. 1991;1:183. [Google Scholar]
- 65.Bordignon E, Cattalini L, Natile G, Scatturi A. J Chem Soc, Chem Commun. 1973:878. [Google Scholar]
- 66.Natile G, Bordignon E, Cattalini L. Inorg Chem. 1976;15:246. [Google Scholar]
- 67.Isab AA, Sadler PJ. Biochim Biophys Acta. 1979;492:322. doi: 10.1016/0005-2795(77)90083-6. [DOI] [PubMed] [Google Scholar]
- 68.Zou J, Guo Z, Parkinson JA, Chen Y, Sadler PJ. Chem Commun. 1999;15:1359. [Google Scholar]
- 69.He XM, Carter DC. Nature. 1992;358:209. doi: 10.1038/358209a0. [DOI] [PubMed] [Google Scholar]
- 70.Marcon G, Messori L, Orioli P, Cinellu MA, Minghetti G. Eur J Biochem. 2003;270:4655. doi: 10.1046/j.1432-1033.2003.03862.x. [DOI] [PubMed] [Google Scholar]
- 71.Engman L, McNaughton M, Gajewska M, Kumar S, Birmingham A, Powis G. Anticancer Drugs. 2006;17:539. doi: 10.1097/00001813-200606000-00007. [DOI] [PubMed] [Google Scholar]
- 72.Wang Y, He Q, Sun RW, Che C, Chiu J. Cancer Res. 2005;65:11553. doi: 10.1158/0008-5472.CAN-05-2867. [DOI] [PubMed] [Google Scholar]
- 73.Wang Y, He QY, Che CM, Chiu JF. Proteomics. 2006;6:131. doi: 10.1002/pmic.200402027. [DOI] [PubMed] [Google Scholar]
- 74.Saggioro D, Rigobello MP, Paloschi L, Folda A, Moggach SA, Parsons S, Ronconi L, Fregona D, Bindoli A. Chem Biol. 2007;14:1128. doi: 10.1016/j.chembiol.2007.08.016. [DOI] [PubMed] [Google Scholar]
- 75.Perry CW, Phillips BJ. Internet J Oncol. 2002:1. [Google Scholar]
- 76.Parkin DM, Bray F, Ferlay J, Pisani P. CA Cancer J Clin. 2005;55:74. doi: 10.3322/canjclin.55.2.74. [DOI] [PubMed] [Google Scholar]
- 77.Bray F, McCarron P, Parkin DM. Breast Cancer Res. 2004;6:229. doi: 10.1186/bcr932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhang M, Coffino P. J Biol Chem. 2004;279:8635. doi: 10.1074/jbc.M310449200. [DOI] [PubMed] [Google Scholar]
- 79.Asher G, Shaul Y. Cell Cycle. 2005;4:1015. doi: 10.4161/cc.4.8.1900. [DOI] [PubMed] [Google Scholar]
- 80.Shringarpure R, Grune T, Mehlhase J, Davies KJ. J Biol Chem. 2003;278:311. doi: 10.1074/jbc.M206279200. [DOI] [PubMed] [Google Scholar]
- 81.Ciechanover A, Orian A, Schwartz AL. Bioessays. 2000;22:442. doi: 10.1002/(SICI)1521-1878(200005)22:5<442::AID-BIES6>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 82.Kisselev AF, Goldberg AL. Chem Biol. 2001;8:739. doi: 10.1016/s1074-5521(01)00056-4. [DOI] [PubMed] [Google Scholar]
- 83.Goldberg AL. Science. 1995;268:522. doi: 10.1126/science.7725095. [DOI] [PubMed] [Google Scholar]
- 84.Ciechanover A. Cell. 1994;79:13. doi: 10.1016/0092-8674(94)90396-4. [DOI] [PubMed] [Google Scholar]
- 85.Hochstrasser M. Curr Opin Cell Biol. 1995;7:215. doi: 10.1016/0955-0674(95)80031-x. [DOI] [PubMed] [Google Scholar]
- 86.Yang Y, Fruh K, Ahn K, Peterson PA. J Biol Chem. 1995;270:27687. doi: 10.1074/jbc.270.46.27687. [DOI] [PubMed] [Google Scholar]
- 87.Rechsteiner M, Realini C, Ustrell V. Biochem J. 2000;345(Pt 1):1. [PMC free article] [PubMed] [Google Scholar]
- 88.Tanahashi N, Murakami Y, Minami Y, Shimbara N, Hendil KB, Tanaka K. J Biol Chem. 2000;275:14336. doi: 10.1074/jbc.275.19.14336. [DOI] [PubMed] [Google Scholar]
- 89.Whitby FG, Masters EI, Kramer L, Knowlton JR, Yao Y, Wang CC, Hill CP. Nature. 2000;408:115. doi: 10.1038/35040607. [DOI] [PubMed] [Google Scholar]
- 90.Groll M, Ditzel L, Lowe J, Stock D, Bochtler M, Bartunik HD, Huber R. Nature. 1997;386:463. doi: 10.1038/386463a0. [DOI] [PubMed] [Google Scholar]
- 91.Lowe J, Stock D, Jap B, Zwickl P, Baumeister W, Huber R. Science. 1995;268:533. doi: 10.1126/science.7725097. [DOI] [PubMed] [Google Scholar]
- 92.Seemuller E, Lupas A, Stock D, Lowe J, Huber R, Baumeister W. Science. 1995;268:579. doi: 10.1126/science.7725107. [DOI] [PubMed] [Google Scholar]
- 93.Groll M, Heinemeyer W, Jager S, Ullrich T, Bochtler M, Wolf DH, Huber R. Proc Natl Acad Sci USA. 1999;96:10976. doi: 10.1073/pnas.96.20.10976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Dou QP, Nam S. Exp Opin Ther Patents. 2000;10:1263. [Google Scholar]
- 95.Hershko A. Curr Opin Cell Biol. 1997;9:788. doi: 10.1016/s0955-0674(97)80079-8. [DOI] [PubMed] [Google Scholar]
- 96.Dou QP, Li B. Drug Resist Updat. 1999;2:215. doi: 10.1054/drup.1999.0095. [DOI] [PubMed] [Google Scholar]
- 97.Wojcik C. Cell, Mol Life Sci. 1999;56:908. doi: 10.1007/s000180050483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Orlowski RZ. Cell Death Differ. 1999;6:303. doi: 10.1038/sj.cdd.4400505. [DOI] [PubMed] [Google Scholar]
- 99.Fenteany G, Standaert RF, Lane WS, Choi S, Corey EJ, Schreiber SL. Science. 1995;268:726. doi: 10.1126/science.7732382. [DOI] [PubMed] [Google Scholar]
- 100.Smith DM, Daniel KG, Wang Z, Guida WC, Chan TH, Dou QP. Proteins. 2004;54:58. doi: 10.1002/prot.10504. [DOI] [PubMed] [Google Scholar]
- 101.Loda M, Cukor B, Tam SW, Lavin P, Fiorentino M, Draetta GF, Jessup JM, Pagano M. Nature Medicine. 1997;3:231. doi: 10.1038/nm0297-231. [DOI] [PubMed] [Google Scholar]
- 102.Kumatori A, Tanaka K, Inamura N, Sone S, Ogura T, Matsumoto T, Tachikawa T, Shin S, Ichihara A. Proc Natl Acad Sci USA. 1990;87:7071. doi: 10.1073/pnas.87.18.7071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Li B, Dou QP. Proc Natl Acad Sci USA. 2000;97:3850. doi: 10.1073/pnas.070047997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Adams J. Drug Discov Today. 2003;8:307. doi: 10.1016/s1359-6446(03)02647-3. [DOI] [PubMed] [Google Scholar]
- 105.Almond JB, Cohen GM. Leukemia. 2002;16:433. doi: 10.1038/sj.leu.2402417. [DOI] [PubMed] [Google Scholar]
- 106.Landis-Piwowar KR, Milacic V, Chen D, Yang H, Zhao Y, Chan TH, Yan B, Dou QP. Drug Resist Updat. 2006;9:263. doi: 10.1016/j.drup.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 107.An B, Goldfarb RH, Siman R, Dou QP. Cell Death Diff. 1998;5:1062. doi: 10.1038/sj.cdd.4400436. [DOI] [PubMed] [Google Scholar]
- 108.Delic J, Masdehors P, Omura S, Cosset JM, Dumont J, Binet JL, Magdelenat H. Br J Cancer. 1998;77:1103. doi: 10.1038/bjc.1998.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Soligo D, Servida F, Delia D, Fontanella E, Lamorte G, Caneva L, Fumiatti R, Deliliers GL. Br J Haematol. 2001;113:126. doi: 10.1046/j.1365-2141.2001.02683.x. [DOI] [PubMed] [Google Scholar]
- 110.Ma M, Yang HH, Parker K, Manyak S, Friedman JM, Altamirano C, Wu ZQ, Borad MJ, Frantzen M, Roussos E, Neeser J, Mikail A, Adams J, Sjak-Shie N, Vescio RA, Berenson JR. Clin Cancer Res. 2003;9:1136. [PubMed] [Google Scholar]
- 111.Imajoh-Ohmi S, Kawaguchi T, Sugiyama S, Tanaka K, Omura S, Kikuchi H. Biochem Biophys Res Commun. 1995;217:1070. doi: 10.1006/bbrc.1995.2878. [DOI] [PubMed] [Google Scholar]
- 112.Omura S, Fujimoto T, Otoguro K, Matsuzaki K, Moriguchi R, Tanaka H, Sasaki Y. J Antibiot (Tokyo) 1991;44:113. doi: 10.7164/antibiotics.44.113. [DOI] [PubMed] [Google Scholar]
- 113.Adams J, Palombella VJ, Sausville EA, Johnson J, Destree A, Lazarus DD, Maas J, Pien CS, Prakash S, Elliott PJ. Cancer Res. 1999;59:2615. [PubMed] [Google Scholar]
- 114.Lightcap ES, McCormack TA, Pien CS, Chau V, Adams J, Elliott PJ. Clin Chem. 2000;46:673. [PubMed] [Google Scholar]
- 115.Tan C, Waldmann TA. Cancer Res. 2002;62:1083. [PubMed] [Google Scholar]
- 116.Orlowski RZ, Eswara JR, Lafond-Walker A, Grever MR, Orlowski M, Dang CV. Cancer Res. 1998;58:4342. [PubMed] [Google Scholar]
- 117.Sun J, Nam S, Lee CS, Li B, Coppola D, Hamilton AD, Dou QP, Sebti SM. Cancer Res. 2001;61:1280. [PubMed] [Google Scholar]
- 118.Orlowski RZ, Dees EC. Breast Cancer Res. 2003;5:1. doi: 10.1186/bcr460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kiyomiya K, Matsuo S, Kurebe M. Cancer Res. 2001;61:2467. [PubMed] [Google Scholar]
- 120.Kapahi P, Takahashi T, Natoli G, Adams SR, Chen Y, Tsien RY, Karin M. J Biol Chem. 2000;275:36062. doi: 10.1074/jbc.M007204200. [DOI] [PubMed] [Google Scholar]
- 121.Chen GC, Guan LS, Hu WL, Wang ZY. Anticancer Res. 2002;22:633. [PubMed] [Google Scholar]
- 122.Wang X, Omura S, Szweda LI, Yang Y, Berard J, Seminaro J, Wu J. Eur J Immunol. 1997;27:2781. doi: 10.1002/eji.1830271106. [DOI] [PubMed] [Google Scholar]
- 123.Pang H, Faber LE. Breast Cancer Res Treat. 2001;70:21. doi: 10.1023/a:1012570204923. [DOI] [PubMed] [Google Scholar]
- 124.Nam S, Smith DM, Dou QP. Cancer Epidemiol Biomarkers Prev. 2001;10:1083. [PubMed] [Google Scholar]
- 125.Piccinini M, Tazartes O, Mezzatesta C, Ricotti E, Bedino S, Grosso F, Dianzani U, Tovo PA, Mostert M, Musso A, Rinaudo MT. Biochem J. 2001;356:835. doi: 10.1042/0264-6021:3560835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Rao S, Porter DC, Chen X, Herliczek T, Lowe M, Keyomarsi K. Proc Natl Acad Sci USA. 1999;96:7797. doi: 10.1073/pnas.96.14.7797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Romero A, Rabinovich MG, Vallejo CT, Perez JE, Rodriguez R, Cuevas MA, Machiavelli M, Lacava JA, Langhi M, Romero-Acuna L. J Clin Oncol. 1994;12:336. doi: 10.1200/JCO.1994.12.2.336. [DOI] [PubMed] [Google Scholar]
- 128.Teicher BA, Ara G, Herbst R, Palombella VJ, Adams J. Clin Cancer Res. 1999;5:2638. [PubMed] [Google Scholar]
- 129.Frankel A, Man S, Elliott P, Adams J, Kerbel RS. Clin Cancer Res. 2000;6:3719. [PubMed] [Google Scholar]
- 130.Ling YH, Liebes L, Jiang JD, Holland JF, Elliott PJ, Adams J, Muggia FM, Perez-Soler R. Clin Cancer Res. 2003;9:1145. [PubMed] [Google Scholar]
- 131.Cusack JC, Jr, Liu R, Houston M, Abendroth K, Elliott PJ, Adams J, Baldwin AS., Jr Cancer Res. 2001;61:3535. [PubMed] [Google Scholar]
- 132.Pham LV, Tamayo AT, Yoshimura LC, Lo P, Ford RJ. J Immunol. 2003;171:88. doi: 10.4049/jimmunol.171.1.88. [DOI] [PubMed] [Google Scholar]
- 133.Crawford LJ, Walker B, Ovaa H, Chauhan D, Anderson KC, Morris TC, Irvine AE. Cancer Res. 2006;66:6379. doi: 10.1158/0008-5472.CAN-06-0605. [DOI] [PubMed] [Google Scholar]
- 134.Ludwig H, Khayat D, Giaccone G, Facon T. Cancer. 2005;104:1794. doi: 10.1002/cncr.21414. [DOI] [PubMed] [Google Scholar]
- 135.Cuasck JC. Cancer Treat Rev. 2003;29:21. doi: 10.1016/s0305-7372(02)00096-8. [DOI] [PubMed] [Google Scholar]
- 136.Orlowski RZ, Stinchcombe TE, Mitchell BS, Shea TC, Baldwin AS, Stahl S, Adams J, Esseltine DL, Elliott PJ, Pien CS, Guerciolini R, Anderson JK, Depcik-Smith ND, Bhagat R, Lehman MJ, Novick SC, O'Connor OA, Soignet SL. J Clin Oncol. 2002;20:4420. doi: 10.1200/JCO.2002.01.133. [DOI] [PubMed] [Google Scholar]
- 137.Kane RC, Farrell AT, Sridhara R, Pazdur R. Clin Cancer Res. 2006;12:2955. doi: 10.1158/1078-0432.CCR-06-0170. [DOI] [PubMed] [Google Scholar]
- 138.Orlowski RZ. Hematology (Am Soc Hematol Educ Program) 2005:220. doi: 10.1182/asheducation-2005.1.220. [DOI] [PubMed] [Google Scholar]
- 139.Dou QP, Goldfarb RH. IDrugs. 2002;5:828. [PubMed] [Google Scholar]
- 140.Papandreou CN, Daliani DD, Nix D, Yang H, Madden T, Wang X, Pien CS, Millikan RE, Tu S, Pagliaro L, Kim J, Adams J, Elliott P, Esseltine D, Petrusich A, Dieringer P, Perez C, Logothetis CJ. J Clin Oncol. 2004;22:2108. doi: 10.1200/JCO.2004.02.106. [DOI] [PubMed] [Google Scholar]
- 141.Yang CH, Gonzalez-Angulo AM, Reuben JM, Booser DJ, Pusztai L, Krishnamurthy S, Esseltine D, Stec J, Broglio KR, Islam R, Hortobagyi GN, Cristofanilli M. Ann Oncol. 2006;17:813. doi: 10.1093/annonc/mdj131. [DOI] [PubMed] [Google Scholar]
- 142.Engel RH, Brown JA, Von Roenn JH, O'Regan RM, Bergan R, Badve S, Rademaker A, Gradishar WJ. Cancer Invest. 2007;25:733. doi: 10.1080/07357900701506573. [DOI] [PubMed] [Google Scholar]
- 143.Schlotter CM, Vogt U, Allgayer H, Brandt B. Breast Cancer Res. 2008;10:211. doi: 10.1186/bcr2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Mitsiades N, Mitsiades CS, Richardson PG, Poulaki V, Tai YT, Chauhan D, Fanourakis G, Gu X, Bailey C, Joseph M, Libermann TA, Schlossman R, Munshi NC, Hideshima T, Anderson KC. Blood. 2003;101:2377. doi: 10.1182/blood-2002-06-1768. [DOI] [PubMed] [Google Scholar]
- 145.Cardoso F, Durbecq V, Laes JF, Badran B, Lagneaux L, Bex F, Desmedt C, Willard-Gallo K, Ross JS, Burny A, Piccart M, Sotiriou C. Mol Cancer Ther. 2006;5:3042. doi: 10.1158/1535-7163.MCT-06-0104. [DOI] [PubMed] [Google Scholar]
- 146.Schmid P, Kühnhardt D, Kiewe P, Lehenbauer-Dehm S, Schippinger W, Greil R, Lange W, Preiss J, Niederle N, Brossart P, Freier W, Kümmel S, Van de Velde H, Regierer A, Possinger K. Ann Oncol. 2008;19:871. doi: 10.1093/annonc/mdm569. [DOI] [PubMed] [Google Scholar]
- 147.Thomas JP, Arzoomanian R, Alberti D, Geiger P, Marnocha R, Tutsch K, Dresen A, Volkman J, Binger K, Kolb M, Feierabend C, Black S, Hampton K, Wilding G. Proc Am Soc Clin Oncol. 2002;21:93a. abstract. [Google Scholar]
- 148.Iqbal S, Lenz HJ, Groshen S, Wei Y, Gandara DR, Lara PN, Jr, Gumerlock P, Doroshow JH, Twardowski P, Synold T. Proc Am Soc Clin Oncol. 2002;21:93a. [Google Scholar]
- 149.Clark JW, Ryan D, Dees C, Eder JP, Winkelmann J, Lynch T, Supko J, Appleman LJ, Fidias P, O'Neil B, Orlowski RZ, Baldwin A, Kinchla N, Zhu A, Esseltine D, Elliott P, Adams J, Kauffman M, Schenkein D, Cusack J. Proc Am Soc Clin Oncol. 2002;21:93a. abstract. [Google Scholar]
- 150.Ryan DP, Eder JP, Winkelmann J, Lynch T, Supko J, Appleman LJ, Fidias P, Enzinger P, Zhu A, Kinchla N, Esseltine D, Baldwin A, Elliott P, Adams J, Kauffman M, Schenkein D, Cusack J. Proc Am Soc Clin Oncol. 2002;21:95a. abstract. [Google Scholar]
- 151.Richardson PG, Barlogie B, Berenson J, Singhal S, Jagannath S, Irwin D, Rajkumar SV, Srkalovic G, Alsina M, Alexanian R, Siegel D, Orlowski RZ, Kuter D, Limentan SA, Lee S, Hideshima T, Esseltine DL, Kauffman M, Adams J, Schenkein DP, Anderson KC. N Engl J Med. 2003;348:2609. doi: 10.1056/NEJMoa030288. [DOI] [PubMed] [Google Scholar]
- 152.Jagannath S, Barlogie B, Berenson J, Siegel D, Irwin D, Richardson PG, Niesvizky R, Alexanian R, Limentani SA, Alsina M, Adams J, Kauffman M, Esseltine DL, Schenkein DP, Anderson KC. Br J Haematol. 2004;127:165. doi: 10.1111/j.1365-2141.2004.05188.x. [DOI] [PubMed] [Google Scholar]
- 153.Bodenner DL, Dedon PC, Keng PC, Borch RF. Cancer Res. 1986;46:2745. [PubMed] [Google Scholar]
- 154.Borch RF, Dedon PC, Gringeri A, Montine TJ. Inhibition of platinum drug toxicity by diethyldithiocarbamate. In: Nicolini M, editor. Platinum and other metal coordination compounds in cancer chemotherapy. Martinus Nijhoff; Boston: 1988. [Google Scholar]
- 155.Huang H, Zhu L, Reid BR, Drobny GP, Hopkins PB. Science. 1995;270:1842. doi: 10.1126/science.270.5243.1842. [DOI] [PubMed] [Google Scholar]
- 156.Milacic V, Chen D, Ronconi L, Landis-Piwowar KR, Fregona D, Dou QP. Cancer Res. 2006;66:10478. doi: 10.1158/0008-5472.CAN-06-3017. [DOI] [PubMed] [Google Scholar]
- 157.Daniel KG, Gupta P, Harbach RH, Guida WC, Dou QP. Biochem Pharmacol. 2004;67:1139. doi: 10.1016/j.bcp.2003.10.031. [DOI] [PubMed] [Google Scholar]
- 158.Daniel KG, Chen D, Orlu S, Cui QC, Miller FR, Dou QP. Breast Cancer Res. 2005;7:R897. doi: 10.1186/bcr1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Lopes UG, Erhardt P, Yao R, Cooper GM. J Biol Chem. 1997;272:12893. doi: 10.1074/jbc.272.20.12893. [DOI] [PubMed] [Google Scholar]
- 160.Troyano A, Fernandez C, Sancho P, de Blas E, Aller P. J Biol Chem. 2001;276:47379. doi: 10.1074/jbc.M104516200. [DOI] [PubMed] [Google Scholar]
- 161.Szweda PA, Friguet B, Szweda LI. Free Radical Biology & Medicine. 2002;33:29. doi: 10.1016/s0891-5849(02)00837-7. [DOI] [PubMed] [Google Scholar]