

Abstract
The myosin regulatory light chain (RLC) wraps around the alpha helical neck region of myosin. This neck region has been proposed to act as a lever arm, amplifying small conformational changes in the myosin head to generate motion. The RLC serves an important structural role, supporting the myosin neck region and a modulatory role, tuning the kinetics of the actin myosin interaction. Given the importance of the RLC, it is not surprising that mutations of the RLC can lead to familial hypertrophic cardiomyopathy (FHC), the leading cause of sudden cardiac death in people under 30. Population studies identified two FHC mutations located near the cationic binding site of the RLC, R58Q and N47K. Although these mutations are close in sequence, they differ in clinical presentation and prognosis with R58Q showing a more severe phenotype. We examined the molecular based changes in myosin that are responsible for the disease phenotype by purifying myosin from transgenic mouse hearts expressing mutant myosins and examining actin filament sliding using the in vitro motility assay. We found that both R58Q and N47K showed reductions in force compared to the wild type that could result in compensatory hypertrophy. Furthermore, we observed a higher ATPase rate and an increased activation at submaximal calcium levels for the R58Q myosin that could lead to decreased efficiency and incomplete cardiac relaxation, potentially explaining the more severe phenotype for the R58Q mutation.
Keywords: motility assay, ATPase, RLC, optical trapping, FHC, calcium binding site
Introduction
Familial hypertrophic cardiomyopathy1 (FHC), the leading cause of sudden cardiac death in people under 30 [1–4], is characterized by myofibrillar disarray and thickening of the left ventricle, papillary muscles, or septum. Individuals bearing FHC mutations sometimes experience shortness of breath or chest pain although often sudden cardiac death occurs with no clinical symptoms. FHC is caused by mutations in cardiac sarcomeric proteins including the myosin heavy chain (MHC), the regulatory (RLC) and essential light chains of myosin, troponins I, T, and C, titin, tropomyosin, actin, and myosin binding protein C (Reviewed in [3, 5]). The clinical presentation and prognosis of the disease depend on the specific mutation.
Several mutations in the myosin regulatory light chain (RLC) have been implicated in FHC (For reviews, see [1, 6]). The RLC wraps around the alpha helical neck of the myosin head by binding to a 35 amino acid IQ motif on the MHC. The neck region of the MHC has been proposed to act as a lever arm, amplifying small conformational changes that originate at the catalytic site in the myosin head into large movements, allowing myosin to generate motion and force [7]. Furthermore, this neck region has been proposed to serve as the compliant element in the myosin crossbridge with the RLC playing a structural role, modulating the stiffness of the lever arm [8]. The RLC also contains a highly conserved phosphorylatable serine [9–12] that plays an important role in the activation and modulation of myosins (Reviewed in [13, 14]).
In addition to a phosphorylation site, the RLC also contains an N-terminal divalent cation binding site [15, 16]. The absence of bound cation in the RLC binding site has been shown to alter the structural properties of the RLC and consequently, the contractile properties of the cation-free myosin [16–18]. Furthermore, binding of nucleotides to the myosin head has been shown to alter the conformation of the RLC [19]. Given the important role of the RLC in muscle contraction, one can hypothesize that FHC mutations in the RLC affect myosin mechanics or kinetics. This report addresses this hypothesis by examining FHC mutant myosins using the in vitro motility assay.
Two of the FHC associated mutations in the RLC [20–22] that have been identified in population studies are located either near (R58Q) or at (N47K) the Ca2+ binding site (Fig. 1). Although these mutations are close to each other in sequence, they differ in both clinical presentation and prognosis, with R58Q showing a more severe phenotype. N47K has been associated with rapidly progressing late onset mid-ventricular hypertrophy and papillary muscle hypertrophy whereas R58Q has been associated with thickening of the left ventricular wall and multiple cases of sudden cardiac death [20–22]. Studies of isolated RLC and myosin reconstituted with mutant RLCs have shown that both R58Q and N47K mutations deactivate RLC for calcium binding however, phosphorylation of R58Q restores this ability [18, 23]. While effects of the mutations have previously been studied in muscle fibers [18, 24], fibers are complicated structures, and it is difficult to dissect the mutation dependent changes in the acto-myosin interaction and how it is regulated by tropomyosin (Tm) and troponin (Tn) at the molecular level. In this study we examine the phenotypic properties of the R58Q and N47K mutations at the level of single molecules by utilizing myosin purified from transgenic mice and employing the in vitro motility assay [25]. We measured the velocity, duty cycle, ATPase activity, isometric force, and calcium sensitivity of both R58Q and N47K mutant myosins. We found several differences in the properties of mutant myosins compared to the wild type (WT) myosin, indicative of the mutation dependent effects in the actin-myosin interaction. Both R58Q and N47K showed a reduction in isometric force that could result in compensatory hypertrophy of the heart. Also, based on our data, we hypothesize that the more severe phenotype of R58Q results from: (1) a decrease in efficiency as evidenced by a higher ATPase rate with a decrease in force and (2) increased rate of activation at submaximal calcium levels that could lead to diastolic dysfunction due to incomplete relaxation of the heart.
Figure 1.

FHC mutations mapped onto the chicken skeletal RLC structure (Accession # 2MYS [7]). The C-terminal region of the myosin heavy chain is shown in yellow and the RLC is shown in cyan. The locations of R58, N47, Mg2+ and the cation binding site are highlighted in blue, red, magenta and green respectively.
Materials and Methods
Protein Preparation
Alpha Cardiac myosin was isolated from the hearts of non-transgenic mice (NTg), transgenic wild type mice expressing the human cardiac RLC (Tg-WT), and transgenic mutant mice expressing the N47K or R58Q RLC mutation, denoted respectively as Tg-N47K and Tg-R58Q. As described in Wang et al. [24], all transgenic mice (WT L2, N47K L6 and R58Q L8 expresses ~100% transgene. Myosin was purified from 5–6 hearts from each group of mice, as described in detail in Szczesna-Cordary et al. [26], The hearts were collected from female and male mice of 4–7 months of age. At least 3 different myosin preparations obtained at different time intervals were used for experiments.
Pig cardiac tropomyosin was purified from pig hearts according to Eisenberg and Kielley [27]. Recombinant human cardiac troponin subunits (TnT, TnI, and TnC) were prepared according to standard methods [28] and the formation of troponin complex was carried out as described in Szczesna et al. [29] and Gomes et al. [30].
Actin was prepared from chicken pectoralis muscle acetone powder using the method of Straub [31] with the modification of Drabikowski et al.[32]. The actin was suspended in actin buffer (25 mM KCl, 1 mM EGTA, 10 mM DTT, 25 mM imidazole, 4 mM MgCl2). TRITC phalloidin labeled actin was prepared by incubating a 1:1 molar ratio of TRITC phalloidin and actin in actin buffer overnight at 4°C.
Unregulated Motility Assay
The quality of the myosin preparation was assessed by measuring the actin filament sliding velocity in the motility assay. Unless otherwise noted, all experiments were performed at 24°C. The average actin filament sliding velocity for NTg murine cardiac myosin at 24°C was 1.2 ± 0.5 μm/s, however, when the flow cell was incubated with 100 μg/ml of NTg myosin and heated to 35°C, the velocity was 4.2 ± 1.3 μm/s, consistent with previously reported values [33]. The in vitro motility assays were performed as previously described with some subtle modifications [26]. Approximately 200 μg of myosin in 50% glycerol was suspended in 1 ml of 10 mM DTT in water and allowed to precipitate for one hour on ice. This step removed the glycerol and, if present, any oxidized myosin molecules unable to form thick filaments. The myosin was then centrifuged at 16,000 × g for 30 minutes at 4°C. The supernatant was then discarded and the pellet was resuspended in 200 μl of myosin buffer (300 mM KCl, 25 mM imidazole, 1 mM EGTA, 4 mM MgCl2, 10 mM DTT). Any damaged myosin heads that were unable to bind and release from actin were removed by mixing the myosin, 1 mM ATP, and 1.1 μM actin and centrifuging in an Airfuge for 30 min at 100,000 × g. The myosin concentration after centrifugation was determined using a Bradford assay (Bio-rad Labs. Hercules, CA) and diluted to the desired concentration in myosin buffer.
Flow cells were constructed by forming a channel between nitrocellulose coated coverslips and a standard glass slide with double stick tape (100 μm width 3 M Corp., St. Paul, MN ). Myosin was adsorbed to the coverslip surface by incubating 30 μl of myosin (100 μg/ml) in myosin buffer for 1 minute. Any remaining surface lacking myosin was blocked by adding 30 μl of 0.5 mg/ml bovine serum albumin (BSA) in myosin buffer followed by a 60 μl wash with actin buffer (25 mM KCl, 25 mM imidazole, 1 mM EGTA, 4 mM MgCl2, 10 mM DTT). As an additional measure to minimize the effects of damaged myosin heads, 30 μl of 1 μM unlabeled actin (myosin: unlabeled actin molar ratios varied from 40:1 to 400:1) in actin buffer was vortexed and added to the flow cell. After incubating for two minutes, the flow cell was washed with 60 μl of actin buffer containing 1 mM ATP and then 120 μl of actin buffer without ATP. 30 μl of 5 nM TRITC labeled actin was then added to the flow cell and allowed to incubate for 1 minute. Motility was initiated by the addition of motility buffer (actin buffer with the addition of 0.5% methyl cellulose, 1 mM ATP, 2 mM dextrose, 160 units glucose oxidase, and 2 μM catalase) to the flow cell.
To determine relative changes in duty cycle defined as the fraction of the myosin biochemical cycle spent attached to actin, actin filament sliding velocities were measured at several myosin surface densities by varying the concentration of myosin used to incubate the flow cell over a range from 0 to 200 μg/ml. The amount of myosin necessary to move actin at maximal velocity provides a qualitative measurement of the myosin duty cycle [17, 34].
Actin-activated myosin ATPase activity
The actin activated myosin ATPase activity was measured as a function of actin concentration. Mouse NTg, Tg-WT, Tg-N47K and Tg-R58Q myosins at a concentration of 1μM were titrated (in triplicates onto 96-well microplates) with increasing concentrations of skeletal muscle actin. ATPases assays were performed in a 120 μl reaction volume in a buffer consisting of 25 mM imidazole, pH 7.0, 4 mM MgCl2, 1 mM EGTA, and 1 mM DTT. The reactions were initiated with the addition of 2.5 mM ATP with mixing in a Jitterbug incubator shaker (Boekel, Feasterville, PA) and allowed to proceed for 15 minutes at 30°C and then terminated by the addition of 5% trichloroacetic acid. Samples were then centrifuged at 18,000 x g for 20 minutes and the 50 μl of supernatant was transferred to a 96-well microplate for determination of inorganic phosphate (Fiske, 1925). Data were analyzed with a double reciprocal plot [35]:
| (Eq. 1) |
where V is the measured ATPase rate, Vmax is the maximal ATPase activity, and Km is the Michaelis-Menten constant.
Frictional Loading Assay
The procedure for the frictional loading assays was identical to the unregulated motility assay except that the initial myosin incubation was done with 100 μg/ml of myosin mixed with an appropriate amount of alpha-actinin (Cytoskeleton Inc., Denver, CO) in myosin buffer. This initial step was allowed to incubate for one minute before the BSA was added. The velocity of the actin filaments versus alpha-actinin concentration was measured as described above. The velocity decreased linearly [36] to a plateau value where a few actin filaments would move at the same velocity regardless of the concentration of alpha-actinin added. These few escaping actin filaments are probably due to inhomogeneities in the alpha-actinin coating on the surface. Since these filaments most likely represent experimental artifact, these filaments were ignored and the standard linear fit, as described by Bing et al. [36], was used to obtain the index of retardation, kr.. The index of retardation was calculated by fitting the data to the linear equation:
| (Eq. 2) |
where m is the slope of the line and kr is the index of retardation.
Regulated Motility Assays
The procedure for regulated motility assays was similar to the unregulated motility assays with some minor modifications. Initially, 100 μg/ml of myosin in myosin buffer was incubated on the flow cell surface. After blocking the surface with BSA and unlabeled actin, TRITC phalloidin labeled actin was added as described earlier. After the TRITC phalloidin labeled actin had bound to the surface for 30 seconds, 30 μl of a mixture of 150 nM tropomyosin and 150 nM troponin in actin buffer was added to the flow cell. After allowing 10 minutes for the tropomyosin and troponin to bind to the actin, 30 μl of appropriate pCa motility buffer (determined using Bathe, a program that balances ionic conditions using binding constants determined by Godt et al. [37]) was added with the inclusion of 75 nM troponin, 75 nM tropomyosin, and an appropriate amount of calcium. The calcium concentration at which half maximal velocity was achieved, the pCa50, and the Hill Coefficients, H, were determined by fitting the data of velocity versus calcium concentration to the equation:
| (Eq. 3) |
where VMax is the maximal velocity and H is the Hill Coefficient.
Microscopy
Rhodamine labeled actin filaments were observed on a Nikon Eclipse TE2000-U microscope (Nikon (Melville, NY)) with standard epifluoresence illumination. Temperature was controlled using an objective heater (Bioptechs Inc., Butler, PA). The images were recorded using video microscopy and captured to a Scion frame grabber (Model AG-5). Using Scion Image (Scion Corp. (Frederick, MD)), 5 to 10 images were captured at an appropriate frame rate (1 to 7 seconds per frame depending on the speed of the actin filament sliding). The filament velocity was determined using the freeware Retrac motility software (http://mc11.mcri.ac.uk/Retrac). Fifteen to thirty moving filaments were analyzed from at least two different areas of the flow cell to ensure that there were no surface artifacts.
Statistics
At least three myosin preparations (each preparation consisting of 5–6 hearts from each group of mice) were used for most of the data collection. Filament motility was recorded in several areas of the flow cell. For each actin filament, the average velocity was measured over 5 to 10 frames. Then, for each flow cell, the velocities of 15 to 30 actin filaments were averaged together. Each point shown in the data plots represents the average velocity of these 15 to 30 actin filaments with error bars given by the standard error of the mean actin filament sliding velocity. Data were fit to the appropriate model using a non-linear least-squares algorithm (SigmaPlot, Systat Software Inc., San Jose, CA). A two-tailed t-test was used to examine the significance of the differences between velocities as determined from the errors in the fits of the data to the model curve. The p value was calculated from the Student’s t-test distribution. The t-tests were corrected for multiple comparisons using the Holm t-test criteria when appropriate.
Results
To examine the effects of N47K and R58Q RLC mutations on the biochemical and mechanical properties of myosin, cardiac myosin was isolated and purified from transgenic mice expressing N47K and R58Q mutations in the RLC driven by the alpha-myosin heavy chain promoter [24]. The myosin purified from N47K and R58Q hearts contained approximately 100% mutant RLC attached to the endogenous mouse alpha-myosin heavy chain [24]. The procedure utilized to purify myosin from transgenic mouse hearts resulted in dephosphorylated RLC (data not shown).
Velocity Measurements
In an in vitro motility assay, fluorescently labeled actin filaments are propelled over a bed of myosin in the presence of ATP. The velocity, V, of myosin induced actin filament translocation in an in-vitro motility assay is given by:
| (Eq. 4) |
Where d is the unitary step size of myosin, kATPase is the actin activated ATPase rate, and f is the duty cycle of the myosin. Changes in actin filament sliding velocity in the in vitro motility assay are indicative of changes in at least one of these parameters. Maximal sliding velocity is achieved when there is at least one myosin head interacting with the actin filament at any given time. A myosin with a higher duty cycle will be able to move an actin filament at maximal velocity with a lower concentration of myosin on the motility assay surface whereas a myosin with a lower duty cycle will require a greater concentration of myosin in order to ensure that at least one myosin head is attached to actin at any given time.
To examine whether the mutants caused changes to the myosin duty cycle and/or actin filament sliding velocity, we measured the sliding velocity of actin as a function of myosin concentration for the WT, N47K, and R58Q myosins (Fig. 2). An actin filament will move at its maximal velocity when incubated with enough myosin to ensure that at least one myosin head is interacting with the actin filament at all times [38–40]. Thus the amount of myosin required to achieve maximal velocity gives a qualitative measurement of the duty cycle [17, 34]. The data show that R58Q has a higher velocity than the WT at lower myosin concentrations ([myosin] < 130 μg/ml) where the velocity is attachment limited. The velocity of N47K is similar to the WT although slightly greater than the WT at low myosin concentrations ([myosin] < 50 μg/ml) and slightly less than the WT at higher myosin concentrations ([myosin] > 100 μg/ml). Furthermore, as can be seen from the normalized plot (Fig. 2b), both R58Q and N47K reach maximal velocity when incubated with approximately 30 μg/ml myosin whereas the WT requires at least 200 μg/ml of myosin to move at maximal velocity, indicative of an increase in duty cycle for the mutant myosins compared to WT [17, 34, 41].
Figure 2.

(A) Raw data and (B) normalized plots showing the velocity of actin filament sliding velocity in the in vitro motility assay as a function of myosin concentration. Both R58Q (long-dashed line) and N47K (short-dashed line) reach maximal velocity before the WT (dotted line), suggesting that R58Q and N47K have increases in duty cycle. The data were fit to the sum of two exponential curves to illustrate the trend of the data. Each point represents the average and standard errors in the means of the velocities of 15 to 30 actin filaments.
According to the standard detachment limited model of actomyosin sliding, once there is a sufficient concentration of myosin on the surface to ensure that at least one head is interacting with the actin at any given time, the actin filament should move at maximal velocity, regardless of the addition of further myosin to the motility assay surface [38, 39]. However, as shown in Fig. 2b, both mutants display a deviation (although more subtle for N47K) from the expected behavior at high myosin concentrations ([myosin] > 50 μg/ml). Both mutants demonstrate a decline in velocity with increasing myosin surface density. In contrast, WT myosin shows the expected rise to maximal velocity without a decline in velocity at higher myosin concentrations.
Based on the equation 4, the observed increases in the duty cycles of R58Q and N47K should result in a decrease of the actin filament sliding velocity. However, we see either no major change (N47K) or increased (R58Q) velocities compared to WT myosin. In order for this to occur, the increase in duty cycle must be accompanied by changes in the ATPase rate or unitary step size (or both). Thus we measured the actin activated ATPase rate of the WT, NTg, N47K, and R58Q myosins.
Actin Activated ATPase Measurements
The maximal ATPase rates, Vmax, values for NTg (0.43 s−1), WT (0.43 s−1), and N47K (0.43 s−1) were identical however, R58Q showed a significant increase in Vmax (0.63 s−1) (Fig. 3, Table 1). This significant increase in ATPase observed for R58Q myosin most likely offsets at least part of the increase in duty cycle observed with R58Q myosin from Fig. 2, giving R58Q an increased velocity compared to the WT. However, the absence of an effect on ATPase rate for N47K myosin suggests that the observed lack of a decrease in the velocity with a parallel increase in the duty cycle produced by N47K myosin most likely result from an increase in myosin unitary step size.
Figure 3.
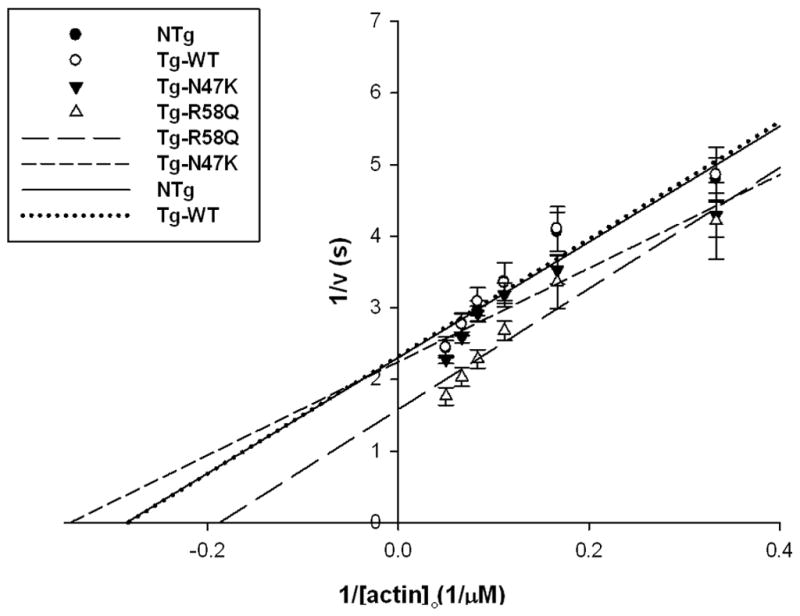
Actin activated ATPase rates of the WT, NTg, N47K, and R58Q. As can be seen WT, NTg, and N47K have similar Vmax values while R58Q showed an increase in actin activated ATPase rates expressed by increased Vmax. Data points are average from 4 independent experiments ± standard error of the mean Data was fit as described in the methods.
Table 1.
| Myosin | ATPase VMax (s−1) | ATPase KM (μM) | kr (μg) | pCa50 | Reg. VMax (μm/s) |
|---|---|---|---|---|---|
| NTg | 0.43 ± 0.03 | 3.5 ± 0.4 | 3.5 ± 0.3 | 6.16 ± 0.10 | 1.41 ± 0.12 |
| WT | 0.43 ± 0.03 | 3.5 ± 0.5 | 3.8 ± 0.2 | 6.21 ± 0.05 | 1.57 ± 0.08 |
| R58Q | 0.63 ± 0.08 | 5.3 ± 0.7 | 2.9 ± 0.2 | 6.66 ± 0.05 | 2.17 ± 0.07 |
| N47K | 0.44 ± 0.03 | 2.9 ± 0.4 | 1.8 ± 0.2 | 6.23 ± 0.04 | 1.00 ± 0.03 |
Frictional Loading Assays
In order to probe whether changes in the isometric force of the mutant myosins could be contributing to the observed disease phenotypes, we measured the isometric force due to the myosin using a frictional loading assay [36]. In this assay, alpha-actinin, a low affinity actin binding protein, is introduced into the in vitro motility assay flow cell. During actin filament sliding, alpha-actinin transiently binds to actin, providing a frictional load opposing the filament sliding. The velocity decreases linearly with alpha-actinin concentration until motility stops as described by Bing et al., [36]. The concentration of alpha-actinin necessary to stop filament motility, known as the index of retardation (kr), provides a measurement of myosin isometric force (i.e. a “stronger” myosin will require a greater frictional load and thus more alpha-actinin in order to stop thin filament sliding). As can be seen in Fig. 4, there is no difference in the index of retardation between the WT (kr = 3.8 ± 0.2 μg) and NTg (kr = 3.5 ± 0.3 μg; p = 0.86). There is however a dramatic reduction in isometric force for N47K (kr = 1.8 ± 0.2 μg; p < 0.01) and a less pronounced reduction observed for R58Q (kr = 2.9 ± 0.2 μg; p< 0.01) compared to the WT.
Figure 4.

Frictional loading assays measuring the isometric force of the WT (dotted line), NTg (solid line), N47K (short-dashed line), and R58Q (long-dashed line) myosins. The amount of alpha-actinin needed to stop filament sliding velocity gives a measurement of the myosin isometric force. The average actin filament sliding velocity and standard errors in the mean sliding velocity are plotted as a function of alpha-actinin added to the motility assay surface. Data was fit as described in the methods. Both R58Q and N47K showed significant reductions in isometric force compared to the WT myosin.
Regulated Motility Assays
Fiber studies of the R58Q mutation in skinned muscle fibers showed a shift towards submaximal calcium activation of ATPase compared to WT, whereas N47K did not show a shift [24]. To examine whether this shift in calcium sensitivity resulted from changes in myosin at the molecular level, the sliding velocity of tropomyosin and troponin decorated thin filaments was measured as a function of added calcium (Fig. 5). Consistent with fiber studies, there is no change in the calcium sensitivity of unloaded sliding velocity, as evidenced in the pCa50, for NTg (pCa50=6.16 ± 0.10; p=0.63), or N47K (pCa50=6.23 ± 0.04; p=0.82) when compared to the WT (pCa50=6.21 ± 0.05) (Fig. 5). However, consistent with fiber studies [24], we observed a leftward shift in the pCa50 of the R58Q (pCa50=6.66 ± 0.05; p<0.01) compared to the WT, indicative of submaximal calcium activation of thin filament sliding.
Figure 5.

Actin filaments were reconstituted with troponin and tropomyosin and the actin filament sliding velocity was measured as a function of added calcium. Data was fit to Hill curves as described in the methods (Eq. 6). Plotted are the (A) raw data and (B) normalized data. As can be seen, R58Q (long-dashed line) shows a significant shift in the pCa50 of activation compared to the WT (dotted line) and NTg (solid line) whereas N47K (short-dashed line) shows no change in the pCa50.
Discussion
In this study the in vitro motility assay was used to examine the mechanical and biochemical properties of myosin isolated from transgenic mice carrying FHC calcium binding site mutations in the RLC, R58Q and N47K. Both mutations have been associated with occurrences of FHC yet they produce different clinical presentations and prognoses. The actin filament velocity, actin activated ATPase, duty cycle, force generating properties, and calcium regulated motility of the myosins were examined to elucidate the mechanical and biochemical properties of the myosins altered by the mutations. The data is summarized in Table 1. A molecular mechanism explaining the more severe disease phenotype associated with the R58Q mutation compared to N47K mutation is also discussed.
At myosin concentrations below 130 μg/ml, the N47K mutant myosin showed similar actin sliding velocity to the WT whereas R58Q showed an increase in sliding velocity compared to WT (Fig. 2). The dependence of velocity on mutant myosin concentration deviated from the predicted trend [17, 34, 41]. According to the conventional detachment limited model of myosin motility, an actin filament in a motility assay will move at maximal velocity if there is at least one head interacting with the filament at any given time. Therefore, once a sufficient density of myosin has been placed on the motility assay surface to ensure that at least one head is interacting with the actin at any time, the actin should move at a constant, maximal velocity, regardless of the addition of more myosin to the surface. For the WT myosin, we observed the expected rise in velocity with myosin concentration to a plateau value at sufficiently high myosin concentrations. However, for the mutant myosins, as the concentration of myosin was increased, actin filament velocity increased to a maximal value and then gradually decreased as the myosin concentration was increased further (although the decrease was less pronounced for N47K). Similar decreases in sliding velocity have been observed using other RLC FHC mutant myosins (J.R.M. unpublished data). To account for the decrease in velocity observed at higher concentrations of mutant myosins, we propose two different, non-exclusive mechanisms. First, it is possible that at a high myosin concentration, there will be an increase in the number of strongly-bound dragging crossbridges since the probability of two heads being attached to actin at any given time increases as the number of myosins on the surface increase. R58Q and N47K both show increases in duty cycle, meaning that the attachment time would be longer, increasing the probability of dragging heads. The second possibility is that the LMM portion of the myosin exerts a frictional drag on sliding actin filaments [42]. The ability of myosin to overcome such a frictional drag would be a function of the isometric force generating capability of the myosin molecule. Consistent with this notion, the mutants, which show reductions in isometric force, have a pronounced decrease in velocity as the myosin concentration is increased. Perhaps, studies with mutant HMM could distinguish between these possibilities.
Since both R58Q and N47K myosins showed similar increases in duty cycle, based on equation 4, one would expect to see similar decreases in velocity for both R58Q and N47K; however, this was not the case. In order for R58Q to show an increase in sliding velocity, the increase in duty cycle must be offset by an increase in ATPase, unitary step size, or both. It appears that the increase in R58Q ATPase activity, at least partially, compensates for the increase in duty cycle resulting in increased velocity of R58Q myosin. This does not, however, preclude changes in the unitary displacement of R58Q myosin. For N47K, the fact that no change was seen for the actin activated ATPase activity compared to WT suggests that the lack of change in velocity can be explained by a reduction in the unitary displacement.
Both R58Q and N47K show a significant increase in duty cycle and consequently one would expect to see an increase in isometric force. However, consistent with studies in skinned papillary muscle fibers [24], both R58Q and N47K show significant reductions in isometric force compared to the WT. The magnitude of the changes in isometric force reported here is somewhat greater compared to the fiber studies. However, it is difficult to directly compare the data derived from an acto-myosin vs. complexed fiber systems as the effect of other filament proteins present in muscle fibers is absent in the in vitro motility assay. For example, the isometric force measurements in the motility assay were done in the absence of tropomyosin and the presence of tropomyosin was shown to alter myosin force production [36, 43, 44].
The isometric force of myosin is given by:
| (Eq. 5) |
where Funi is the unitary force due to a single crossbridge, n is the number of myosin molecules available to interact with the actin, f is the duty cycle, k is the crossbridge stiffness, and d is the myosin step size. The observed reduction in force for both R58Q and N47K, with a concurrent increase in duty cycle, suggests that the reduction in force must come from reductions in either the number of myosin molecules available to interact with the actin filament, the unitary force or both. It is possible that the mutations could cause a preferential alignment of myosin on the motility assay surface, altering the number of myosin molecules available to interact with actin. However, we consider the latter possibility unlikely as both, RLC depletion and phosphorylation of myosin, considered much more drastic alterations to the myosin structure than a one amino acid replacement, do not change the number of available heads (J.R. Moore, unpublished observation). Thus, it appears that the reduction in isometric force observed for both N47K and R58Q myosin could originate from a change in the unitary force produced by myosin. Decreased unitary force for R58Q and N47K myosins could explain decreased maximal force per cross-section of muscle observed in Tg-R58Q and Tg-N47K skinned papillary muscle fibers [24]. These changes when placed in vivo would lead to compensatory hypertrophy of R58Q and N47K mutated hearts.
Regulated motility assays demonstrated no shift in the pCa50 for N47K myosin whereas R58Q myosin showed a clear shift in the pCa50 towards submaximal calcium activation. This result is consistent with previous skinned muscle fiber studies that showed a leftward shift towards submaximal calcium activation only for R58Q but not N47K [24]. Two different, non-mutually exclusive mechanisms, could explain this result. First, it has been suggested that R58Q may indirectly affect (decrease) the release rate of calcium from troponin C, increasing the Ca2+ sensitivity of force generation [24]. Such a shift would reduce the energy barrier to thin filament activation by allowing myosin heads to bind to actin more easily. A decrease in the calcium release rate from troponin C would result in prolonged force transients which, in fact, were observed in intact R58Q papillary muscle fibers from transgenic mice [24]. Another possible explanation stems from the higher ATPase rate observed in this study for R58Q myosin. Since phosphate release is the rate limiting step of the myosin ATPase cycle, it is possible that the R58Q causes an acceleration of the kinetics of phosphate release, which would tend to cause an increase in the rate of transition from a weakly bound to a strongly bound state. An increase in the rate of strong binding crossbridges would allow for the activation of the thin filament at submaximal calcium levels and the observed shift in pCa50 [45]. It is also worth noting that the observed shift in pCa50 for R58Q observed here was greater than the shift observed in previous studies using skinned papillary muscle fibers [24]. As explained earlier, this difference most likely results from the ability of the motility system to dissect the effects of mutations on the regulated actomyosin interaction from broader cellular based changes due to the hypertrophic response.
Furthermore, both R58Q and N47K have been shown to abolish calcium binding to the cationic binding site of the RLC [18, 23] suggesting that the mutations disrupt RLC structure and hence the mechanical properties of the lever arm, which is consistent with our data. A mutant of the calcium binding loop of skeletal myosin, D47A, was also found to have impaired calcium and magnesium binding [46]. Muscle fibers containing the D47A mutant RLC displayed a reduction in stiffness, leading the authors to propose that the change in stiffness results from either changes in the stiffness of the myosin crossbridges or the number of interacting crossbridges due to increased detachment kinetics; however, their experiment could not conclusively distinguish between these possibilities. As suggested by Sherwood et al. [47] using an optical trap, the D47A containing RLC most likely decreased the crossbridge stiffness and unitary step size of myosin. Consistent with this observation, our data suggests that the crossbridge stiffness of R58Q and N47K myosins are altered by the FHC mutations. Furthermore, it has been suggested that the FHC-inducing E22K mutation of the RLC, which is also located near the RLC calcium binding site, alters the conformation of the RLC by changing the intramolecular hydrogen bonding abilities of the RLC [48]. Levine et al. [48] suggested that the E22K mutation, by changing the conformation of the RLC, could affect the ability of the RLC to support the alpha-helical neck region of the myosin heavy chain, consistent with our observations. Single molecule experiments using an optical trap will be able to distinguish whether changes in the crossbridge stiffness or unitary step size (or both) are responsible for the observed effects. In addition, changes to the stiffness of the lever arm could change the efficiency of force transmission between the lever arm and the myosin head, altering the kinetics of ADP release, the force sensitive step in the myosin ATPase cycle (Reviewed in [49]). Force dependent effects on the kinetics of ADP release could affect the myosin attachment time, altering the duty cycle, consistent with the observed changes in duty cycle for the mutants. Both FHC mutations are located in the region of the RLC that interacts with the hinge region of the myosin lever arm which not only contributes to the communication between the lever arm and the catalytic domain of the myosin head but also links the myosin head with the myosin rod [7]. In the crystal structure this region of MHC makes a sharp bend and interacts with the N-lobe of the RLC [7]. Introducing a positively charged lysine residue through the N47K mutation or removing the positively charged arginine residue through the R58Q mutation may significantly alter the electrostatics of the RLC-MHC hinge interaction affecting the ability of myosin cross-bridge to interact with actin.
Implications for FHC
Both R58Q and N47K mutant myosins displayed a reduction in isometric force which could lead to compensatory hypertrophy. The more extreme phenotype for R58Q could stem from several factors: 1) The fact that R58Q shows an increase in ATPase compared to WT and N47K and a reduction in force compared to the WT suggests that the efficiency of the heart might be drastically reduced. ATP consumption would be increased with little increase in force leading to inefficient energy utilization by the R58Q heart. The inefficient ATP utilization would cause an accumulation of metabolites (i.e. ADP and inorganic phosphate) that would change the free energy of ATP hydrolysis [14, 50]. This is consistent with the observed alterations in cardiac muscle energetics seen with mice harboring another FHC mutation, R403Q located in β-myosin heavy chain [51]. Several biophysical studies of FHC mutations have shown a similar increase in enzymatic activity [52–54] with the apparent “gain of function” being responsible for cardiac hypertrophy. This altered ATP utilization in R58Q hearts could explain the difference in phenotypic severity of the individuals harboring the R58Q mutation compared to N47K positive patients. 2) The increased ATPase rate of R58Q compared to the WT and N47K could lead to a decrease in the total amount of free ATP in the cell [51]. There are several ATP dependent processes in the heart involved in calcium sequestering and re-equilibration of ionic balance after myofibril contraction (i.e. the sarcoplasmic reticulum calcium ATPase, the cellular calcium ATPase, and the sodium ATPase) [55] that can become affected by the reduction of available ATP [52]. A reduction of the ATPase activities of these pumps involved in calcium sequestering would keep the heart activated longer during calcium transients, reducing the diastolic interval, leading to diastolic dysfunction. 3) Our data suggests that R58Q, under conditions of steady state calcium, is activated at a lower calcium concentration than the WT and N47K. Since the cardiomyocyte would remain activated at submaximal calcium levels in vivo during calcium transients, this could lead to incomplete cardiac relaxation during diastole, placing an increased work load on the heart, leading to diastolic dysfunction [50, 56]. This effect of incomplete relaxation would be more pronounced during periods of exercise when the diastolic interval is shorter. Consistent with our data, skinned fiber studies by Wang et al. [24] also observed a shift towards submaximal calcium activation in force/ATPase – pCa relationships with R58Q myosin, which led them to propose that increased calcium sensitivity in R58Q fibers is part of the reason for the more extreme phenotype of R58Q compared to N47K [24]. In support of these hypotheses, the in vivo examination of R58Q hearts demonstrates global diastolic dysfunction and decreased contractile efficiency in R58Q hearts compared to controls (personal communication of D. Szczesna-Cordary).
Conclusions and Prospects
The data presented here shows that FHC mutations in the RLC can lead to changes in the mechanical and kinetic properties of myosin, changing the workload of the heart. The heart is known to respond to alterations in workload via several signal transduction cascades that lead to a hypertrophic response that, if left unchecked, could lead to cardiomyopathy [56, 57]. Since the prognosis and clinical presentation of FHC depends on the specific mutation, the knowledge of the mutation mediated effects on force and motion generation may lead to the development of target specific therapeutic measures.
Acknowledgments
We would like to thank Tanya Mealy for technical support and Zhibing Lu for help with Figure 1. This work was supported by NIH-HL077280 and AHA 0435434T (to J.M.), NIH-HL071778 (to D. S-C.), and by AHA 0815704D (to M.G.).
Abbreviations used
- FHC
Familial hypertrophic cardiomyopathy
- RLC
Regulatory light chain
- MHC
Myosin heavy chain
- Tg
Transgenic
- NTg
Non-transgenic
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Szczesna D. Regulatory light chains of striated muscle myosin. Structure, function and malfunction. Curr Drug Targets Cardiovasc Haematol Disord. 2003 Jun;3(2):187–97. doi: 10.2174/1568006033481474. [DOI] [PubMed] [Google Scholar]
- 2.Richard P, Charron P, Carrier L, Ledeuil C, Cheav T, Pichereau C, et al. Hypertrophic cardiomyopathy: distribution of disease genes, spectrum of mutations, and implications for a molecular diagnosis strategy. Circulation. 2003 May 6;107(17):2227–32. doi: 10.1161/01.CIR.0000066323.15244.54. [DOI] [PubMed] [Google Scholar]
- 3.Morita H, Seidman J, Seidman CE. Genetic causes of human heart failure. J Clin Invest. 2005 Mar;115(3):518–26. doi: 10.1172/JCI200524351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Maron BJ. The young competitive athlete with cardiovascular abnormalities: causes of sudden death, detection by preparticipation screening, and standards for disqualification. Card Electrophysiol Rev. 2002 Feb;6(1–2):100–3. doi: 10.1023/a:1017903709361. [DOI] [PubMed] [Google Scholar]
- 5.Tardiff JC. Sarcomeric proteins and familial hypertrophic cardiomyopathy: linking mutations in structural proteins to complex cardiovascular phenotypes. Heart Fail Rev. 2005 Sep;10(3):237–48. doi: 10.1007/s10741-005-5253-5. [DOI] [PubMed] [Google Scholar]
- 6.Poetter K, Jiang H, Hassanzadeh S, Master SR, Chang A, Dalakas MC, et al. Mutations in either the essential or regulatory light chains of myosin are associated with a rare myopathy in human heart and skeletal muscle. Nat Genet. 1996 May;13(1):63–9. doi: 10.1038/ng0596-63. [DOI] [PubMed] [Google Scholar]
- 7.Rayment I, Rypniewski WR, Schmidt-Base K, Smith R, Tomchick DR, Benning MM, et al. Three-dimensional structure of myosin subfragment-1: a molecular motor. Science. 1993 Jul 2;261(5117):50–8. doi: 10.1126/science.8316857. [DOI] [PubMed] [Google Scholar]
- 8.Howard J, Spudich JA. Is the lever arm of myosin a molecular elastic element? Proc Natl Acad Sci U S A. 1996 Apr 30;93(9):4462–4. [PubMed] [Google Scholar]
- 9.Korn ED, Atkinson MA, Brzeska H, Hammer JA, 3rd, Jung G, Lynch TJ. Structure-function studies on Acanthamoeba myosins IA, IB, and II. J Cell Biochem. 1988 Jan;36(1):37–50. doi: 10.1002/jcb.240360105. [DOI] [PubMed] [Google Scholar]
- 10.Perrie WT, Smillie LB, Perry SB. A phosphorylated light-chain component of myosin from skeletal muscle. Biochem J. 1973 Sep;135(1):151–64. doi: 10.1042/bj1350151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chacko S, Conti MA, Adelstein RS. Effect of phosphorylation of smooth muscle myosin on actin activation and Ca2+ regulation. Proc Natl Acad Sci U S A. 1977 Jan;74(1):129–33. doi: 10.1073/pnas.74.1.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sobieszek A. Ca-linked phosphorylation of a light chain of vertebrate smooth-muscle myosin. Eur J Biochem. 1977 Mar 1;73(2):477–83. doi: 10.1111/j.1432-1033.1977.tb11340.x. [DOI] [PubMed] [Google Scholar]
- 13.Sweeney HL, Bowman BF, Stull JT. Myosin light chain phosphorylation in vertebrate striated muscle: regulation and function. Am J Physiol. 1993 May;264(5 Pt 1):C1085–95. doi: 10.1152/ajpcell.1993.264.5.C1085. [DOI] [PubMed] [Google Scholar]
- 14.Cooke R. Modulation of the actomyosin interaction during fatigue of skeletal muscle. Muscle Nerve. 2007 Dec;36(6):756–77. doi: 10.1002/mus.20891. [DOI] [PubMed] [Google Scholar]
- 15.Collins JH. Myoinformatics report: myosin regulatory light chain paralogs in the human genome. J Muscle Res Cell Motil. 2006;27(1):69–74. doi: 10.1007/s10974-006-9057-6. [DOI] [PubMed] [Google Scholar]
- 16.Reinach FC, Nagai K, Kendrick-Jones J. Site-directed mutagenesis of the regulatory light-chain Ca2+/Mg2+ binding site and its role in hybrid myosins. Nature. 1986 Jul 3–9;322(6074):80–3. doi: 10.1038/322080a0. [DOI] [PubMed] [Google Scholar]
- 17.Szczesna-Cordary D, Jones M, Moore JR, Watt J, Kerrick WG, Xu Y, et al. Myosin Regulatory Light Chain E22K Mutation Results in Decreased Cardiac Intracellular Calcium and Force Transients. Faseb J. 2007 Jul 2; doi: 10.1096/fj.07-8630com. [DOI] [PubMed] [Google Scholar]
- 18.Szczesna-Cordary D, Guzman G, Ng SS, Zhao J. Familial hypertrophic cardiomyopathy-linked alterations in Ca2+ binding of human cardiac myosin regulatory light chain affect cardiac muscle contraction. The Journal of biological chemistry. 2004 Jan 30;279(5):3535–42. doi: 10.1074/jbc.M307092200. [DOI] [PubMed] [Google Scholar]
- 19.Mazhari SM, Selser CT, Cremo CR. Novel sensors of the regulatory switch on the regulatory light chain of smooth muscle Myosin. The Journal of biological chemistry. 2004 Sep 17;279(38):39905–14. doi: 10.1074/jbc.M407062200. [DOI] [PubMed] [Google Scholar]
- 20.Andersen PS, Havndrup O, Bundgaard H, Moolman-Smook JC, Larsen LA, Mogensen J, et al. Myosin light chain mutations in familial hypertrophic cardiomyopathy: phenotypic presentation and frequency in Danish and South African populations. J Med Genet. 2001 Dec;38(12):E43. doi: 10.1136/jmg.38.12.e43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Flavigny J, Richard P, Isnard R, Carrier L, Charron P, Bonne G, et al. Identification of two novel mutations in the ventricular regulatory myosin light chain gene (MYL2) associated with familial and classical forms of hypertrophic cardiomyopathy. J Mol Med. 1998 Mar;76(3–4):208–14. doi: 10.1007/s001090050210. [DOI] [PubMed] [Google Scholar]
- 22.Kabaeva ZT, Perrot A, Wolter B, Dietz R, Cardim N, Correia JM, et al. Systematic analysis of the regulatory and essential myosin light chain genes: genetic variants and mutations in hypertrophic cardiomyopathy. Eur J Hum Genet. 2002 Nov;10(11):741–8. doi: 10.1038/sj.ejhg.5200872. [DOI] [PubMed] [Google Scholar]
- 23.Szczesna D, Ghosh D, Li Q, Gomes AV, Guzman G, Arana C, et al. Familial hypertrophic cardiomyopathy mutations in the regulatory light chains of myosin affect their structure, Ca2+ binding, and phosphorylation. The Journal of biological chemistry. 2001 Mar 9;276(10):7086–92. doi: 10.1074/jbc.M009823200. [DOI] [PubMed] [Google Scholar]
- 24.Wang Y, Xu Y, Kerrick WG, Wang Y, Guzman G, Diaz-Perez Z, et al. Prolonged Ca2+ and force transients in myosin RLC transgenic mouse fibers expressing malignant and benign FHC mutations. J Mol Biol. 2006 Aug 11;361(2):286–99. doi: 10.1016/j.jmb.2006.06.018. [DOI] [PubMed] [Google Scholar]
- 25.Kron SJ, Spudich JA. Fluorescent actin filaments move on myosin fixed to a glass surface. Proc Natl Acad Sci U S A. 1986 Sep;83(17):6272–6. doi: 10.1073/pnas.83.17.6272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Szczesna-Cordary D, Jones M, Moore JR, Watt J, Kerrick WG, Xu Y, et al. Myosin regulatory light chain E22K mutation results in decreased cardiac intracellular calcium and force transients. Faseb J. 2007 Dec;21(14):3974–85. doi: 10.1096/fj.07-8630com. [DOI] [PubMed] [Google Scholar]
- 27.Eisenberg E, Kielley WW. Troponin-tropomyosin complex. Column chromatographic separation and activity of the three, active troponin components with and without tropomyosin present. The Journal of biological chemistry. 1974 Aug 10;249(15):4742–8. [PubMed] [Google Scholar]
- 28.Potter JD. Preparation of troponin and its subunits. Methods Enzymol. 1982;85 Pt B:241–63. doi: 10.1016/0076-6879(82)85024-6. [DOI] [PubMed] [Google Scholar]
- 29.Szczesna D, Zhang R, Zhao J, Jones M, Guzman G, Potter JD. Altered regulation of cardiac muscle contraction by troponin T mutations that cause familial hypertrophic cardiomyopathy. The Journal of biological chemistry. 2000 Jan 7;275(1):624–30. doi: 10.1074/jbc.275.1.624. [DOI] [PubMed] [Google Scholar]
- 30.Gomes AV, Liang J, Potter JD. Mutations in human cardiac troponin I that are associated with restrictive cardiomyopathy affect basal ATPase activity and the calcium sensitivity of force development. The Journal of biological chemistry. 2005 Sep 2;280(35):30909–15. doi: 10.1074/jbc.M500287200. [DOI] [PubMed] [Google Scholar]
- 31.Straub FB. Actin. In Stud Inst Med Chem Univ Szeged. 1942;2:3–16. [Google Scholar]
- 32.Drabikowski W, Gergely J. The effect of the temperature of extraction and of tropomyosin on the viscosity of actin. In: Gergely J, editor. Biochemistry of Muscle Contraction. Boston: Little, Brown, & Co.; 1964. pp. 125–31. [Google Scholar]
- 33.Lowey S, Lesko LM, Rovner AS, Hodges AR, White SL, Low RB, et al. Functional Effects of the Hypertrophic Cardiomyopathy R403Q Mutation Are Different in an {alpha}- or {beta}-Myosin Heavy Chain Backbone. The Journal of biological chemistry. 2008 Jul 18;283(29):20579–89. doi: 10.1074/jbc.M800554200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hooft AM, Maki EJ, Cox KK, Baker JE. An accelerated state of myosin-based actin motility. Biochemistry. 2007 Mar 20;46(11):3513–20. doi: 10.1021/bi0614840. [DOI] [PubMed] [Google Scholar]
- 35.Lineweaver H, Burk D. The Determination of Enzyme Dissociation Constants. J Amer Chem Society. 1934;56:658–66. [Google Scholar]
- 36.Bing W, Knott A, Marston SB. A simple method for measuring the relative force exerted by myosin on actin filaments in the in vitro motility assay: evidence that tropomyosin and troponin increase force in single thin filaments. Biochem J. 2000 Sep 15;350 Pt 3:693–9. [PMC free article] [PubMed] [Google Scholar]
- 37.Godt RE, Lindley BD. Influence of temperature upon contractile activation and isometric force production in mechanically skinned muscle fibers of the frog. J Gen Physiol. 1982 Aug;80(2):279–97. doi: 10.1085/jgp.80.2.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Harris DE, Warshaw DM. Smooth and skeletal muscle myosin both exhibit low duty cycles at zero load in vitro. The Journal of biological chemistry. 1993 Jul 15;268(20):14764–8. [PubMed] [Google Scholar]
- 39.Uyeda TQ, Kron SJ, Spudich JA. Myosin step size. Estimation from slow sliding movement of actin over low densities of heavy meromyosin. J Mol Biol. 1990 Aug 5;214(3):699–710. doi: 10.1016/0022-2836(90)90287-V. [DOI] [PubMed] [Google Scholar]
- 40.Moore JR, Krementsova EB, Trybus KM, Warshaw DM. Myosin V exhibits a high duty cycle and large unitary displacement. J Cell Biol. 2001 Nov 12;155(4):625–35. doi: 10.1083/jcb.200103128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Reck-Peterson SL, Tyska MJ, Novick PJ, Mooseker MS. The yeast class V myosins, Myo2p and Myo4p, are nonprocessive actin-based motors. J Cell Biol. 2001 May 28;153(5):1121–6. doi: 10.1083/jcb.153.5.1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Guo B, Guilford WH. The tail of myosin reduces actin filament velocity in the in vitro motility assay. Cell Motil Cytoskeleton. 2004 Dec;59(4):264–72. doi: 10.1002/cm.20040. [DOI] [PubMed] [Google Scholar]
- 43.Haeberle JR, Hemric ME. A model for the coregulation of smooth muscle actomyosin by caldesmon, calponin, tropomyosin, and the myosin regulatory light chain. Can J Physiol Pharmacol. 1994 Nov;72(11):1400–9. doi: 10.1139/y94-202. [DOI] [PubMed] [Google Scholar]
- 44.VanBuren P, Palmiter KA, Warshaw DM. Tropomyosin directly modulates actomyosin mechanical performance at the level of a single actin filament. Proc Natl Acad Sci U S A. 1999 Oct 26;96(22):12488–93. doi: 10.1073/pnas.96.22.12488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gorga JA, Fishbaugher DE, VanBuren P. Activation of the calcium-regulated thin filament by myosin strong binding. Biophys J. 2003 Oct;85(4):2484–91. doi: 10.1016/s0006-3495(03)74671-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Diffee GM, Patel JR, Reinach FC, Greaser ML, Moss RL. Altered kinetics of contraction in skeletal muscle fibers containing a mutant myosin regulatory light chain with reduced divalent cation binding. Biophys J. 1996 Jul;71(1):341–50. doi: 10.1016/S0006-3495(96)79231-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sherwood JJ, Waller GS, Warshaw DM, Lowey S. A point mutation in the regulatory light chain reduces the step size of skeletal muscle myosin. Proc Natl Acad Sci U S A. 2004 Jul 27;101(30):10973–8. doi: 10.1073/pnas.0401699101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Levine RJ, Yang Z, Epstein ND, Fananapazir L, Stull JT, Sweeney HL. Structural and functional responses of mammalian thick filaments to alterations in myosin regulatory light chains. J Struct Biol. 1998;122(1–2):149–61. doi: 10.1006/jsbi.1998.3980. [DOI] [PubMed] [Google Scholar]
- 49.Nyitrai M, Geeves MA. Adenosine diphosphate and strain sensitivity in myosin motors. Philos Trans R Soc Lond B Biol Sci. 2004 Dec 29;359(1452):1867–77. doi: 10.1098/rstb.2004.1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ventura-Clapier R, Garnier A, Veksler V. Energy metabolism in heart failure. J Physiol. 2004 Feb 15;555(Pt 1):1–13. doi: 10.1113/jphysiol.2003.055095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Spindler M, Saupe KW, Christe ME, Sweeney HL, Seidman CE, Seidman JG, et al. Diastolic dysfunction and altered energetics in the alphaMHC403/+ mouse model of familial hypertrophic cardiomyopathy. J Clin Invest. 1998 Apr 15;101(8):1775–83. doi: 10.1172/JCI1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tyska MJ, Hayes E, Giewat M, Seidman CE, Seidman JG, Warshaw DM. Single-molecule mechanics of R403Q cardiac myosin isolated from the mouse model of familial hypertrophic cardiomyopathy. Circ Res. 2000 Apr 14;86(7):737–44. doi: 10.1161/01.res.86.7.737. [DOI] [PubMed] [Google Scholar]
- 53.Palmiter KA, Tyska MJ, Haeberle JR, Alpert NR, Fananapazir L, Warshaw DM. R403Q and L908V mutant beta-cardiac myosin from patients with familial hypertrophic cardiomyopathy exhibit enhanced mechanical performance at the single molecule level. J Muscle Res Cell Motil. 2000;21(7):609–20. doi: 10.1023/a:1005678905119. [DOI] [PubMed] [Google Scholar]
- 54.Keller DI, Coirault C, Rau T, Cheav T, Weyand M, Amann K, et al. Human homozygous R403W mutant cardiac myosin presents disproportionate enhancement of mechanical and enzymatic properties. J Mol Cell Cardiol. 2004 Mar;36(3):355–62. doi: 10.1016/j.yjmcc.2003.12.006. [DOI] [PubMed] [Google Scholar]
- 55.Kammermeier H. High energy phosphate of the myocardium: concentration versus free energy change. Basic Res Cardiol. 1987;82 Suppl 2:31–6. doi: 10.1007/978-3-662-11289-2_3. [DOI] [PubMed] [Google Scholar]
- 56.Piano MR, Bondmass M, Schwertz DW. The molecular and cellular pathophysiology of heart failure. Heart Lung. 1998 Jan-Feb;27(1):3–19. 20–1. doi: 10.1016/s0147-9563(98)90063-2. [DOI] [PubMed] [Google Scholar]
- 57.Heineke J, Molkentin JD. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat Rev Mol Cell Biol. 2006 Aug;7(8):589–600. doi: 10.1038/nrm1983. [DOI] [PubMed] [Google Scholar]