

Abstract
Pulmonary fibrosis and architectural remodeling of tissues can severely disrupt lung function, often with fatal consequences. The etiology of pulmonary fibrotic diseases is varied, with an array of triggers including allergens, chemicals, radiation and environmental particles. However, the cause of one of the most common pulmonary fibrotic conditions, idiopathic pulmonary fibrosis (IPF), is still unclear. This review examines common mechanisms of pulmonary wound-healing responses following lung injury, and highlights the pathogenesis of some of the most widespread pulmonary fibrotic diseases. A three phase model of wound repair is reviewed that includes; (1) injury; (2) inflammation; and (3) repair. In most pulmonary fibrotic conditions dysregulation at one or more of these phases has been reported. Chronic inflammation can lead to an imbalance in the production of chemokines, cytokines, growth factors, and disrupt cellular recruitment. These changes coupled with excessive pro-fibrotic IL-13 and/or TGFβ1 production can turn a well-controlled healing response into a pathogenic fibrotic response. Endogenous regulatory mechanisms are discussed including novel areas of therapeutic intervention. Restoring homeostasis to these dysregulated healing responses, or simply neutralizing the key pro-fibrotic mediators may prevent or slow the progression of pulmonary fibrosis.
Introduction
Following injury it is paramount that tissue architecture is restored to regain normal organ function. Acute inflammatory responses that result from infection or injury can disrupt epithelial and endothelial integrity leading to edema, the recruitment of leukocytes and angiogenesis. The resolution of inflammation through apoptotic and phagocytic pathways often leaves minimal damage and restores normal tissue architecture. However, common to most fibrotic conditions is the presence of a persistent irritant, which can be known agents, such as allergens, toxic chemicals, radiation, or other persistent irritants or unknown factors that trigger idiopathic pulmonary fibrosis (IPF). Indeed, a dysregulated healing response can gradually evolve into a pathogenic fibrotic response when important checkpoints are missed and inflammation becomes unrelenting. These processes can result in a local milieu rich in chemokines, pro-inflammatory, angiogenic, and fibrogenic cytokines, growth factors and tissue destructive enzymes.1, 2, 3 This mélange of dysregulated processes can result in an increased accumulation of extracellular matrix (ECM) components and fibrotic lesions. Concurrent inflammation, tissue destruction and tissue regeneration can present a “perfect storm” of damage and regeneration.
A tightly regulated repair response following tissue injury is therefore critical. A well-coordinated influx of cells replace resident tissue cells, supply essential nutrients, and reform the tissue during a regenerative period. In some cases, this is followed by a period of fibroplasia, with too much extracellular matrix deposition and connective tissue formation. These events are often associated with vascular diseases and can give rise to many clinical conditions such as atherosclerosis, cirrhosis, scleroderma, asthma, and various types of pulmonary fibrosis. The regenerative process following tissue damage, despite having common mechanisms, can lead to various organ-specific disorders. This review will focus on pulmonary fibrotic conditions and, if known, present common regulatory mechanisms across diseases.
The prevalence and incidence of pulmonary fibrotic diseases are hard to estimate, given the vast array of clinical conditions. IPF affecting 30 in 100 0004 with 34 000 new cases annually5 and allergic asthma, affecting one in five in the United States; (http://www.cdc.gov/nchs/fastats/asthma.htm) although not always leading to airway remodeling and fibrosis, which are two of the most common pulmonary fibrotic diseases. In addition, there are many other fibrotic diseases of the lung including cystic lung disease, scleroderma, radiation and chemotherapy-induced fibrosis, granulomatous lung disease, sarcoidosis and environmental, and smoking-associated COPD. These fibrotic conditions are frequently fatal, with a median survival time following diagnosis of 3–5 years in the case of IPF.6
Mechanisms of wound healing and fibrosis
A wound-healing response is often described as having three distinct phases—injury, inflammation and repair (Figure 1 ). Although not all pulmonary fibrotic conditions follow this simple paradigm, it has been a useful model to elucidate the common and divergent mechanisms of pulmonary fibrosis.
Figure 1.
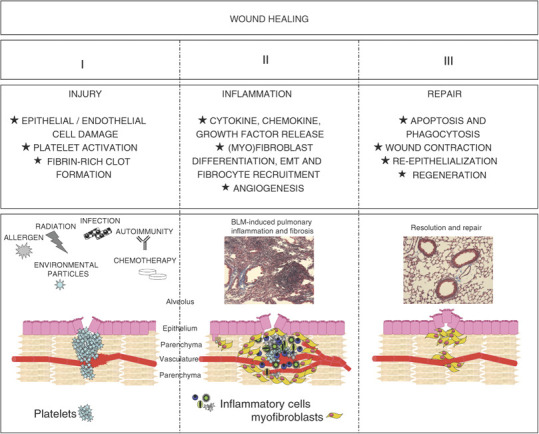
Phases of wound healing. A three-phase injury and wound-healing model describes distinct phases of a successful response. (1) Injury; many agents can cause pulmonary injury, including environmental particles, allergens, infectious agents, chemotherapy and radiation. Disruption of epithelial and endothelial cells initiate an anti-fibrinolytic cascade, temporarily plugging the affected tissue. (2) Inflammation; circulating inflammatory cells and fibrocytes are recruited to the injured site through chemokine gradients, supplying fibroblast-activating cytokines and growth factors. Neo-vascularization provides access to damaged areas and a steady stream of inflammatory, anti-inflammatory, and phagocytic cells. (3) Fibroblasts contract and decrease the size of the wound. Inflammatory cells and α-SMA+ myofibroblasts undergo apoptosis, terminating collagen deposition, and are cleared by phagocytic cells. Epithelial and endothelial cells are replaced and tissue architecture is restored.
Phase I: injury
Injury caused by autoimmune or allergic reactions, environmental particulates, infection or mechanical damage often results in the disruption of normal tissue architecture, initiating a healing response. Inflammation following insult, can also contribute to cellular damage and tissue destruction. Damaged epithelial and endothelial cells must be replaced to maintain barrier function and integrity and prevent blood loss, respectively. Acute damage to endothelial cells leads to the release of inflammatory mediators and initiation of an anti-fibrinolytic coagulation cascade,7 temporarily plugging the damaged vessel with a platelet and fibrin-rich clot. Lung homogenates, epithelial cells or BAL fluid8 from IPF patients express greater levels of the platelet-differentiating factor, X-box-binding protein-1, compared with COPD and control patients,9 suggesting that clot-forming responses are continuously activated. In addition, thrombin (a serine protease required to convert fibrinogen into fibrin) is also readily detected within the lung and intra-alveolar spaces of several pulmonary fibrotic conditions,10, 11, 12 further confirming the activation of the clotting pathway. Thrombin can also directly activate fibroblasts,13 increasing proliferation and promoting fibroblast differentiation into collagen-producing myofibroblasts.14, 15 Damage to the airway epithelium, specifically alveolar pneumocytes16 can evoke a similar anti-fibrinolytic cascade and lead to interstitial edema, areas of acute inflammation and separation of the epithelium from the basement membrane.17
Platelet recruitment, degranulation and clot formation rapidly progress into a phase of vasodilation with increased permeability,18 allowing the extravasation and direct recruitment of leukocytes to the injured site. The basement membrane, which forms the ECM underlying the epithelium and endothelium of parenchymal tissue, precludes direct access to the damaged tissue. To disrupt this physical barrier, zinc-dependent endopeptidases, also called matrix metalloproteinases (MMPs), cleave one or more ECM constituents allowing extravasation of cells into, and out of, damaged sites. Specifically, MMP-2 (gelatinase A, Type N collagenase) and MMP-9 (Gelatinase B, Type IV collagenase) cleave type N collagens and gelatin, two important constituents of the basement membrane.19, 20, 21 In the majority of studies, but not all,22 MMP-2 and MMP-9 are upregulated23, 24, 25, 26 highlighting the tissue destructive and regenerative processes common in fibrotic conditions.
The precise function of MMP-2 and MMP-9 was elegantly demonstrated in a model of allergic airway inflammation and remodeling with MMP-2−/−, MMP-9−/− and MMP-2−/−MMP-9−/− double knockout mice.27, 28 In these studies, the authors demonstrated that MMP-2, and more importantly MMP-9, were required for successful egression and clearance of inflammatory cells out of the inflamed tissue and into the airspaces. In the absence of these MMPs, cells were trapped within the parenchyma of the lung and were not able to move into the airspaces, which resulted in fatal asphyxiation.
The activities of MMPs are controlled by several mechanisms including transcriptional regulation, proenzyme regulation, and specific tissue inhibitors of MMPs. The balance between MMPs and the various inhibitory mechanisms can regulate inflammation and determine the net amount of collagen deposited during the healing response.4
Phase II: inflammation
Once access to the site of tissue damage has been achieved, chemokine gradients recruit inflammatory cells. Neutrophils, eosinophils,29 lymphocytes, and macrophages are observed at sites of acute injury with cell debris and areas of necrosis cleared by phagocytes. The influence of specific inflammatory cells on downstream fibrosis, particularly in IPF, is controversial (30, 31, 32 and was recently reviewed33). One school of thought stems from the observation that anti-inflammatory agents have little efficacy in the treatment of IPF34, 35, 36 and usual interstitial pneumonia patients. Based on these observations, many investigators have suggested that inflammation per se may not be a contributing factor in fibrosis. However, we believe the controversy reflects our limited knowledge and insight into the causative agent(s) and mechanisms involved in IPF. The timing of inflammatory events may determine the role played by the inflammatory process. Early inflammation that is diminished at the later stages of disease may promote wound healing and may contribute to fibrosis. For example the early recruitment of eosinophils, neutrophils, lymphocytes, and macrophages providing inflammatory cytokines and chemokines can contribute to local TGFβ and IL-13.37, 38, 39, 40, 41 However, following the initial insult and wave of inflammatory cells, a late-stage recruitment of inflammatory cells may assist in phagocytosis, clear cell debris, and control excessive cellular proliferation, which together may contribute to normal healing. Thus late-stage inflammation may in fact serve an anti-fibrotic role and could be required for successful resolution of wound-healing responses. For example a late-phase inflammatory profile rich in phagocytic macrophages,42 assisting in fibroblast clearance, in addition to IL-10-secreting regulatory T cells, suppressing local chemokine production and TGFβ,43 may prevent excessive fibroblast activation. Thus, the absence of inflammation observed in IPF patients,36 and interpretation that inflammation is not involved, may simply be a matter of timing. Indeed, corticosteroids that inhibit endogenous suppressive and phagocytic pathways may even be detrimental. However, It should not be forgotten that the mechanisms leading to pulmonary fibrosis are diverse, with immeasurable genetic, environmental and immunological interactions regulating the entire process.
The nature of the insult or causative agent often dictates the character of the ensuing inflammatory response. For example, exogenous stimuli like pathogen-associated molecular patterns (PAMPs) are recognized by pathogen recognition receptors, such as toll-like receptors and NOD-like receptors, and influence the response of innate cells to invading pathogens.44 Endogenous danger signals45 can also influence local innate cells and orchestrate the inflammatory cascade. For immunologists, classifying the type of immune response into Type-1 (Th1 cells, IFNγ, TNFα, and IgG2 antibody responses, generally considered pro-inflammatory) Type 2 (Th2 cells, IL-4, IL-5, IL-13, and IgE, generally considered as a wound-healing response) and type 17 (Th17 cells, recently associated with pro-inflammatory conditions) based upon the T helper cell-dominant cytokine responses, although often oversimplifying, allows for easier discussion.
The nature of the inflammatory response dramatically influences resident tissue cells and the ensuing inflammatory cells. Inflammatory cells themselves also propagate further inflammation through the secretion of chemokines, cytokines, and growth factors. Many cytokines are involved throughout a wound-healing and fibrotic response, with specific groups of genes activated in various conditions. For example, chronic allergic airway disease in asthmatics is commonly associated with elevated type-2 cytokine profiles (IL-4, IL-5, IL-13, IL-9, IL-346) whereas IPF patients more frequently present pro-inflammatory cytokine profiles (IL-1α, IL-1β, TNFα, TGFβ, and platelet-derived growth factors (PDGF)47). Among many cytokines in various pulmonary fibrotic conditions, IL-4, IL-13, and TGF-β have received significant attention. Each of these cytokines can exhibit significant pro-fibrotic activity,48, 49, 50, 51 acting through the recruitment, activation and proliferation of fibroblasts, macrophages, and myofibroblasts.2
Type-2 inflammatory responses: pro-fibrotic IL-4 and IL-13
IL-4, the archetypal type-2 cytokine, has been firmly established as a pro-fibrotic cytokine and is elevated in IPF,52 cryptogenic fibrosing alveolitis,53 radiation-induced pneumonitis and pulmonary fibrosis54 as well as liver fibrosis following infection with Schistomsoma mansoni. 55 IL-4 receptors are present on lung fibroblasts49 with IL-4 signaling increasing extra cellular matrix proteins and collagen deposition. Surprisingly, some studies have suggested that IL-4 is superior to TGF-β1 at inducing collagen synthesis from fibroblasts.49 Indirect mechanisms of IL-4 include its ability to promote the alternative activation of macrophages (AA-Mac), identified by the expression of arginase,56 Fizz-1,57 Ym-1,58 and mannose receptors.59 Macrophages in general have long been associated with pulmonary fibrosis. However, the precise mechanisms and functions of AA-Macs in pulmonary fibrosis are only now being dissected. AA-Macs can produce TGF-β, PDGF60 and, through arginase upregulation, modulate polyamine and proline biosynthesis, cell growth, and collagen formation.61 AA-Macs have been isolated and cultured from the bronchoalveolar lavage (BAL) of IPF patients,62 with culture supernatants from these AA-Macs significantly increasing collagen production by normal human fibroblasts in a CCL18-dependent manner. Animal studies have also identified the involvement of AA-Macs in several models of fibrosis, including mice overexpressing human TGFβ in the lung,63 in human and animal studies of dystrophic muscle fibrosis64 and in multiple organ fibrosis following infection of IFNγR−/− mice with γ herpes virus.65 Although not identified as AA-Macs, macrophages in general have long been appreciated in human66 and animal models of pulmonary fibrosis.67, 68, 69 Together these data suggested that direct secretion of TGFβ, PDGF and proline by AA-Macs are just a few of the many ways in which AA-Macs influence the progression of pulmonary fibrosis.
Finally, one of the most renowned properties of IL-4 is its ability to promote the differentiation of T cells into Th2 cells, providing a source of many type-2 cytokines in this inflammatory axis (IL-5, IL-9, IL-13, and IL-21). The Th2 cytokines interact in dramatic ways propagating wound healing and potentially pro-fibrotic responses. For example, IL-5 mobilizes, matures, and recruits eosinophils,70 with IL-4 promoting TGF-β production from eosinophils.37 IL-5 can also augment IL-13 production and increase IL-13-dependent fibrosis.71 IL-9 can selectively recruit and activate mast cells,72 with mast-cell-derived chymase increasing TGFβ activity and contributing to pulmonary fibrosis.73 Mast cells can also promote fibroblast proliferation, collagen, and MMP production,74 and may be involved in subepithelial fibrosis following allergen challenge.75 IL-21 can also amplify Th2 pulmonary responses and IL-13-associated fibrosis by upregulating IL-4/IL-13 receptor expression. Mice deficient in the IL-21R showed reduced IL-13-dependent fibrosis following S. mansoni infection76 and reduced IL-13-mediated AHR in a murine model of asthma, suggesting it may be an important regulator of Th2-driven remodeling in the lung.77
IL-13 shares many properties with IL-4, due to common receptor subunits (IL-4Rα), signal transduction pathways and transcription factors (STAT-6). However, recent animal studies have identified IL-4Rα-78 and STAT-679-independent IL-13-associated responses, which may involve IL-13 signaling through IL-13Rα2.80, 81, 82 Despite the common properties between IL-4 and IL-13, IL-13 has been identified as a key fibrogenic cytokine in many fibrotic conditions (83, reviewed in 51) and can function independently of TGF-β.84 IL-13 can trigger the differentiation of fibroblasts into α-smooth muscle actin (α-SMA) expressing myofibroblasts and PDGF-producing cells85 with significant mitogenic properties. Interestingly, IL-13-mediated differentiation of fibroblasts into myofibroblasts is refractory to steroid inhibition, which may explain why steroids are not effective at inhibiting fibrosis.
Pro-fibrotic IL-13 has been widely studied in animal models, where gain of function experiments using a novel transgenic approach (overexpressing IL-13), led to subepithelial fibrosis accompanied by eosinophilic inflammation and mucus production,86 comparable to allergen-induced airway responses. Similarly, loss of function studies, blocking or germ line deletion of IL-13 but not IL-4, reduced collagen deposition following exposure to aspergillus,87 OVA,88 bleomycin89, 90 and FITC.91 It is important to note that the pro-fibrotic properties of IL-13 are not restricted to the lung, because hepatic fibrosis, following infection with Schistomsoma mansoni is also significantly decreased following IL-13 blockade.91
In vitro culture of normal human fibroblasts with normal human epithelial cells, which were pre-treated with IL-13, produced significantly more TGF-β, soluble and fibrillar collagen,92 supporting the notion that IL-13 can both directly and indirectly promote collagen production by fibroblasts. Indeed, fibroblasts isolated from IPF patients93 and allergic asthmatics94 demonstrate a hyper-responsiveness to IL-13, as well as TGFβ and CCL-2, with significant interplay between these three mediators.93 Several animal studies also propose a model where IL-13, through various receptor subunits80, 81 can induce plasminogen activator and MMP-9, enhancing the release of active TGFβ95 and subsequent fibrosis. Together these human and animal studies indicate a coordinated and potentially combined effect of IL-13 and TGFβ on fibroblast activation and collagen deposition.40
Involvement of TGFβ in pulmonary fibrosis
TGFβ is derived from most cell lineages derived from the bone marrow96 including T cells, macrophages,97 eosinophils, and neutrophils98 and is one of the most widely studied pro-fibrotic cytokines. The potent activity of TGFβ is regulated at the post-transcriptional level by a latency-associated protein (LAP), which keeps TGFβ in an inactive state. Dissociation of TGFβ from LAP is achieved by many agents commonly found in fibrotic conditions, including cathepsins, plasmin,99 calpain,100 thromombospondin,96 integrin αvβ6,101 and MMPs.102 These agents trigger the release of biologically active TGFβ. Once active, TGFβ is incredibly pleiotropic with growth and chemotactic properties, stimulating fibroblast proliferation and the synthesis of extracellular matrix proteins,50 recruiting inflammatory cells through MCP-1 (CCL2)103 and suppressing T-cell responses. The various and often opposing functions of TGFβ are likely explained by its various sources and cellular targets.104 The inhibitory and suppressive properties of TGFβ were reviewed elsewhere,105, 106, 107 whereas this review focuses on the pro-fibrotic properties of TGFβ.
Similar to the approach employed to study IL-13-mediated fibrosis in the lung,86 active TGFβ has also been overexpressed in the lungs of mice, with the development of severe interstitial and pleural fibrosis, consisting of excess collagen deposition, extracellular matrix proteins, fibronectin, elastin, and the presence of myofibroblasts.108 Interestingly, unlike IL-13 overexpression, TGFβ did not recruit inflammatory cells or enhance mucus secretion in the lung, suggesting that TGFβ can directly induce fibrosis in the absence of significant inflammation. Inhibiting TGFβ activity, by interfering with SMAD-mediated signaling,109 significantly reduced dermal,110, 111 renal,112, 113, 114 ocular,115 and pulmonary fibrosis.116, 117 As mentioned above, TGFβ-independent84, 118, 119 as well as TGFβ- and IL-13-combined mechanisms can contribute to wound healing and fibrosis. Knowledge of the precise interactions and non-redundant compensatory pathways in addition to disease-specific dominance of IL-13 and/or TGFβ could significantly improve therapeutic options.
Chemokine cocktails in the fibrotic lung
Cytokine-producing cells are efficiently recruited to sites of injury through chemokine gradients. Many chemokine gradients develop during wound-healing responses, each recruiting specific chemokine receptor-bearing cells; however, the CC and CXC chemokine families have received considerable attention in fibrotic responses. For example, eosinophils bearing CCR3 and following CCL11 (Eotaxin) gradients and neutrophils, macrophages and monocytes, bearing CCR2 and following IL-8 (KC in mice), IL-17, CCL2 (MCP-1) and CCL3 (MIP1α) gradients120 have all been implicated in pulmonary fibrosis. CCL2, CCL3, and CCL11 are themselves upregulated in pulmonary fibrotic conditions,121, 122, 123, 124, 125, 126, 127 with gene-deficient animal models confirming their importance.124, 128, 129 However, a previously underappreciated circulating cell, the fibrocyte, expressing CCR2,130 CCR3,131 CCR5,132 and CCR7,131 as well as CXCR4,133 represents a significant population of collagen-producing cells.134 The discovery of a rapid, ready, and plentiful supply of collagen-producing fibrocytes from the bone marrow adds a new dimension to pulmonary wound repair and fibrosis.135, 136 Currently, there are three potential origins of α-SMA+ myofibroblasts in lung fibrosis; (1) resident interstitial fibroblasts differentiating into collagen-secreting and extracellular matrix producing cells; (2) a process of epithelial to mesenchymal transformation (EMT) where local epithelial cells adopt fibroblast-like properties and (3) the extravasation of circulating fibrocytes, originating from the bone marrow and differentiating in the tissue into myofibroblasts.137
During chronic injury, endothelial cells enter a process of vasculogenesis (de-novo blood vessel formation) and angiogenesis (budding of new capillary branches from existing blood vessels),138 laying down dense vascular beds permeating fibrotic and regenerative tissue. Angiogenesis can be controlled by several angiogenic factors including vascular endothelial growth factor (VEGF), fibroblast growth factor, TGFβ, PDGF, angiopoietin 1 (Ang1) and a vast array of cytokines139 and chemokines.137
In particular, CXC chemokines identified as angiogenic or angiostatic by their amino terminus, 3-aa sequence (Glu-Leu-Arg), known as the ELR motif, regulate the degree of neo-vascularization and remodeling. In general ELR+CXC chemokines (CXCL1, 2, 3, 5, and 8), which bind to CXCR2, are angiogenic and ELR−CXC chemokines (CXCL4, 9, 10, and 11), which bind to CXCR3, are angiostatic. BAL fluid from IPF patients is rich in ELR+CXC chemokines with a relative downregulation of ELR−CXC chemokines.139, 140, 141 Imbalanced ELR+ and ELR− CXC chemokine levels have also been observed in animal models of pulmonary fibrosis,142, 143, 144 confirming observations made in patients.
In summary, inflammation and the recruitment of circulating granulocytes, lymphocytes, monocytes, macrophages, and fibrocytes, presents a continuous supply of pro- and anti-fibrotic players, vital for efficient wound repair but potentially deadly when not adequately controlled. Every step of this pathway requires negative feedback loops that evoke significant control over the entire process. An imbalance in chemokine production coupled with dysregulated cellular recruitment can result in an excess of pro-fibrotic IL-13 or TGFβ, a surplus of myofibroblasts, and can convert a normal wound-healing response into a pathological fibrotic reaction.
Phase III: tissue repair and contraction
The closing phase of wound healing consists of an orchestrated cellular re-organization guided by a fibrin-rich scaffold formation, wound contraction, closure and re-epithelialization. The vast majority of studies elucidating the processes involved in this phase of wound repair have come from dermal wound studies and in vitro systems. For this reason, we will extrapolate these studies to the lung, where possible.
Myofibroblast-derived collagens and α-SMA form the provisional extracellular matrix, with macrophage, platelet, and fibroblast-derived fibronectin145, 146 forming a fibrin scaffold. Collectively, these structures are commonly referred to as granulation tissues. Primary fibroblasts or alveolar macrophages147 isolated from IPF patients produce significantly more fibronectin and α-SMA than control fibroblasts,148 indicative of a state of heightened fibroblast activation. Interestingly, IPF patients undergoing steroid treatment had similar elevated levels of macrophage-derived fibronectin as IPF patients without treatment. Thus, similar to steroid resistant IL-13-mediated myofibroblast differentiation,85 macrophage-derived fibronectin release147 also appears to be resistant to steroid treatment, providing another reason why steroid treatment may be ineffective. From animal models, fibronectin149, 150 appears to be required for the development of pulmonary fibrosis, as mice with a specific deletion of extra type III domain of fibronectin (EDA) developed significantly less fibrosis following bleomycin administration148 compared with their wild-type counterparts.
In addition to fibronectin, the provisional extracellular matrix consists of glycoproteins (such as PDGF151), glycosaminoglycans (such as Hyaluronic acid152), proteoglycans,153 and elastin.154, 155 Growth factor and TGFβ-activated fibroblasts migrate along the extracellular matrix network and repair the wound. Within skin wounds, TGFβ also induces a contractile response, regulating the orientation of collagen fibers.156 Fibroblast to myofibroblast differentiation, as discussed above, also creates stress fibers and the neo-expression of α-SMA,157 both of which confer the high contractile activity158 within myofibroblasts. The attachment of myofibroblasts to the extracellular matrix at specialized sites called the “fibronexus” or “super mature focal adhesions” pull the wound together, reducing the size of the lesion during the contraction phase.159 The degree of extracellular matrix laid down and, the quantity of activated myofibroblasts160 determines the amount of collagen deposition. To this end, the balance of MMPs to TIMPs161, 162, 163 and collagens to collagenases vary throughout the response, shifting from pro-synthesis and increased collagen deposition, towards a controlled balance, with no net increase in collagen. For successful wound healing, this balance often occurs when fibroblasts undergo apoptosis, inflammation begins to subside, and granulation tissue recedes, leaving a collagen-rich lesion. The removal of inflammatory cells and especially α-SMA+ myofibroblasts is essential to terminate collagen deposition.164 Interestingly, in IPF patients, the removal of fibroblasts can be delayed, with cells resistant to apoptotic signals,165, 166, 167 despite the observation of elevated levels of the apoptosis inductor9 and FAS-signaling molecules.164 This relative resistance to apoptosis may potentially underlie this fibrotic disease.160, 168 However, it is important to note that several studies have also observed increased rates of collagen-secreting fibroblast and epithelial cell169 apoptosis in IPF,170 suggesting that yet another balance requires monitoring—that of fibroblast apoptosis and fibroblast proliferation. The signals which initiate fibroblast apoptosis in IPF, are not very well understood with several factors postulated, such as cytokine imbalances, genetic causes, and constitutive anti-apoptotic pathways160, 165, 170, 171 similar to some cancerous cells.
From skin studies, re-epithelialization of the wound site re-establishes barrier function and allows encapsulated cellular re-organization. Several in vitro and in vivo 172 models, using human173 or rat174 epithelial cells grown over a collagen matrix, or tracheal wounds in vivo, have identified significant stages of cell migration, proliferation,175 and cell spreading. Rapid and dynamic motility and proliferation, with epithelial restitution from the edges of the denuded area172, 173, 176 occur within hours of the initial wound. In addition, sliding sheets of epithelial cells can migrate over the injured area177, 178 assisting wound coverage. Several factors can regulate re-epithelialization with serum-derived TGFα174 or MMP-7179, 180 (which itself is regulated by TIMP-1181) appearing to play significant roles.
Collectively, the degree of inflammation, angiogenesis, and amount of extracellular matrix deposition all contribute to the net collagen deposition and ultimately whether a fibrotic lesion develops. Therapeutic intervention, interfering with fibroblast activation, proliferation or apoptosis requires a thorough understanding and appreciation of all of the phases of wound repair. Although these three phases are often presented sequentially, during chronic or repeated injury these processes function in parallel, placing significant demands on regulatory mechanisms.
Etiology and pathogenesis of common pulmonary fibrotic diseases
Alleviating symptoms is the primary concern of patients presenting pulmonary fibrosis. Understanding the etiology of pulmonary fibrosis can provide long-term symptomatic relief and possible reversal of the disease. To this end, there are currently several well-known risk factors associated with pulmonary fibrosis that will be described below. In many cases, animal models have obvious advantages in studying the regulatory mechanisms in pulmonary fibrosis and airway remodeling.
Cystic fibrosis and cystic lung disease
With regard to etiology, cystic fibrosis (CF) is unique among pulmonary fibrotic conditions and can be attributed to a single gene mutation making it the most common monogenic disease of Caucasians, affecting 1 in 2,500–4,000.182
CF transmembrane conductance regulator (CFTR),183 is the genetic “Achilles heal” responsible for the disease. The CFTR protein product is a chloride channel protein found in the membrane of cells lining the lungs, as well as the liver, pancreas, intestines, reproductive tract, and skin.184, 185, 186 However, the leading cause of mortality in humans with CF is lung disease.184 In addition to direct effects of CFTR mutations, resulting in deficient cAMP-mediated chloride secretion across epithelia and dysfunctional mucus regulation, CF patients are prone to progressive pulmonary damage, submucosal inflammation, and increased susceptibility to bacterial infection.187 Long-term aerosolized antibiotics may limit bacterial colonization;188, 189, 190 however, a consequence of chronic infection is recurring lung injury, chronic inflammation,191, 192 airway remodeling,193 and fibrosis. The chronic inflammatory response, in particular the neutrophilic response, is a significant feature driving pathology in CF. Gaggar et al. 194 recently identified that neutrophil elastase, an enzyme that is significantly elevated in BAL fluid from CF patients, can promote pro-MMP9 and inhibit TIMP-1, thereby disrupting the protease/anti-protease balance.194, 195, 196 In addition, epithelial cell regeneration and repair may also be disrupted, accounting for altered lung physiology in CF.197 Several cftr −/− mice have been developed with varying degrees of lung disease, identifying CF-modifying genes within different founding lines.198, 199, 200 The monumental task of developing a mouse that spontaneously develops lung disease was achieved, allowing the pathophysiological dissection of murine CF. cftr −/− mice develop parenchymal interstitial thickening and fibrosis with granulocyte influx, fibroblast infiltration and the deposition of matrix proteins.199 The development of a mouse model of CF has allowed interesting studies addressing anti-inflammatory responses,201 the involvement of modifier genes,202, 203 the impact of bacterial infection204, 205 and multi-organ complications.206
Radiation and chemotherapy-induced lung injury
Thoracic radiation therapy (RT) is used to treat lung, eosophageal, breast, and lymphoid cancers. However, a common dose-limiting complication of RT is the development of pulmonary interstitial injury and inflammation, often referred to as radiation pneumonitis and emergence of fibrotic foci.207, 208, 209 Multiple mechanisms have been identified in RT-induced fibrosis, including alveolar damage,210 increased reactive oxygen species (ROS) and the toxic effects of ROS on parenchymal cells,211, 212 disruption of proliferation-associated transcription factors,213 and the influx of inflammatory cells, such as macrophages and lymphocytes.214, 215 Furthermore, dysregulated pro-inflammatory and pro-fibrotic cytokines, TGFβ, IL-6, MMPs,216, 217, 218, 219, 220 and chemokines,221 in addition to reduced anti-inflammatory cytokines following radiation222 can further exacerbate the inflammatory and wound-healing response. Animal models have revealed genetic determinants of RT-induced fibrosis213, 223 corresponding with similar genotype-related associations in humans.224 Collectively, RT of the thoracic region can cause significant damage to radiation-sensitive alveolar regions of the lung invoking a dysregulated inflammatory cascade, rich in pro-inflammatory and pro-fibrotic mediators. Dysregulated chemokines, transcription factors, and anti-inflammatory pathways can further compound this uncontrolled response, leading to pulmonary fibrosis.
Similar to radiation therapy, chemotherapy can cause lung injury with variable consequences depending on dose rate, duration, pre-existing lung disease, and concomitant steroid use.225, 226 The Streptomyces verticullatus-derived antibiotic, bleomycin (BLM),227 is effective against squamous cell carcinomas and skin tumors;228 however, like RT, an unfavorable side effect involves inflammatory and fibrotic responses in the lung. BLM-induced inflammation occurs in up to 46% of patients treated229 with complications in the lung and skin due to a lack of the endogenous bleomycin-inactivating enzyme, bleomycin hydrolase, in these tissues.230
Our understanding of BLM-induced fibrosis has been assisted by the development of animal models, which reproduce many, but not all, of the characteristics of the human disease.230 BLM can directly cause cell death231 and reduce O2 into free radicals, causing DNA breakage.232 Depending upon the route of administration, epithelial and endothelial cells are some of the earliest cells affected,233 causing a leukocyte-rich inflammatory response. Blockade of this inflammatory response in animal models, with anti-CD11 Ab-inhibiting cellular extravasation, dramatically reduced pulmonary collagen and fibrosis, demonstrating the significant contribution of inflammatory cells on the resulting fibrotic response.234 The inflammatory cytokines, TNFα,235 IL-1β,236 IL-6237 and pro-fibrotic TGFβ238, 239 are accompanied by FAS-L-expressing cells, leading to more apoptosis.16, 240 Blockade of TNFα, IL-1, FAS-Ligand or TGFβ can reduce the inflammatory and resultant fibrotic response following BLM administration.235, 240, 241, 242 Thus, TNFα, IL-1, IL-6, and TGFβ are some of the possibly many mediators involved in BLM-induced fibrosis. The BLM model has been used to dissect the involvement of many cytokines in the pulmonary fibrotic response. The involvement of type-2 cytokines is less clear, with IL-4 and IL-5 playing no significant role,243, 244, 245 whereas IL-13, either directly90 or indirectly through TGFβ,80, 81 contributes to the fibrotic response. There is also evidence that Type-1 cytokines are involved,246 with fewer inflammatory cells, lung hydroxyproline content, weight loss, and mortality observed in IFNγ−/− mice.247 Blocking the IFNγ-promoting cytokine IL-12 or germ line deletion of IL-12248 yielded similar results.248 BLM, although invoking a significant inflammatory response, can also promote fibroblast proliferation249 and TGFβ production from endothelial cells250 directly. Thus, BLM appears to have multiple properties, directly causing cell death and apoptosis, invoking an inflammatory response and promoting fibroblast proliferation and TGFβ production. For these reasons, the mouse model of BLM-induced fibrosis provides a great tool to dissect the relative contribution of the many pathways, cells, and mediators involved in drug-induced fibrosis.
Asthma and allergic airway inflammation
The number of individuals suffering from allergic airway inflammation and asthma has seen an unprecedented growth over the past 30 years, particularly within the urban areas of both developed and developing countries.251 Allergic asthma is a polygenic disease,252 characterized by allergen-specific IgE and IgG1, airway and interstitial eosinophilia, mucus secretion and airway hyper-reactivity.253 Chronic asthma with repeated bouts of allergen exposure and dysregulated inflammation at mucosal surfaces can lead to goblet cell hyperplasia, smooth muscle hypertrophy and hyperplasia, angiogenesis and ultimately subepithelial fibrosis.254, 255, 256, 257
CD4+ Th2 cells orchestrate many aspects of the allergic inflammation, driven by dendritic cell or basophil-derived IL-4 and IL-25.258, 259, 260, 261, 262, 263 Activation and egression of cytokine-secreting Th2 cells into the interstitium and mucosal surfaces of the lung propagate local cellular influx. More specifically, Th2-derived cytokines, IL-5 and IL-9, mobilize, mature and recruit eosinophils and mast cells70, 264, 265 into the tissue and airspaces, and these cells are typically found in biopsies of asthmatic individuals. TGFβ is also significantly elevated in human asthmatics41, 266, 267, 268, 269, 270 with the degree of subepithelial fibrosis correlating with a loss of forced expiratory volume (FEV1). These observations of increased eosinophils, TGFβ and subepithelial fibrosis led Flood-page et al. 271 to study the specific cellular source of TGFβ. Indeed, 86% of TGFβ mRNA+ cells in the bronchial mucosa of asthmatics were eosinophils, distinguishing eosinophils as a significant source of pro-fibrotic TGFβ in the allergic lung.39 Furthermore, several studies have identified correlated collagen deposition with increased numbers of tissue eosinophils and myofibroblasts19, 38 as well as the expression of submucosal MMP9 and MMP12.272
These observations have led to several clinical trials and treatment regimens using anti-IL-5 antibodies to block tissue eosinophilia with few successes. Treatment of allergic asthmatic patients, as well as atopic dermatitis patients,273 with anti-IL-5 antibodies (mepolizumab) led to significant reductions in tissue eosinophilia,271, 274 despite no change in late-phase cutaneous allergic reactions. Most striking was a reduced thickness and density of the extracellular matrix (tenascin, lumican, and procollagen III (COL3A)) following anti-IL-5 treatment, suggesting that IL-5-mediated tissue eosinophilia was indeed responsible for ECM deposition. However, despite these encouraging results, the precise role and involvement of eosinophils in human asthma is debated, with many clinical trials of anti-IL-5 mAb reporting little to no clinical improvement.275, 276
Animal studies, using either IL-5-deficient mice277 or eosinophil-ablated mice278, 279 have supported a significant role for eosinophils, with reduced airway remodeling, including peri-bronchial fibrosis and smooth muscle thickness, in addition to several other features of allergic asthma following chronic airway exposure. Similarly, blocking TGFβ280 or interfering with TGFβ signaling281 could also significantly attenuate airway remodeling following chronic allergen exposure.
Taken together, animal models have demonstrated a clear role for eosinophils and eosinophil-derived TGFβ in airway damage and remodeling. Human studies, however, have produced a spectrum of results and require additional studies, with well-defined end points to address the role of IL-5 and eosinophils in the progression and resolution of subepithelial fibrosis in asthmatic airways.
IL-13 may also be a damaging cytokine in allergic individuals. Many of the pathological conditions identified in allergic asthmatics can be traced to IL-13. For example, IL-13 can mediate goblet cell hyperplasia in local epithelia282 and increase mucus production92 that can block the small airways.283, 284 IL-13 can also promote epithelial repair,285, 286 fibroblast growth,85, 287 EMT,288 and collagen deposition.92 Beyond the airway epithelium, IL-13 also causes smooth muscle hyperplasia289 and subepithelial fibrosis.88 Similar to mechanisms proposed using the bleomycin model, IL-13 can synergize with and promote pro-fibrotic TGFβ290, 291 eotaxin production,40 and TIMP expression.292 Thus, within the context of allergic asthma, eosinophils, TGFβ, and IL-13 may all contribute to airway remodeling and pulmonary fibrosis.
Less common pulmonary fibrotic conditions with known etiologies
Environmental particulates from smoking or occupational exposure can have toxic effects on the mucosal surfaces of the lung. For example, jobs that involve mining or that expose workers to asbestos, metal dusts, or silica dust can cause pulmonary fibrosis.293 Agricultural workers can also be affected,293 with exposure to organic and inorganic substances,294, 295 fumes,296 or moldy hay297 causing allergic inflammation and fibrosis, often referred to as Farmer's Lung.298, 299, 300 Granulomatous lung disease and sarcoidosis is less common, with a global incidence of 16.5–19/100,000.301 These diseases are significantly influenced by genetic and environmental factors. To date, the causative agents have not been identified.302 An alveolar macrophage gene-transcript profile303 that is similar to Mycobacterium tuberculosis infection has led to the hypothesis that bacteria may be involved. However, to date, bacteria have not been isolated from sarcoidosis patients. Chronic inflammation and the development of inflammatory cell-rich pulmonary granulomas,304, 305 rich in type-1 cytokines and chemokines122, 306, 307, 308, 309 and T cells310 can dramatically disrupt parenchymal architecture, endothelial cells and the alveolar spaces of sarcoidosis patients. Immunohistochemical analysis of human and animal lung biopsies and post-mortem histological sections have identified elevated collagen and fibronectin in granulomas of sarcoidosis patients.305 Furthermore, co-expression of pro-fibrotic TGFβ within the granulomas was also observed in sarcoidosis granulomas.311, 312, 313 Despite these varying etiologies, recurring lung injury and inflammation314 is common to many of these fibrotic conditions, and may broadly underlie the pathogenesis of pulmonary fibrosis.
Idiopathic pulmonary fibrosis
When all known causes of interstitial lung disease and fibrosis have been ruled out, the condition is referred to as “idiopathic” (of unknown origin) pulmonary fibrosis (IPF). Despite an unknown etiology, there are a number of conditions and risk factors associated with the disease including; smoking,315 farming, and occupational hazards316, 317 and viral, and bacterial infections.318, 319, 320 Furthermore, in one study, IPF patients had a greater propensity to develop primary lung cancer, compared with non-IPF patients with chronic lung disease or patients without lung disease.321 Reports of familial aggregation of IPF also suggest that there may be a genetic component to IPF.322, 323 As mentioned above, the incidence of IPF in the United States is 30/100 0004 and 34 000 new cases annually,5 with a similar increasing incidence of IPF in the United Kingdom.324
IPF is characterized by usual interstitial pneumonitis325 and progressive interstitial fibrosis caused by excessive extracellular matrix deposition. Regions of fibroblast and myofibroblast accumulation, specifically between the vascular endothelium and alveolar epithelium disrupt the architecture of the lung, giving a “honeycomb” appearance.321 The pathogenesis of IPF has been debated for many years with two different schools of thought. One group suggests an inflammatory stimulus is involved, with recurring inflammation leading to immunopathology, tissue destruction and the propagation of a wound-healing response.32, 36, 326, 327 Others suggest a slightly different pathogenic mechanism in which an initial or absent inflammatory stage is quickly followed by an uncontrolled wound-healing response.30, 328 Central to the argument negating the dominant role of inflammation is the inefficiency of corticosteroids and other anti-inflammatory agents to control IPF34, 35 despite some reports of enhanced survival.329 Furthermore, the ability of epithelial cell-derived TGFβ330, 331 to invoke a fibrotic cascade with increased interstitial collagen and fibroblast proliferation in the absence of inflammation further support these views. We believe the controversy reflects our limited knowledge and insight into the causative agent(s) and pathogenesis of IPF. A common, and accepted view is the early role of inflammatory events, initiating a wound-healing response. Whether the dysregulated wound-healing response continues in the absence of subsequent inflammation or not has yet to be clarified. Continuous chemokine and cytokine production47 in diagnosed IPF patients indicates that damage and subsequent inflammation may be ongoing.
The cytokine profile from biopsy or BAL-derived cells or BAL fluid of IPF patients is rich in pro-inflammatory cytokines; IL-1β,332, 333 IL-8,122 IL-18,334 TNFα,335 MCP-1122, 336as well as Type-2 cytokines, and their receptors.337, 338 The mixed cytokine profile, derived primarily from inflammatory cells67 and leukocytes,33, 339 can have significant effects on all aspects of wound healing including vascular remodeling, myofibroblast differentiation, EMT, TGFβ, and IL-13 production. In addition to the direct fibroblast-activating properties of TGFβ and IL-13, co-expression of these two cytokines in IPF has been observed.93 Fibroblast hyperplasia170 and the reduced expression of apoptotic mechanisms (bcl-2 and membrane FAS-L)340 in IPF can further augment the fibrotic response. Collectively, a cascade of failed regulatory mechanisms and hyper-secretion of cytokines, chemokines and growth factors,47 culminates in an out-of-control fibroblast-mediated wound-healing response. Physiologically, IPF can dramatically compromise oxygen diffusion, lung function341 and is typically a fatal disease.
Regulation of pulmonary fibrosis
It is becoming clear that an imbalance of stimulatory cytokines, chemokines, and growth factors likely over-activate resident parenchymal and circulating cells and may underlie the over exuberant wound-healing responses that lead to fibrosis. In a normally controlled cellular response, negative feed back loops, anti-inflammatory molecules, inhibitory receptors, and apoptotic pathways operate to fine tune and terminate responses once a desired outcome is achieved. Common to many pulmonary fibrotic conditions with both known and unknown etiologies may be a break down in these regulatory mechanisms, resulting in an excessive inflammatory cascade, neo-vascularization, uncontrolled fibroblast activation, and fibrosis. In this section of the review we will highlight some of these endogenous regulatory mechanisms that either operate endogenously or can be exploited therapeutically to counter balance the uncontrolled responses.
Regulation of inflammatory responses: Tregs and IL-10
T cells with the primary function of attenuating immune cell activation and proliferation are frequently referred to as regulatory T cells (Treg). Although the specific details of antigen specificity, precise mechanisms of suppression, and distinguishing features continue to grow, their role in fibrotic responses have been under studied. However, given their ability to dampen inflammatory responses, the ability of Tregs to interfere with upstream events and slow the progression of fibrosis has been implied. In particular Treg-derived IL-10, and other surface molecules342 although not exclusively derived from Tregs, can function as a general immunosuppressant343 and control fibrosis.342 Polymorphisms in the signal sequence of the IL-10 gene have been identified in IPF patients, corresponding to reduced IL-10 production, suggesting that endogenous anti-inflammatory mechanisms may be impaired in this condition. Supporting this notion, loss of function studies using LPS-induced lung injury and fibrosis in IL-10-deficient mice led to significantly stronger inflammatory responses with greater subepithelial thickening and extracelular matrix protein content.344 In gain of function studies, induction of IL-10 significantly reduced collagen deposition following bleomycin administration in murine models.345 Following IL-10 gene delivery BAL fluid TNFα and neutrophil-derived MPO levels were significantly reduced, with another similar study observing reduced macrophage-derived TGFβ,43 suggesting that IL-10 inhibits inflammatory cell recruitment.345 Corroborating these findings, Dosanjh et al. 346 reported higher levels of IL-8, with reduced IL-10 levels in the BAL fluid of cystic fibrosis patients. Thus, IL-10 can attenuate the inflammatory events upstream of the fibrotic pathway. In addition to suppressing inflammatory events, IL-10 can act directly on fibroblasts, reducing TGFβ-induced collagen production.345 Following lung injury in a rat model of radiation-induced fibrosis, pneumocytes in the epithelial layer of the lung had reduced expression of IL-10, compared to control lungs, which may permit greater local inflammation.222 Thus, IL-10 immunotherapy, with a sound understanding of timing and when to dampen inflammatory events may hold promise for pulmonary fibrotic conditions.347 Beyond the lung, IL-10 has been shown to regulate kidney348 and liver349 inflammation, and fibrosis.350 Therapeutically manipulating IL-10, in particular endogenous IL-10-producing cells which may be present but in too low frequencies to significantly halt the inflammatory onslaught, may be a useful avenue to pursue. This has been demonstrated successfully in models of allergic airway inflammation, where a reduction in airway and tissue inflammation, mucus production, and airway hyper-responsiveness was observed.351
IL-13Rα2 and LAP: endogenous attenuators of fibrosis
As discussed throughout this review, TGFβ and IL-1351, 84, 85, 352, 353 are dominant pro-fibrotic cytokines, activating fibroblasts, and promoting differentiation into α-SMA-producing myofibroblasts and collagen production. Thus, tight regulation and fine-tuning of these two potent molecules is essential. Two molecules that can serve this very purpose are the IL-13Rα2, an endogenous decoy receptor that attenuates IL-13 activity and, as discussed earlier, LAP, a latency-associated protein, which keeps TGFβ in an inactive state. To our knowledge there are no studies to date reporting the specific induction of endogenous LAP to attenuate TGFβ bioactivity; however, introduction of exogenous recombinant LAP could theoretically be used to attenuate TGFβ activity354 (similar to anti-TGFβ antibody280). Upregulation of a TGFβ-binding protein, endoglin, however has been observed in animal models of renal fibrosis.355 Exploiting this pathway to attenuate TGFβ may be another option. Although the exact mechanism is unknown, the introduction of taurine and niacin into the diet of small rodents attenuates BLM-induced fibrosis, apparently by reducing TGFβ production,356 suggesting that dietary supplements may be useful therapeutics. Disrupting TGFβ-associated ROS357 or other downstream TGFβ-signaling pathways358 also hold promise.
IL-13Rα2 is expressed predominantly by non-hematopoietic cells (unpublished observations) and attenuates IL-13 activity in vivo.359 Given that IL-13 can act at multiple stages of the inflammatory and wound-healing response,51, 92, 284, 288, 358, 360 it comes as little surprise that attenuation of IL-13 can have profound effects on the degree of pulmonary inflammation and fibrosis in many pulmonary disease models.88, 90, 361 Several methods of IL-13 attenuation have been described, including neutralizing Abs,87 treatment with sIL-13Rα2,359, 362, 363, 364 or targeting the IL-13Rα2-expressing cells. The conclusions from all of these studies indicate that targeting the IL-13 pathway holds great promise for the treatment of fibrosis.
Resetting the imbalance
An imbalance of cytokines, chemokines or cells can disrupt many downstream processes (Figure 2 ). For example, an imbalance between collagen-catabolizing MMPs and their specific inhibitors, TIMPs, can result in excessive collagen breakdown. However, they can also promote TGFβ activation in peripheral cells.365, 366, 367 Increased TGFβ can further feed back to induce more MMPs368 and promote EMT.368 Thus, a breakdown in one process (MMP production) can quickly catalyze and disrupt other regulatory mechanisms (TGFβ responses). Within mammalian systems, a refined balance between “on” and “off” signals is critical to maintain homeostasis. In a dysregulated wound-healing response several key mechanisms appear to be off balance (Figure 2).
Inflammation “vs.” Immunosuppression. Excessive or recurring inflammatory events can cause excessive wound-healing responses that lead to the development of fibrosis. Either eliminating the causative agent, such as allergen avoidance, or treatments with anti-inflammatory agents such as corticosteroids may help restore the balance.
MMP “vs.” TIMP. MMPs can disrupt the basement membrane and allow the influx of inflammatory cells. Inhibiting MMP activity could be detrimental in immunity and in the process of re-epithelialization;369 however, in pathological fibrotic responses, neutralization of specific MMPs either with small molecules,157 inhibitors157 or by influencing TIMP expression may help restore this imbalance.
Fibroblast apoptosis “vs.” proliferation. The late-stage apoptosis of fibroblasts is required for successful wound healing and termination of collagen deposition. As mentioned above, resistance to apoptosis has been observed in fibroblasts from IPF patients.165, 166, 167 Restoring fibroblast apoptotic pathways or selectively depleting fibroblasts at the appropriate time may help slow the progression of fibrosis. Modulating local cytokine and growth factor levels could also influence fibroblast proliferation and activation indirectly.
ELR+ “vs.” ELR − CXC chemokines. The prolonged induction ELR+ chemokines, due to inflammatory signals can lead to excessive vascularization. Anti-angiogenic therapy,370 an area actively pursued in cancer therapy was recently investigated in fibrotic conditions.371 Inhibiting VEGF or promoting endostatin and anastellin (endogenous inhibitors of angiogenesis) may limit inflammation and the recruitment of myofibroblasts. Neutralizing angiogenic ELR+ CXC chemokines or enhancing angiostatic ELR− CXC chemokines,372, 373, 374 in combination with other therapeutic interventions, may also dramatically halt the inflammatory cascade and avoid the requirements for angiogenesis.
Figure 2.
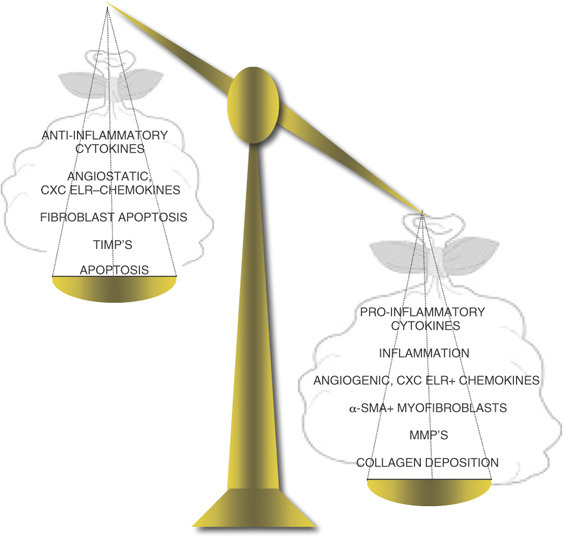
Imbalanced wound-healing response. For successful wound healing, a regulated response is maintained through negative feedback loops and a balance of catabolising and regenerative processes. Several imbalances may develop and lead a normal healing response into a fibrotic cascade. Excessive inflammation and the production of inflammatory and fibroblast-activating cytokines, through a breakdown in anti-inflammatory mechanisms can develop. Over-production of angiogenic CXC ELR+ chemokines, the recruitment of fibrocytes and increased frequency of α-SMA+ cells in the injury site can result in too much collagen deposition. Resetting the balance with targeted therapeutics (i.e., cytokine-blocking antibodies) may help slow the progression of fibrosis.
Conclusion
Pulmonary wound repair is an extremely dynamic process intersecting immunology, structural biology, and airway physiology. For successful repair a collaborative effort between these systems is essential. Dysregulation in one response can have ripple effects on others and progressively turn a well-choreographed healing response into a fibrotic lesion. Vascular damage must be quickly repaired with a fibrin-rich clot. This is followed by an influx of inflammatory cells. Chronic or recurring inflammation requires rapid resolution to avert immunopathology while providing the necessary cellular participants. Parenchymal cells that are responsive to inflammatory cues must proliferate and migrate into the damaged area, restore tissue architecture, with the inflammatory cells ultimately undergoing apoptosis to prevent excessive collagen deposition.
In pulmonary fibrotic disease states, the development and progression of the healing response has slipped out of control, disrupting many delicate balances. As discussed in this review, there are a number of compensatory and redundant processes, which all contribute to proficient healing and remodeling. With this in mind, and despite significant advances in our understanding of these pathways and balances, there is a lack of therapeutic intervention and new therapies for pulmonary fibrosis. Further studies are required to elucidate the roles of the many mediators (cytokines, chemokines and growth factors) observed in both human and animal models of pulmonary fibrosis. Greater still, pre-clinical and clinical investigations with chemokine receptor antagonists, angiogenesis inhibitors and Abs to the pro-fibrotic molecules IL-13 and TGFβ are required. A combined effort by clinicians and lab researchers across platforms and disciplines could make this a climbable mountain.
Disclosure
The author declared no conflict of interest.
Acknowledgments
This review was improved by peer-review and funded by the Intramural Research Program at the NIH/NIAID. Owing to space and word limitations, we apologize to the many researchers whose work we have not mentioned in this review, but who have significantly contributed to our current understanding of pulmonary fibrosis. We also thank Dr Allen Cheever for helpful comments.
Footnotes
Supplementary information The online version of this article (doi:10.1038/mi.2008.85) contains supplementary material, which is available to authorized users.
Acknowledgments
PowerPoint slide for Fig. 1
PowerPoint slide for Fig. 2
References
- 1.Wynn TA. Common and unique mechanisms regulate fibrosis in various fibroproliferative diseases. J. Clin. Invest. 2007;117,:524–529. doi: 10.1172/JCI31487. 1:CAS:528:DC%2BD2sXis12hsbY%3D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wynn TA. Fibrotic disease and the T(H)1/T(H)2 paradigm. Nat. Rev. Immunol. 2004;4,:583–594. doi: 10.1038/nri1412. 1:CAS:528:DC%2BD2cXmtFWktr0%3D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown RA. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat. Rev. Mol. Cell Biol. 2002;3,:349–363. doi: 10.1038/nrm809. 1:CAS:528:DC%2BD38XjsFOgsbw%3D. [DOI] [PubMed] [Google Scholar]
- 4.Pardo A, Selman M. Matrix metalloproteases in aberrant fibrotic tissue remodeling. Proc. Am. Thorac. Soc. 2006;3,:383–388. doi: 10.1513/pats.200601-012TK. 1:CAS:528:DC%2BD28XmtVOktrs%3D. [DOI] [PubMed] [Google Scholar]
- 5.Raghu G, Weycker D, Edelsberg J, Bradford WZ, Oster G. Incidence and prevalence of idiopathic pulmonary fibrosis. Am. J. Resp. Crit. Care Med. 2006;174,:810–816. doi: 10.1164/rccm.200602-163OC. [DOI] [PubMed] [Google Scholar]
- 6.Daniil ZD. A histologic pattern of nonspecific interstitial pneumonia is associated with a better prognosis than usual interstitial pneumonia in patients with cryptogenic fibrosing alveolitis. Am. J. Resp. Crit. Care Med. 1999;160,:899–905. doi: 10.1164/ajrccm.160.3.9903021. [DOI] [PubMed] [Google Scholar]
- 7.Chambers RC. Role of coagulation cascade proteases in lung repair and fibrosis. Eur. Respir. J. Suppl. 2003;44,:33s–35s. doi: 10.1183/09031936.03.00001003. 1:CAS:528:DC%2BD3sXovFCju7s%3D. [DOI] [PubMed] [Google Scholar]
- 8.Hamada N. The role of high mobility group box1 in pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2008;39,:440–447. doi: 10.1165/rcmb.2007-0330OC. 1:CAS:528:DC%2BD1cXht1Srs7%2FF. [DOI] [PubMed] [Google Scholar]
- 9.Korfei M. Epithelial endoplasmic reticulum stress and apoptosis in sporadic idiopathic pulmonary fibrosis. Am. J. Resp. Crit. Care Med. 2008;178,:838–846. doi: 10.1164/rccm.200802-313OC. 1:CAS:528:DC%2BD1cXhtlaisLfK. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dik WA, Zimmermann LJ, Naber BA, Janssen DJ, van Kaam AH, Versnel MA. Thrombin contributes to bronchoalveolar lavage fluid mitogenicity in lung disease of the premature infant. Pediatr. Pulmonol. 2003;35,:34–41. doi: 10.1002/ppul.10219. [DOI] [PubMed] [Google Scholar]
- 11.Hernandez-Rodriguez NA. Role of thrombin in pulmonary fibrosis. Lancet. 1995;346,:1071–1073. doi: 10.1016/S0140-6736(95)91744-6. [DOI] [PubMed] [Google Scholar]
- 12.Gabazza EC. Thrombin in the airways of asthmatic patients. Lung. 1999;177,:253–262. doi: 10.1007/PL00007645. [DOI] [PubMed] [Google Scholar]
- 13.Chen LB, Buchanan JM. Mitogenic activity of blood components. I. Thrombin and prothrombin. Proc. Natl. Acad. Sci. USA. 1975;72,:131–135. doi: 10.1073/pnas.72.1.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bogatkevich GS, Tourkina E, Silver RM, Ludwicka-Bradley A. Thrombin differentiates normal lung fibroblasts to a myofibroblast phenotype via the proteolytically activated receptor-1 and a protein kinase C-dependent pathway. J. Biol. Chem. 2001;276,:45184–45192. doi: 10.1074/jbc.M106441200. 1:CAS:528:DC%2BD3MXovFensLs%3D. [DOI] [PubMed] [Google Scholar]
- 15.Chambers RC, Dabbagh K, McAnulty RJ, Gray AJ, Blanc-Brude OP, Laurent GJ. Thrombin stimulates fibroblast procollagen production via proteolytic activation of protease-activated receptor 1. Biochem. J. 1998;333(Part 1):121–127. doi: 10.1042/bj3330121. 1219564, 1:CAS:528:DyaK1cXkslyisb0%3D, 9639571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kuwano K. Essential roles of the Fas-Fas ligand pathway in the development of pulmonary fibrosis. J. Clin. Invest. 1999;104,:13–19. doi: 10.1172/JCI5628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vaccaro CA, Brody JS, Snider GL. Alveolar wall basement membranes in bleomycin-induced pulmonary fibrosis. Am. Rev. Respir. Dis. 1985;132,:905–912. doi: 10.1164/arrd.1985.132.4.905. [DOI] [PubMed] [Google Scholar]
- 18.McKeown, S., Richter, A.G., O'Kane, C., McAuley, D.F. & Thickett, D.R. Matrix metalloproteinase expression and abnormal lung permeability are important determinants of outcome in IPF. Eur. Respir. J.32, (2008). (E-pub ahead of print). [DOI] [PubMed]
- 19.Hoshino M, Nakamura Y, Sim J, Shimojo J, Isogai S. Bronchial subepithelial fibrosis and expression of matrix metalloproteinase-9 in asthmatic airway inflammation. J. Allergy Clin. Immunol. 1998;104,:783–788. doi: 10.1016/S0091-6749(98)70018-1. [DOI] [PubMed] [Google Scholar]
- 20.Murphy G, Docherty AJ. The matrix metalloproteinases and their inhibitors. Am. J. Respir. Cell Mol. Biol. 1992;7,:120–125. doi: 10.1165/ajrcmb/7.2.120. [DOI] [PubMed] [Google Scholar]
- 21.Corbel M, Belleguic C, Boichot E, Lagente V. Involvement of gelatinases (MMP-2 and MMP-9) in the development of airway inflammation and pulmonary fibrosis. Cell Biol. Toxicol. 2002;18,:51–61. doi: 10.1023/A:1014471213371. [DOI] [PubMed] [Google Scholar]
- 22.Ruiz V. Unbalanced collagenases/TIMP-1 expression and epithelial apoptosis in experimental lung fibrosis. Am. J. Physiol. 2003;285,:L1026–L1036. doi: 10.1152/ajplung.00183.2003. [DOI] [PubMed] [Google Scholar]
- 23.Gadek JE. Collagenase in the lower respiratory tract of patients with idiopathic pulmonary fibrosis. N. Engl. J. Med. 1979;301,:737–742. doi: 10.1056/NEJM197910043011401. [DOI] [PubMed] [Google Scholar]
- 24.O'Connor C, Odlum C, Van Breda A, Power C, Fitzgerald MX. Collagenase and fibronectin in bronchoalveolar lavage fluid in patients with sarcoidosis. Thorax. 1988;43,:393–400. doi: 10.1136/thx.43.5.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Oggionni T. Time course of matrix metalloproteases and tissue inhibitors in bleomycin-induced pulmonary fibrosis. Eur. J. Histochem. 2006;50,:317–325. [PubMed] [Google Scholar]
- 26.O'Connor CM, FitzGerald MX. Matrix metalloproteases and lung disease. Thorax. 1994;49,:602–609. doi: 10.1136/thx.49.6.602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Corry DB. Decreased allergic lung inflammatory cell egression and increased susceptibility to asphyxiation in MMP2-deficiency. Nat Immunol. 2002;3,:347–353. doi: 10.1038/ni773. 1:CAS:528:DC%2BD38Xis1Kksbs%3D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Corry DB. Overlapping and independent contributions of MMP2 and MMP9 to lung allergic inflammatory cell egression through decreased CC chemokines. FASEB. J. 2004;18,:995–997. doi: 10.1096/fj.03-1412fje. 1:CAS:528:DC%2BD2cXkslWit7s%3D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Erjefalt JS, Sundler F, Persson CG. Eosinophils, neutrophils, and venular gaps in the airway mucosa at epithelial removal-restitution. Am. J. Resp. Crit. Care Med. 1996;153,:1666–1674. doi: 10.1164/ajrccm.153.5.8630618. [DOI] [PubMed] [Google Scholar]
- 30.Gauldie J. Pro: Inflammatory mechanisms are a minor component of the pathogenesis of idiopathic pulmonary fibrosis. Am. J. Resp. Crit. Care Med. 2002;165,:1205–1206. doi: 10.1164/rccm.2202054. [DOI] [PubMed] [Google Scholar]
- 31.Piguet PF. Inflammation in idiopathic pulmonary fibrosis. Am. J. Resp. Crit. Care Med. 2003;167,:1037. doi: 10.1164/ajrccm.167.7.952. [DOI] [PubMed] [Google Scholar]
- 32.Strieter RM. Con: Inflammatory mechanisms are not a minor component of the pathogenesis of idiopathic pulmonary fibrosis. Am. J. Resp. Crit. Care Med. 2002;165,:1206–1207. doi: 10.1164/rccm.2202055. [DOI] [PubMed] [Google Scholar]
- 33.Bringardner BD, Baran CP, Eubank TD, Marsh CB. The role of inflammation in the pathogenesis of idiopathic pulmonary fibrosis. Antioxid Redox Signal. 2008;10,:287–301. doi: 10.1089/ars.2007.1897. 1:CAS:528:DC%2BD1cXht1ShsA%3D%3D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Davies, H.R., Richeldi, L. & Walters, E.H. Immunomodulatory agents for idiopathic pulmonary fibrosis. Cochrane Database Syst. Rev. CD003134 (2003). [DOI] [PubMed]
- 35.Richeldi, L., Davies, H.R., Ferrara, G. & Franco, F. Corticosteroids for idiopathic pulmonary fibrosis. Cochrane Database Syst. Rev. CD002880 (2003). [DOI] [PMC free article] [PubMed]
- 36.Selman M, King TE, Pardo A. Idiopathic pulmonary fibrosis: prevailing and evolving hypotheses about its pathogenesis and implications for therapy. Ann. Intern. Med. 2001;134,:136–151. doi: 10.7326/0003-4819-134-2-200101160-00015. [DOI] [PubMed] [Google Scholar]
- 37.Elovic AE. IL-4-dependent regulation of TGF-alpha and TGF-beta1 expression in human eosinophils. J. Immunol. 1998;160,:6121–6127. [PubMed] [Google Scholar]
- 38.Minshall EM. Eosinophil-associated TGF-beta1 mRNA expression and airways fibrosis in bronchial asthma. Am. J. Respir. Cell Mol. Biol. 1997;17,:326–333. doi: 10.1165/ajrcmb.17.3.2733. [DOI] [PubMed] [Google Scholar]
- 39.Ohno I. Transforming growth factor beta 1 (TGF beta 1) gene expression by eosinophils in asthmatic airway inflammation. Am. J. Respir. Cell Mol. Biol. 1996;15,:404–409. doi: 10.1165/ajrcmb.15.3.8810646. [DOI] [PubMed] [Google Scholar]
- 40.Wenzel SE. TGF-beta and IL-13 synergistically increase eotaxin-1 production in human airway fibroblasts. J. Immunol. 2002;169,:4613–4619. doi: 10.4049/jimmunol.169.8.4613. [DOI] [PubMed] [Google Scholar]
- 41.Zagai U, Dadfar E, Lundahl J, Venge P, Skold CM. Eosinophil cationic protein stimulates TGF-beta1 release by human lung fibroblasts in vitro. Inflammation. 2007;30,:153–160. doi: 10.1007/s10753-007-9032-4. 1:CAS:528:DC%2BD2sXhtFGgsrzN. [DOI] [PubMed] [Google Scholar]
- 42.Moodley Y. Macrophage recognition and phagocytosis of apoptotic fibroblasts is critically dependent on fibroblast-derived thrombospondin 1 and CD36. Am. J. Pathol. 2003;162,:771–779. doi: 10.1016/S0002-9440(10)63874-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nakagome K, Dohi M, Okunishi K, Tanaka R, Miyazaki J, Yamamoto K. In vivo IL-10 gene delivery attenuates bleomycin induced pulmonary fibrosis by inhibiting the production and activation of TGF-beta in the lung. Thorax. 2006;61,:886–894. doi: 10.1136/thx.2005.056317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Janeway CA, Jr, Medzhitov R. Innate immune recognition. Annu. Rev. Immunol. 2002;20,:197–216. doi: 10.1146/annurev.immunol.20.083001.084359. [DOI] [PubMed] [Google Scholar]
- 45.Matzinger P. The danger model: a renewed sense of self. Science. 2002;296,:301–305. doi: 10.1126/science.1071059. [DOI] [PubMed] [Google Scholar]
- 46.Holgate ST. Pathogenesis of asthma. Clin. Exp. Allergy. 2008;38,:872–897. doi: 10.1111/j.1365-2222.2008.02971.x. 1:CAS:528:DC%2BD1cXotVOht7Y%3D. [DOI] [PubMed] [Google Scholar]
- 47.Agostini C, Gurrieri C. Chemokine/cytokine cocktail in idiopathic pulmonary fibrosis. Proc. Am. Thorac Soc. 2006;3,:357–363. doi: 10.1513/pats.200601-010TK. 1:CAS:528:DC%2BD28XmtVOksbc%3D. [DOI] [PubMed] [Google Scholar]
- 48.Fertin C, Nicolas JF, Gillery P, Kalis B, Banchereau J, Maquart FX. Interleukin-4 stimulates collagen synthesis by normal and scleroderma fibroblasts in dermal equivalents. Cell Mol. Biol. 1991;37,:823–829. [PubMed] [Google Scholar]
- 49.Sempowski GD, Beckmann MP, Derdak S, Phipps RP. Subsets of murine lung fibroblasts express membrane-bound and soluble IL-4 receptors. Role of IL-4 in enhancing fibroblast proliferation and collagen synthesis. J. Immunol. 1994;152,:3606–3614. [PubMed] [Google Scholar]
- 50.Strutz F. TGF-beta 1 induces proliferation in human renal fibroblasts via induction of basic fibroblast growth factor (FGF-2) Kidney Int. 2001;59,:579–592. doi: 10.1046/j.1523-1755.2001.059002579.x. [DOI] [PubMed] [Google Scholar]
- 51.Wynn TA. IL-13 effector functions. Annu. Rev. Immunol. 2003;21,:425–456. doi: 10.1146/annurev.immunol.21.120601.141142. 1:CAS:528:DC%2BD3sXjtF2isrY%3D. [DOI] [PubMed] [Google Scholar]
- 52.Emura M, Nagai S, Takeuchi M, Kitaichi M, Izumi T. In vitro production of B cell growth factor and B cell differentiation factor by peripheral blood mononuclear cells and bronchoalveolar lavage T lymphocytes from patients with idiopathic pulmonary fibrosis. Clin. Exp. Immunol. 1990;82,:133–139. doi: 10.1111/j.1365-2249.1990.tb05416.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wallace WA, Ramage EA, Lamb D, Howie SE. A type 2 (Th2-like) pattern of immune response predominates in the pulmonary interstitium of patients with cryptogenic fibrosing alveolitis (CFA) Clin. Exp. Immunol. 1995;101,:436–441. doi: 10.1111/j.1365-2249.1995.tb03131.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Buttner C. Local production of interleukin-4 during radiation-induced pneumonitis and pulmonary fibrosis in rats: macrophages as a prominent source of interleukin-4. Am. J. Respir. Cell Mol. Biol. 1997;17,:315–325. doi: 10.1165/ajrcmb.17.3.2279. [DOI] [PubMed] [Google Scholar]
- 55.Booth M. Periportal fibrosis in human Schistosoma mansoni infection is associated with low IL-10, low IFN-gamma, high TNF-alpha, or low RANTES, depending on age and gender. J. Immunol. 2004;172,:1295–1303. doi: 10.4049/jimmunol.172.2.1295. [DOI] [PubMed] [Google Scholar]
- 56.Pauleau AL, Rutschman R, Lang R, Pernis A, Watowich SS, Murray PJ. Enhancer-mediated control of macrophage-specific arginase I expression. J. Immunol. 2004;172,:7565–7573. doi: 10.4049/jimmunol.172.12.7565. [DOI] [PubMed] [Google Scholar]
- 57.Liu T. Regulation of found in inflammatory zone 1 expression in bleomycin-induced lung fibrosis: role of IL-4/IL-13 and mediation via STAT-6. J. Immunol. 2004;173,:3425–3431. doi: 10.4049/jimmunol.173.5.3425. [DOI] [PubMed] [Google Scholar]
- 58.Lee E, Yook J, Haa K, Chang HW. Induction of Ym1/2 in mouse bone marrow-derived mast cells by IL-4 and identification of Ym1/2 in connective tissue type-like mast cells derived from bone marrow cells cultured with IL-4 and stem cell factor. Immunol. Cell Biol. 2005;83,:468–474. doi: 10.1111/j.1440-1711.2005.01352.x. 1:CAS:528:DC%2BD2MXhtFeqtrnK. [DOI] [PubMed] [Google Scholar]
- 59.Martinez-Pomares L. Analysis of mannose receptor regulation by IL-4, IL-10, and proteolytic processing using novel monoclonal antibodies. J. Leukoc. Biol. 2003;73,:604–613. doi: 10.1189/jlb.0902450. 1:CAS:528:DC%2BD3sXjslOmtr8%3D. [DOI] [PubMed] [Google Scholar]
- 60.Song E, Ouyang N, Horbelt M, Antus B, Wang M, Exton MS. Influence of alternatively and classically activated macrophages on fibrogenic activities of human fibroblasts. Cell Immunol. 2000;204,:19–28. doi: 10.1006/cimm.2000.1687. 1:CAS:528:DC%2BD3cXmslKgu7c%3D. [DOI] [PubMed] [Google Scholar]
- 61.Hesse M. Differential regulation of nitric oxide synthase-2 and arginase-1 by type 1/type 2 cytokines in vivo: granulomatous pathology is shaped by the pattern of L-arginine metabolism. J. Immunol. 2001;167,:6533–6544. doi: 10.4049/jimmunol.167.11.6533. [DOI] [PubMed] [Google Scholar]
- 62.Prasse A. A vicious circle of alveolar macrophages and fibroblasts perpetuates pulmonary fibrosis via CCL18. Am. J. Resp. Crit. Care Med. 2006;173,:781–792. doi: 10.1164/rccm.200509-1518OC. 1:CAS:528:DC%2BD28Xms1Cgtr4%3D. [DOI] [PubMed] [Google Scholar]
- 63.Pulichino AM. Identification of transforming growth factor beta1-driven genetic programs of acute lung fibrosis. Am. J. Respir. Cell Mol. Biol. 2008;39,:324–336. doi: 10.1165/rcmb.2007-0186OC. 1:CAS:528:DC%2BD1cXhtV2is7bE. [DOI] [PubMed] [Google Scholar]
- 64.Vidal B. Fibrinogen drives dystrophic muscle fibrosis via a TGFbeta/alternative macrophage activation pathway. Genes Dev. 2008;22,:1747–1752. doi: 10.1101/gad.465908. 1:CAS:528:DC%2BD1cXot1egu74%3D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gangadharan B. Murine gammaherpesvirus-induced fibrosis is associated with the development of alternatively activated macrophages. J. Leukoc. Biol. 2008;84,:50–58. doi: 10.1189/jlb.0507270. 1:CAS:528:DC%2BD1cXot1Oksbk%3D. [DOI] [PubMed] [Google Scholar]
- 66.Emad A, Emad Y. Increased granulocyte-colony stimulating factor (G-CSF) and granulocyte-macrophage colony stimulating factor (GM-CSF) levels in BAL fluid from patients with sulfur mustard gas-induced pulmonary fibrosis. J. Aerosol. Med. 2007;20,:352–360. doi: 10.1089/jam.2007.0590. 1:CAS:528:DC%2BD2sXhtVyjtrfO. [DOI] [PubMed] [Google Scholar]
- 67.Baran CP. Important roles for macrophage colony-stimulating factor, CC chemokine ligand 2, and mononuclear phagocytes in the pathogenesis of pulmonary fibrosis. Am. J. Resp. Crit. Care Med. 2007;176,:78–89. doi: 10.1164/rccm.200609-1279OC. 1:CAS:528:DC%2BD2sXot1Kms7c%3D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ishida Y. Essential roles of the CC chemokine ligand 3-CC chemokine receptor 5 axis in bleomycin-induced pulmonary fibrosis through regulation of macrophage and fibrocyte infiltration. Am. J. Pathol. 2007;170,:843–854. doi: 10.2353/ajpath.2007.051213. 1:CAS:528:DC%2BD2sXjs1Wls7g%3D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Trujillo G, O'Connor EC, Kunkel SL, Hogaboam CM. A novel mechanism for CCR4 in the regulation of macrophage activation in bleomycin-induced pulmonary fibrosis. Am. J. Pathol. 2008;172,:1209–1221. doi: 10.2353/ajpath.2008.070832. 1:CAS:528:DC%2BD1cXmtFOntb4%3D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Takatsu K, Nakajima H. IL-5 and eosinophilia. Curr. Opin. Immunol. 2008;20,:288–294. doi: 10.1016/j.coi.2008.04.001. 1:CAS:528:DC%2BD1cXns1ahtrY%3D. [DOI] [PubMed] [Google Scholar]
- 71.Reiman RM. Interleukin-5 (IL-5) augments the progression of liver fibrosis by regulating IL-13 activity. Infect Immun. 2006;74,:1471–1479. doi: 10.1128/IAI.74.3.1471-1479.2006. 1:CAS:528:DC%2BD28XmtFSqt7k%3D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Eklund KK, Ghildyal N, Austen KF, Stevens RL. Induction by IL-9 and suppression by IL-3 and IL-4 of the levels of chromosome 14-derived transcripts that encode late-expressed mouse mast cell proteases. J. Immunol. 1993;151,:4266–4273. [PubMed] [Google Scholar]
- 73.Tomimori Y. Involvement of mast cell chymase in bleomycin-induced pulmonary fibrosis in mice. Eur. J. Pharmacol. 2003;478,:179–185. doi: 10.1016/j.ejphar.2003.08.050. 1:CAS:528:DC%2BD3sXot12isL4%3D. [DOI] [PubMed] [Google Scholar]
- 74.Garbuzenko E. Human mast cells stimulate fibroblast proliferation, collagen synthesis and lattice contraction: a direct role for mast cells in skin fibrosis. Clin. Exp. Allergy. 2002;32,:237–246. doi: 10.1046/j.1365-2222.2002.01293.x. [DOI] [PubMed] [Google Scholar]
- 75.Masuda T. Mast cells play a partial role in allergen-induced subepithelial fibrosis in a murine model of allergic asthma. Clin. Exp. Allergy. 2003;33,:705–713. doi: 10.1046/j.1365-2222.2003.01588.x. [DOI] [PubMed] [Google Scholar]
- 76.Pesce J. The IL-21 receptor augments Th2 effector function and alternative macrophage activation. J. Clin. Invest. 2006;116,:2044–2055. doi: 10.1172/JCI27727. 1:CAS:528:DC%2BD28XmvV2lu70%3D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Frohlich A. IL-21 receptor signaling is integral to the development of Th2 effector responses in vivo. Blood. 2007;109,:2023–2031. doi: 10.1182/blood-2006-05-021600. 1:CAS:528:DC%2BD2sXjtFeqtbY%3D. [DOI] [PubMed] [Google Scholar]
- 78.Webb DC, Mahalingam S, Cai Y, Matthaei KI, Donaldson DD, Foster PS. Antigen-specific production of interleukin (IL)-13 and IL-5 cooperate to mediate IL-4Ralpha-independent airway hyperreactivity. Eur. J. Immunol. 2003;33,:3377–3385. doi: 10.1002/eji.200324178. 1:CAS:528:DC%2BD3sXpvFCmsL8%3D. [DOI] [PubMed] [Google Scholar]
- 79.Blease K. Stat6-deficient mice develop airway hyperresponsiveness and peribronchial fibrosis during chronic fungal asthma. Am. J. Pathol. 2002;160,:481–490. doi: 10.1016/S0002-9440(10)64867-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Fichtner-Feigl S. Induction of IL-13 triggers TGF-beta1-dependent tissue fibrosis in chronic 2,4,6-trinitrobenzene sulfonic acid colitis. J. Immunol. 2007;178,:5859–5870. doi: 10.4049/jimmunol.178.9.5859. [DOI] [PubMed] [Google Scholar]
- 81.Fichtner-Feigl S, Young CA, Kitani A, Geissler EK, Schlitt HJ, Strober W. IL-13 Signaling via IL-13Ralpha(2) Induces Major Downstream Fibrogenic Factors Mediating Fibrosis in Chronic TNBS Colitis. Gastroenterology. 2008;135,:2003–2013. doi: 10.1053/j.gastro.2008.08.055. 1:CAS:528:DC%2BD1MXht1ertrs%3D. [DOI] [PubMed] [Google Scholar]
- 82.Shimamura T, Fujisawa T, Husain SR, Kioi M, Nakajima A, Puri RK. Novel role of IL-13 in fibrosis induced by nonalcoholic steatohepatitis and its amelioration by IL-13R-directed cytotoxin in a rat model. J. Immunol. 2008;181,:4656–4665. doi: 10.4049/jimmunol.181.7.4656. [DOI] [PubMed] [Google Scholar]
- 83.Munitz A, Brandt EB, Mingler M, Finkelman FD, Rothenberg ME. Distinct roles for IL-13 and IL-4 via IL-13 receptor alpha1 and the type II IL-4 receptor in asthma pathogenesis. Proc. Natl. Acad. Sci. USA. 2008;105,:7240–7245. doi: 10.1073/pnas.0802465105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kaviratne M. IL-13 activates a mechanism of tissue fibrosis that is completely TGF-beta independent. J. Immunol. 2004;173,:4020–4029. doi: 10.4049/jimmunol.173.6.4020. [DOI] [PubMed] [Google Scholar]
- 85.Ingram JL, Rice AB, Geisenhoffer K, Madtes DK, Bonner JC. IL-13 and IL-1beta promote lung fibroblast growth through coordinated up-regulation of PDGF-AA and PDGF-Ralpha. FASEB J. 2004;18,:1132–1134. doi: 10.1096/fj.03-1492fje. 1:CAS:528:DC%2BD2cXlslKktro%3D. [DOI] [PubMed] [Google Scholar]
- 86.Zhu Z. Pulmonary expression of interleukin-13 causes inflammation, mucus hypersecretion, subepithelial fibrosis, physiologic abnormalities, and eotaxin production. J. Clin. Invest. 1999;103,:779–788. doi: 10.1172/JCI5909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Blease K, Jakubzick C, Westwick J, Lukacs N, Kunkel SL, Hogaboam CM. Therapeutic effect of IL-13 immunoneutralization during chronic experimental fungal asthma. J. Immunol. 2001;166,:5219–5224. doi: 10.4049/jimmunol.166.8.5219. [DOI] [PubMed] [Google Scholar]
- 88.Yang G. Anti-IL-13 monoclonal antibody inhibits airway hyperresponsiveness, inflammation and airway remodeling. Cytokine. 2004;28,:224–232. doi: 10.1016/j.cyto.2004.08.007. 1:CAS:528:DC%2BD2cXhtVWisbjK. [DOI] [PubMed] [Google Scholar]
- 89.Belperio JA. Interaction of IL-13 and C10 in the pathogenesis of bleomycin-induced pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2002;27,:419–427. doi: 10.1165/rcmb.2002-0009OC. [DOI] [PubMed] [Google Scholar]
- 90.Jakubzick C. Therapeutic attenuation of pulmonary fibrosis via targeting of IL-4- and IL-13-responsive cells. J. Immunol. 2003;171,:2684–2693. doi: 10.4049/jimmunol.171.5.2684. [DOI] [PubMed] [Google Scholar]
- 91.Kolodsick JE. Protection from fluorescein isothiocyanate-induced fibrosis in IL-13-deficient, but not IL-4-deficient, mice results from impaired collagen synthesis by fibroblasts. J. Immunol. 2004;172,:4068–4076. doi: 10.4049/jimmunol.172.7.4068. [DOI] [PubMed] [Google Scholar]
- 92.Malavia NK, Mih JD, Raub CB, Dinh BT, George SC. IL-13 induces a bronchial epithelial phenotype that is profibrotic. Respir. Res. 2008;9,:27. doi: 10.1186/1465-9921-9-27. 1:CAS:528:DC%2BD1cXntlertrk%3D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Murray LA. Hyper-responsiveness of IPF/UIP fibroblasts: interplay between TGFbeta1, IL-13 and CCL2. Int. J. Biochem. Cell Biol. 2008;40,:2174–2182. doi: 10.1016/j.biocel.2008.02.016. 1:CAS:528:DC%2BD1cXovF2jtb8%3D. [DOI] [PubMed] [Google Scholar]
- 94.Kraft M, Lewis C, Pham D, Chu HW. IL-4, IL-13, and dexamethasone augment fibroblast proliferation in asthma. J. Allergy Clin. Immunol. 2001;107,:602–606. doi: 10.1067/mai.2001.113760. [DOI] [PubMed] [Google Scholar]
- 95.Lee CG. Interleukin-13 induces tissue fibrosis by selectively stimulating and activating transforming growth factor beta(1) J. Exp. Med. 2001;194,:809–821. doi: 10.1084/jem.194.6.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Letterio JJ, Roberts AB. Regulation of immune responses by TGF-beta. Annu. Rev. Immunol. 1998;16,:137–161. doi: 10.1146/annurev.immunol.16.1.137. [DOI] [PubMed] [Google Scholar]
- 97.Assoian RK. Expression and secretion of type beta transforming growth factor by activated human macrophages. Proc. Natl. Acad. Sci. USA. 1987;84,:6020–6024. doi: 10.1073/pnas.84.17.6020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Grotendorst GR, Smale G, Pencev D. Production of transforming growth factor beta by human peripheral blood monocytes and neutrophils. J. Cell Physiol. 1989;140,:396–402. doi: 10.1002/jcp.1041400226. [DOI] [PubMed] [Google Scholar]
- 99.Khalil N, Corne S, Whitman C, Yacyshyn H. Plasmin regulates the activation of cell-associated latent TGF-beta 1 secreted by rat alveolar macrophages after in vivo bleomycin injury. Am. J. Respir. Cell Mol. Biol. 1996;15,:252–259. doi: 10.1165/ajrcmb.15.2.8703482. [DOI] [PubMed] [Google Scholar]
- 100.Gressner AM, Lahme B, Roth S. Attenuation of TGF-beta-induced apoptosis in primary cultures of hepatocytes by calpain inhibitors. Biochem. Biophys. Res. Commun. 1997;231,:457–462. doi: 10.1006/bbrc.1996.5777. [DOI] [PubMed] [Google Scholar]
- 101.Munger JS. The integrin alpha v beta 6 binds and activates latent TGF beta 1: a mechanism for regulating pulmonary inflammation and fibrosis. Cell. 1999;96,:319–328. doi: 10.1016/S0092-8674(00)80545-0. [DOI] [PubMed] [Google Scholar]
- 102.Yu Q, Stamenkovic I. Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-beta and promotes tumor invasion and angiogenesis. Genes Dev. 2000;14,:163–176. [PMC free article] [PubMed] [Google Scholar]
- 103.Szardening-Kirchner, C., Konrad, L., Hauck, E.W., Haag, S.M., Eickelberg, O. & Weidner, W. Upregulation of mRNA expression of MCP-1 by TGF-beta1 in fibroblast cells from Peyronie??s disease. World J. Urol. (2008) (E-pub ahead of print). [DOI] [PubMed]
- 104.Coker RK. Transforming growth factors-beta 1, -beta 2, and -beta 3 stimulate fibroblast procollagen production in vitro but are differentially expressed during bleomycin-induced lung fibrosis. Am. J. Pathol. 1997;150,:981–991. [PMC free article] [PubMed] [Google Scholar]
- 105.Chen W, Perruche S, Li J. CD4+CD25+ T regulatory cells and TGF-beta in mucosal immune system: the good and the bad. Curr. Med. Chem. 2007;14,:2245–2249. doi: 10.2174/092986707781696591. [DOI] [PubMed] [Google Scholar]
- 106.Huber S, Schramm C. TGF-beta and CD4+CD25+ regulatory T cells. Front Biosci. 2006;11,:1014–1023. doi: 10.2741/1859. [DOI] [PubMed] [Google Scholar]
- 107.Wahl SM, Swisher J, McCartney-Francis N, Chen W. TGF-beta: the perpetrator of immune suppression by regulatory T cells and suicidal T cells. J. Leukoc. Biol. 2004;76,:15–24. doi: 10.1189/jlb.1103539. 1:CAS:528:DC%2BD2cXlsVOisrs%3D. [DOI] [PubMed] [Google Scholar]
- 108.Sime PJ, Xing Z, Graham FL, Csaky KG, Gauldie J. Adenovector-mediated gene transfer of active transforming growth factor-beta1 induces prolonged severe fibrosis in rat lung. J. Clin. Invest. 1997;100,:768–776. doi: 10.1172/JCI119590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Flanders KC. Smad3 as a mediator of the fibrotic response. Int. J. Exp. Pathol. 2004;85,:47–64. doi: 10.1111/j.0959-9673.2004.00377.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Flanders KC. Mice lacking Smad3 are protected against cutaneous injury induced by ionizing radiation. Am. J. Pathol. 2002;160,:1057–1068. doi: 10.1016/S0002-9440(10)64926-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Lakos G. Targeted disruption of TGF-beta/Smad3 signaling modulates skin fibrosis in a mouse model of scleroderma. Am. J. Pathol. 2004;165,:203–217. doi: 10.1016/S0002-9440(10)63289-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Border WA. Natural inhibitor of transforming growth factor-beta protects against scarring in experimental kidney disease. Nature. 1992;360,:361–364. doi: 10.1038/360361a0. [DOI] [PubMed] [Google Scholar]
- 113.Inazaki K. Smad3 deficiency attenuates renal fibrosis, inflammation,and apoptosis after unilateral ureteral obstruction. Kidney Int. 2004;66,:597–604. doi: 10.1111/j.1523-1755.2004.00779.x. [DOI] [PubMed] [Google Scholar]
- 114.Sato M, Muragaki Y, Saika S, Roberts AB, Ooshima A. Targeted disruption of TGF-beta1/Smad3 signaling protects against renal tubulointerstitial fibrosis induced by unilateral ureteral obstruction. J. Clin. Invest. 2003;112,:1486–1494. doi: 10.1172/JCI200319270. 1:CAS:528:DC%2BD3sXptFWqt7k%3D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Stramer BM, Austin JS, Roberts AB, Fini ME. Selective reduction of fibrotic markers in repairing corneas of mice deficient in Smad3. J. Cell Physiol. 2005;203,:226–232. doi: 10.1002/jcp.20215. 1:CAS:528:DC%2BD2MXit1Squrk%3D. [DOI] [PubMed] [Google Scholar]
- 116.Bonniaud P. Smad3 null mice develop airspace enlargement and are resistant to TGF-beta-mediated pulmonary fibrosis. J. Immunol. 2004;173,:2099–2108. doi: 10.4049/jimmunol.173.3.2099. [DOI] [PubMed] [Google Scholar]
- 117.Zhao J. Smad3 deficiency attenuates bleomycin-induced pulmonary fibrosis in mice. Am. J. Physiol. 2002;282,:L585–L593. doi: 10.1152/ajpcell.00547.2001. [DOI] [PubMed] [Google Scholar]
- 118.Ashcroft GS. Mice lacking Smad3 show accelerated wound healing and an impaired local inflammatory response. Nat. Cell Biol. 1999;1,:260–266. doi: 10.1038/12971. 1:CAS:528:DyaK1MXlvFChu7w%3D. [DOI] [PubMed] [Google Scholar]
- 119.Ma LJ. Transforming growth factor-beta-dependent and -independent pathways of induction of tubulointerstitial fibrosis in beta6(−/−) mice. Am. J. Pathol. 2003;163,:1261–1273. doi: 10.1016/S0002-9440(10)63486-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Proudfoot AE. Chemokine receptors: multifaceted therapeutic targets. Nat. Rev. Immunol. 2002;2,:106–115. doi: 10.1038/nri722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Blease K, Mehrad B, Lukacs NW, Kunkel SL, Standiford TJ, Hogaboam CM. Antifungal and airway remodeling roles for murine monocyte chemoattractant protein-1/CCL2 during pulmonary exposure to Asperigillus fumigatus conidia. J. Immunol. 2001;166,:1832–1842. doi: 10.4049/jimmunol.166.3.1832. [DOI] [PubMed] [Google Scholar]
- 122.Car BD, Meloni F, Luisetti M, Semenzato G, Gialdroni-Grassi G, Walz A. Elevated IL-8 and MCP-1 in the bronchoalveolar lavage fluid of patients with idiopathic pulmonary fibrosis and pulmonary sarcoidosis. Am. J. Resp. Crit. Care Med. 1994;149(3 Part 1):655–659. doi: 10.1164/ajrccm.149.3.8118632. 1:STN:280:DyaK2c7mtlCitA%3D%3D, 8118632. [DOI] [PubMed] [Google Scholar]
- 123.Emad A, Emad Y. Relationship between eosinophilia and levels of chemokines (CCL5 and CCL11) and IL-5 in bronchoalveolar lavage fluid of patients with mustard gas-induced pulmonary fibrosis. J. Clin. Immunol. 2008;28,:298–305. doi: 10.1007/s10875-007-9109-8. [DOI] [PubMed] [Google Scholar]
- 124.Huaux F. Role of Eotaxin-1 (CCL11) and CC chemokine receptor 3 (CCR3) in bleomycin-induced lung injury and fibrosis. Am. J. Pathol. 2005;167,:1485–1496. doi: 10.1016/S0002-9440(10)61235-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Puxeddu I, Bader R, Piliponsky AM, Reich R, Levi-Schaffer F, Berkman N. The CC chemokine eotaxin/CCL11 has a selective profibrogenic effect on human lung fibroblasts. J. Allergy Clin. Immunol. 2006;117,:103–110. doi: 10.1016/j.jaci.2005.08.057. 1:CAS:528:DC%2BD28XktlSh. [DOI] [PubMed] [Google Scholar]
- 126.Rose CE, Jr, Sung SS, Fu SM. Significant involvement of CCL2 (MCP-1) in inflammatory disorders of the lung. Microcirculation. 2003;10,:273–288. doi: 10.1080/mic.10.3-4.273.288. [DOI] [PubMed] [Google Scholar]
- 127.Zhang K, Gharaee-Kermani M, Jones ML, Warren JS, Phan SH. Lung monocyte chemoattractant protein-1 gene expression in bleomycin-induced pulmonary fibrosis. J. Immunol. 1994;153,:4733–4741. [PubMed] [Google Scholar]
- 128.Gharaee-Kermani M, McCullumsmith RE, Charo IF, Kunkel SL, Phan SH. CC-chemokine receptor 2 required for bleomycin-induced pulmonary fibrosis. Cytokine. 2003;24,:266–276. doi: 10.1016/j.cyto.2003.08.003. 1:CAS:528:DC%2BD3sXovVeiurg%3D. [DOI] [PubMed] [Google Scholar]
- 129.Wu Y, Li YY, Matsushima K, Baba T, Mukaida N. CCL3-CCR5 axis regulates intratumoral accumulation of leukocytes and fibroblasts and promotes angiogenesis in murine lung metastasis process. J. Immunol. 2008;181,:6384–6393. doi: 10.4049/jimmunol.181.9.6384. [DOI] [PubMed] [Google Scholar]
- 130.Moore BB. CCR2-mediated recruitment of fibrocytes to the alveolar space after fibrotic injury. Am. J. Pathol. 2005;166,:675–684. doi: 10.1016/S0002-9440(10)62289-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Abe R, Donnelly SC, Peng T, Bucala R, Metz CN. Peripheral blood fibrocytes: differentiation pathway and migration to wound sites. J. Immunol. 2001;166,:7556–7562. doi: 10.4049/jimmunol.166.12.7556. [DOI] [PubMed] [Google Scholar]
- 132.van Deventer HW. C-C chemokine receptor 5 on pulmonary fibrocytes facilitates migration and promotes metastasis via matrix metalloproteinase 9. Am. J. Pathol. 2008;173,:253–264. doi: 10.2353/ajpath.2008.070732. 1:CAS:528:DC%2BD1cXptVKjsrk%3D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Phillips RJ. Circulating fibrocytes traffic to the lungs in response to CXCL12 and mediate fibrosis. J. Clin. Invest. 2004;114,:438–446. doi: 10.1172/JCI200420997. 1:CAS:528:DC%2BD2cXmtlyns7s%3D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Quan TE, Cowper S, Wu SP, Bockenstedt LK, Bucala R. Circulating fibrocytes: collagen-secreting cells of the peripheral blood. Int. J. Biochem. Cell Biol. 2004;36,:598–606. doi: 10.1016/j.biocel.2003.10.005. 1:CAS:528:DC%2BD2cXhvFSmurY%3D. [DOI] [PubMed] [Google Scholar]
- 135.Bucala R, Spiegel LA, Chesney J, Hogan M, Cerami A. Circulating fibrocytes define a new leukocyte subpopulation that mediates tissue repair. Mol. Med. 1994;1,:71–81. doi: 10.1007/BF03403533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Schmidt M, Sun G, Stacey MA, Mori L, Mattoli S. Identification of circulating fibrocytes as precursors of bronchial myofibroblasts in asthma. J. Immunol. 2003;171,:380–389. doi: 10.4049/jimmunol.171.1.380. [DOI] [PubMed] [Google Scholar]
- 137.Strieter RM, Gomperts BN, Keane MP. The role of CXC chemokines in pulmonary fibrosis. J. Clin. Invest. 2007;117,:549–556. doi: 10.1172/JCI30562. 1:CAS:528:DC%2BD2sXis12htr8%3D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Semenza GL. Vasculogenesis, angiogenesis, and arteriogenesis: mechanisms of blood vessel formation and remodeling. J. Cell Biochem. 2007;102,:840–847. doi: 10.1002/jcb.21523. 1:CAS:528:DC%2BD2sXhtlSisLzE. [DOI] [PubMed] [Google Scholar]
- 139.Keane MP. The CXC chemokines, IL-8 and IP-10, regulate angiogenic activity in idiopathic pulmonary fibrosis. J. Immunol. 1997;159,:1437–1443. [PubMed] [Google Scholar]
- 140.Antoniou KM. Different angiogenic activity in pulmonary sarcoidosis and idiopathic pulmonary fibrosis. Chest. 2006;130,:982–988. doi: 10.1378/chest.130.4.982. [DOI] [PubMed] [Google Scholar]
- 141.Burdick MD. CXCL11 attenuates bleomycin-induced pulmonary fibrosis via inhibition of vascular remodeling. Am. J. Resp. Crit. Care Med. 2005;171,:261–268. doi: 10.1164/rccm.200409-1164OC. [DOI] [PubMed] [Google Scholar]
- 142.Keane MP. IFN-gamma-inducible protein-10 attenuates bleomycin-induced pulmonary fibrosis via inhibition of angiogenesis. J. Immunol. 1999;163,:5686–5692. [PubMed] [Google Scholar]
- 143.Keane MP. Neutralization of the CXC chemokine, macrophage inflammatory protein-2, attenuates bleomycin-induced pulmonary fibrosis. J. Immunol. 1999;162,:5511–5518. [PubMed] [Google Scholar]
- 144.Peao MN, Aguas AP, de Sa CM, Grande NR. Neoformation of blood vessels in association with rat lung fibrosis induced by bleomycin. Anat. Rec. 1994;238,:57–67. doi: 10.1002/ar.1092380108. [DOI] [PubMed] [Google Scholar]
- 145.Driscoll KE, Maurer JK, Poynter J, Higgins J, Asquith T, Miller NS. Stimulation of rat alveolar macrophage fibronectin release in a cadmium chloride model of lung injury and fibrosis. Toxicol. Appl. Pharmacol. 1992;116,:30–37. doi: 10.1016/0041-008X(92)90141-E. [DOI] [PubMed] [Google Scholar]
- 146.Kadler KE, Hill A, Canty-Laird EG. Collagen fibrillogenesis: fibronectin, integrins, and minor collagens as organizers and nucleators. Curr. Opin. Cell Biol. 2008;20,:495–501. doi: 10.1016/j.ceb.2008.06.008. 1:CAS:528:DC%2BD1cXhtVyks7zF. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Lacronique JG, Rennard SI, Bitterman PB, Ozaki T, Crystal RG. Alveolar macrophages in idiopathic pulmonary fibrosis have glucocorticoid receptors, but glucocorticoid therapy does not suppress alveolar macrophage release of fibronectin and alveolar macrophage derived growth factor. Am. Rev. Respir. Dis. 1984;130,:450–456. doi: 10.1164/arrd.1984.130.3.450. [DOI] [PubMed] [Google Scholar]
- 148.Muro AF. An essential role for fibronectin extra type III domain A in pulmonary fibrosis. Am. J. Resp. Crit. Care Med. 2008;177,:638–645. doi: 10.1164/rccm.200708-1291OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Hernnas J, Nettelbladt O, Bjermer L, Sarnstrand B, Malmstrom A, Hallgren R. Alveolar accumulation of fibronectin and hyaluronan precedes bleomycin-induced pulmonary fibrosis in the rat. Eur. Respir. J. 1992;5,:404–410. [PubMed] [Google Scholar]
- 150.Lazenby AJ, Crouch EC, McDonald JA, Kuhn C., III Remodeling of the lung in bleomycin-induced pulmonary fibrosis in the rat. An immunohistochemical study of laminin, type IV collagen, and fibronectin. Am. Rev. Respir. Dis. 1990;142,:206–214. doi: 10.1164/ajrccm/142.1.206. [DOI] [PubMed] [Google Scholar]
- 151.Iwano M, Plieth D, Danoff TM, Xue C, Okada H, Neilson EG. Evidence that fibroblasts derive from epithelium during tissue fibrosis. J. Clin. Invest. 2002;110,:341–350. doi: 10.1172/JCI0215518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Venkatesan N, Roughley PJ, Ludwig MS. Proteoglycan expression in bleomycin lung fibroblasts: role of transforming growth factor-beta(1) and interferon-gamma. Am. J. Physiol. 2002;283,:L806–L814. doi: 10.1152/ajplung.00061.2002. [DOI] [PubMed] [Google Scholar]
- 153.Bensadoun ES, Burke AK, Hogg JC, Roberts CR. Proteoglycan deposition in pulmonary fibrosis. Am. J. Resp. Crit. Care Med. 1996;154(6 Part 1):1819–1828. doi: 10.1164/ajrccm.154.6.8970376. 1:STN:280:DyaK2s7isFaqtQ%3D%3D, 8970376. [DOI] [PubMed] [Google Scholar]
- 154.McGowan SE. Extracellular matrix and the regulation of lung development and repair. FASEB J. 1992;6,:2895–2904. doi: 10.1096/fasebj.6.11.1644255. [DOI] [PubMed] [Google Scholar]
- 155.Parra ER, Kairalla RA, de Carvalho CR, Capelozzi VL. Abnormal deposition of collagen/elastic vascular fibres and prognostic significance in idiopathic interstitial pneumonias. Thorax. 2007;62,:428–437. doi: 10.1136/thx.2006.062687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Farahani RM, Kloth LC. The hypothesis of “biophysical matrix contraction”: wound contraction revisited. Int. Wound J. 2008;5,:477–482. doi: 10.1111/j.1742-481X.2007.00402.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Mirastschijski U, Haaksma CJ, Tomasek JJ, Agren MS. Matrix metalloproteinase inhibitor GM 6001 attenuates keratinocyte migration, contraction and myofibroblast formation in skin wounds. Exp. Cell Res. 2004;299,:465–475. doi: 10.1016/j.yexcr.2004.06.007. 1:CAS:528:DC%2BD2cXntlOhsLc%3D. [DOI] [PubMed] [Google Scholar]
- 158.Hinz B. Masters and servants of the force: the role of matrix adhesions in myofibroblast force perception and transmission. Eur. J. Cell Biol. 2006;85,:175–181. doi: 10.1016/j.ejcb.2005.09.004. 1:CAS:528:DC%2BD28Xkt1Oltbc%3D. [DOI] [PubMed] [Google Scholar]
- 159.Singer II. Fibronexus formation is an early event during fibronectin-induced restoration of more normal morphology and substrate adhesion patterns in transformed hamster fibroblasts. J. Cell Sci. 1982;56,:1–20. doi: 10.1242/jcs.56.1.1. [DOI] [PubMed] [Google Scholar]
- 160.Thannickal VJ, Horowitz JC. Evolving concepts of apoptosis in idiopathic pulmonary fibrosis. Proc. Am. Thorac. Soc. 2006;3,:350–356. doi: 10.1513/pats.200601-001TK. 1:CAS:528:DC%2BD28XmtVOksbk%3D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Garcia-Alvarez J. Tissue inhibitor of metalloproteinase-3 is up-regulated by transforming growth factor-beta1 in vitro and expressed in fibroblastic foci in vivo in idiopathic pulmonary fibrosis. Exp. Lung Res. 2006;32,:201–214. doi: 10.1080/01902140600817481. 1:CAS:528:DC%2BD28XhtVWrt7rO. [DOI] [PubMed] [Google Scholar]
- 162.Gill SE, Parks WC. Metalloproteinases and their inhibitors: regulators of wound healing. Int. J. Biochem. Cell Biol. 2008;40,:1334–1347. doi: 10.1016/j.biocel.2007.10.024. 1:CAS:528:DC%2BD1cXlslGlu7w%3D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Selman M. TIMP-1, -2, -3, and -4 in idiopathic pulmonary fibrosis. A prevailing nondegradative lung microenvironment? Am. J. Physiol. 2000;279,:L562–L574. doi: 10.1152/ajplung.2000.279.3.L562. [DOI] [PubMed] [Google Scholar]
- 164.Fattman CL. Apoptosis in pulmonary fibrosis: too much or not enough? Antioxid Redox Signal. 2008;10,:379–385. doi: 10.1089/ars.2007.1907. 1:CAS:528:DC%2BD1cXht1Shtg%3D%3D. [DOI] [PubMed] [Google Scholar]
- 165.Moodley YP. Comparison of the morphological and biochemical changes in normal human lung fibroblasts and fibroblasts derived from lungs of patients with idiopathic pulmonary fibrosis during FasL-induced apoptosis. J. Pathol. 2004;202,:486–495. doi: 10.1002/path.1531. 1:CAS:528:DC%2BD2cXjsFegu7k%3D. [DOI] [PubMed] [Google Scholar]
- 166.Buhling F. Altered expression of membrane-bound and soluble CD95/Fas contributes to the resistance of fibrotic lung fibroblasts to FasL induced apoptosis. Respir. Res. 2005;6,:37. doi: 10.1186/1465-9921-6-37. 1:CAS:528:DC%2BD2MXhtVahsrrL. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Maeyama T. Upregulation of Fas-signalling molecules in lung epithelial cells from patients with idiopathic pulmonary fibrosis. Eur. Respir. J. 2001;17,:180–189. doi: 10.1183/09031936.01.17201800. [DOI] [PubMed] [Google Scholar]
- 168.Moodley YP. Inverse effects of interleukin-6 on apoptosis of fibroblasts from pulmonary fibrosis and normal lungs. Am. J. Respir. Cell Mol. Biol. 2003;29,:490–498. doi: 10.1165/rcmb.2002-0262OC. [DOI] [PubMed] [Google Scholar]
- 169.Plataki M, Koutsopoulos AV, Darivianaki K, Delides G, Siafakas NM, Bouros D. Expression of apoptotic and antiapoptotic markers in epithelial cells in idiopathic pulmonary fibrosis. Chest. 2005;127,:266–274. doi: 10.1378/chest.127.1.266. [DOI] [PubMed] [Google Scholar]
- 170.Ramos C. Fibroblasts from idiopathic pulmonary fibrosis and normal lungs differ in growth rate, apoptosis, and tissue inhibitor of metalloproteinases expression. Am. J. Respir. Cell Mol. Biol. 2001;24,:591–598. doi: 10.1165/ajrcmb.24.5.4333. [DOI] [PubMed] [Google Scholar]
- 171.Frankel SK. TNF-alpha sensitizes normal and fibrotic human lung fibroblasts to Fas-induced apoptosis. Am. J. Respir. Cell Mol. Biol. 2006;34,:293–304. doi: 10.1165/rcmb.2005-0155OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Erjefalt JS, Erjefalt I, Sundler F, Persson CG. In vivo restitution of airway epithelium. Cell Tissue Res. 1995;281,:305–316. doi: 10.1007/BF00583399. [DOI] [PubMed] [Google Scholar]
- 173.Zahm JM. Cell migration and proliferation during the in vitro wound repair of the respiratory epithelium. Cell Motil Cytoskeleton. 1997;37,:33–43. doi: 10.1002/(SICI)1097-0169(1997)37:1. [DOI] [PubMed] [Google Scholar]
- 174.Kheradmand F, Folkesson HG, Shum L, Derynk R, Pytela R, Matthay MA. Transforming growth factor-alpha enhances alveolar epithelial cell repair in a new in vitro model. Am. J. Physiol. 1994;267(6 Part 1):L728–L738. doi: 10.1152/ajplung.1994.267.6.L728. 1:CAS:528:DyaK2MXivFKnurk%3D, 7810677. [DOI] [PubMed] [Google Scholar]
- 175.Chilosi M. Abnormal re-epithelialization and lung remodeling in idiopathic pulmonary fibrosis: the role of deltaN-p63. Lab Invest. 2002;82,:1335–1345. doi: 10.1097/01.LAB.0000032380.82232.67. 1:CAS:528:DC%2BD38Xns1Clsrc%3D. [DOI] [PubMed] [Google Scholar]
- 176.Erjefalt JS, Persson CG. Airway epithelial repair: breathtakingly quick and multipotentially pathogenic. Thorax. 1997;52,:1010–1012. doi: 10.1136/thx.52.11.1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Farooqui R, Fenteany G. Multiple rows of cells behind an epithelial wound edge extend cryptic lamellipodia to collectively drive cell-sheet movement. J. Cell Sci. 2005;118(Part 1):51–63. doi: 10.1242/jcs.01577. 1:CAS:528:DC%2BD2MXhsVCrtbo%3D, 15585576. [DOI] [PubMed] [Google Scholar]
- 178.Zhao M, Song B, Pu J, Forrester JV, McCaig CD. Direct visualization of a stratified epithelium reveals that wounds heal by unified sliding of cell sheets. FASEB J. 2003;17,:397–406. doi: 10.1096/fj.02-0610com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Dunsmore SE. Matrilysin expression and function in airway epithelium. J. Clin. Invest. 1998;102,:1321–1331. doi: 10.1172/JCI1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.McGuire JK, Li Q, Parks WC. Matrilysin (matrix metalloproteinase-7) mediates E-cadherin ectodomain shedding in injured lung epithelium. Am. J. Pathol. 2003;162,:1831–1843. doi: 10.1016/S0002-9440(10)64318-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Chen P. Tissue inhibitor of metalloproteinase-1 moderates airway re-epithelialization by regulating matrilysin activity. Am. J. Pathol. 2008;172,:1256–1270. doi: 10.2353/ajpath.2008.070891. 1:CAS:528:DC%2BD1cXmtFOnur4%3D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Goss CH, Rosenfeld M. Update on cystic fibrosis epidemiology. Curr. Opin. Pulm Med. 2004;10,:510–514. doi: 10.1097/01.mcp.0000138994.46519.72. [DOI] [PubMed] [Google Scholar]
- 183.Anderson MP. Demonstration that CFTR is a chloride channel by alteration of its anion selectivity. Science. 1991;253,:202–205. doi: 10.1126/science.1712984. [DOI] [PubMed] [Google Scholar]
- 184.Jacquot J, Tabary O, Clement A. Hyperinflammation in airways of cystic fibrosis patients: what??s new? Expert Rev. Mol. Diagn. 2008;8,:359–363. doi: 10.1586/14737159.8.4.359. [DOI] [PubMed] [Google Scholar]
- 185.Koch C, Hoiby N. Diagnosis and treatment of cystic fibrosis. Respiration. 2000;67,:239–247. doi: 10.1159/000029503. [DOI] [PubMed] [Google Scholar]
- 186.Ratjen F. Recent advances in cystic fibrosis. Paediatr Respir Rev. 2008;9,:144–148. doi: 10.1016/j.prrv.2008.01.004. [DOI] [PubMed] [Google Scholar]
- 187.Saiman L. Microbiology of early CF lung disease. Paediatr Respir Rev. 2004;5(Suppl A):S367–S369. doi: 10.1016/S1526-0542(04)90065-6. 14980298. [DOI] [PubMed] [Google Scholar]
- 188.Hansen CR, Pressler T, Koch C, Hoiby N. Long-term azitromycin treatment of cystic fibrosis patients with chronic Pseudomonas aeruginosa infection; an observational cohort study. J. Cyst. Fibros. 2005;4,:35–40. doi: 10.1016/j.jcf.2004.09.001. 1:CAS:528:DC%2BD2MXitFCjtLk%3D. [DOI] [PubMed] [Google Scholar]
- 189.Pai VB, Nahata MC. Efficacy and safety of aerosolized tobramycin in cystic fibrosis. Pediatr. Pulmonol. 2001;32,:314–327. doi: 10.1002/ppul.1125. [DOI] [PubMed] [Google Scholar]
- 190.Westerman EM, Le Brun PP, Touw DJ, Frijlink HW, Heijerman HG. Effect of nebulized colistin sulphate and colistin sulphomethate on lung function in patients with cystic fibrosis: a pilot study. J. Cyst. Fibros. 2004;3,:23–28. doi: 10.1016/j.jcf.2003.12.005. 1:CAS:528:DC%2BD2cXisFOgu7o%3D. [DOI] [PubMed] [Google Scholar]
- 191.Nichols D, Chmiel J, Berger M. Chronic inflammation in the cystic fibrosis lung: alterations in inter- and intracellular signaling. Clin. Rev. Allergy Immunol. 2008;34,:146–162. doi: 10.1007/s12016-007-8039-9. 1:CAS:528:DC%2BD1cXkvFamsrk%3D. [DOI] [PubMed] [Google Scholar]
- 192.Soferman R. Immunopathophysiologic mechanisms of cystic fibrosis lung disease. Isr. Med. Assoc. J. 2006;8,:44–48. [PubMed] [Google Scholar]
- 193.Regamey N. Increased airway smooth muscle mass in children with asthma, cystic fibrosis, and non-cystic fibrosis bronchiectasis. Am. J. Resp. Crit. Care Med. 2008;177,:837–843. doi: 10.1164/rccm.200707-977OC. [DOI] [PubMed] [Google Scholar]
- 194.Gaggar A. Matrix metalloprotease-9 dysregulation in lower airway secretions of cystic fibrosis patients. Am. J. Physiol. 2007;293,:L96–L104. doi: 10.1152/ajplung.00492.2006. [DOI] [PubMed] [Google Scholar]
- 195.Delacourt C. Imbalance between 95 kDa type IV collagenase and tissue inhibitor of metalloproteinases in sputum of patients with cystic fibrosis. Am. J. Resp. Crit. Care Med. 1995;152,:765–774. doi: 10.1164/ajrccm.152.2.7633740. [DOI] [PubMed] [Google Scholar]
- 196.Sagel SD, Kapsner RK, Osberg I. Induced sputum matrix metalloproteinase-9 correlates with lung function and airway inflammation in children with cystic fibrosis. Pediatr. Pulmonol. 2005;39,:224–232. doi: 10.1002/ppul.20165. [DOI] [PubMed] [Google Scholar]
- 197.Hajj R, Lesimple P, Nawrocki-Raby B, Birembaut P, Puchelle E, Coraux C. Human airway surface epithelial regeneration is delayed and abnormal in cystic fibrosis. J. Pathol. 2007;211,:340–350. doi: 10.1002/path.2118. 1:CAS:528:DC%2BD2sXisFyksLk%3D. [DOI] [PubMed] [Google Scholar]
- 198.Clarke LL, Grubb BR, Gabriel SE, Smithies O, Koller BH, Boucher RC. Defective epithelial chloride transport in a gene-targeted mouse model of cystic fibrosis. Science. 1992;257,:1125–1128. doi: 10.1126/science.257.5073.1125. [DOI] [PubMed] [Google Scholar]
- 199.Kent G. Lung disease in mice with cystic fibrosis. J. Clin. Invest. 1997;100,:3060–3069. doi: 10.1172/JCI119861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Snouwaert JN. An animal model for cystic fibrosis made by gene targeting. Science. 1992;257,:1083–1088. doi: 10.1126/science.257.5073.1083. [DOI] [PubMed] [Google Scholar]
- 201.Bensalem N. Down-regulation of the anti-inflammatory protein annexin A1 in cystic fibrosis knock-out mice and patients. Mol. Cell Proteomics. 2005;4,:1591–1601. doi: 10.1074/mcp.M500019-MCP200. 1:CAS:528:DC%2BD2MXhtFeltb3N. [DOI] [PubMed] [Google Scholar]
- 202.Haston CK, McKerlie C, Newbigging S, Corey M, Rozmahel R, Tsui LC. Detection of modifier loci influencing the lung phenotype of cystic fibrosis knockout mice. Mamm. Genome. 2002;13,:605–613. doi: 10.1007/s00335-002-2190-7. 1:CAS:528:DC%2BD38Xptlehtro%3D. [DOI] [PubMed] [Google Scholar]
- 203.Salvatore F, Scudiero O, Castaldo G. Genotype-phenotype correlation in cystic fibrosis: the role of modifier genes. Am. J. Med. Genet. 2002;111,:88–95. doi: 10.1002/ajmg.10461. [DOI] [PubMed] [Google Scholar]
- 204.Davidson DJ, Webb S, Teague P, Govan JR, Dorin JR. Lung pathology in response to repeated exposure to Staphylococcus aureus in congenic residual function cystic fibrosis mice does not increase in response to decreased CFTR levels or increased bacterial load. Pathobiology. 2004;71,:152–158. doi: 10.1159/000076470. [DOI] [PubMed] [Google Scholar]
- 205.Stotland PK, Radzioch D, Stevenson MM. Mouse models of chronic lung infection with Pseudomonas aeruginosa: models for the study of cystic fibrosis. Pediatr. Pulmonol. 2000;30,:413–424. doi: 10.1002/1099-0496(200011)30:5. [DOI] [PubMed] [Google Scholar]
- 206.Durie PR, Kent G, Phillips MJ, Ackerley CA. Characteristic multiorgan pathology of cystic fibrosis in a long-living cystic fibrosis transmembrane regulator knockout murine model. Am. J. Pathol. 2004;164,:1481–1493. doi: 10.1016/S0002-9440(10)63234-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Burkhardt A. Alveolitis and collapse in the pathogenesis of pulmonary fibrosis. Am. Rev. Respir. Dis. 1989;140,:513–524. doi: 10.1164/ajrccm/140.2.513. [DOI] [PubMed] [Google Scholar]
- 208.Carver JR. American Society of Clinical Oncology clinical evidence review on the ongoing care of adult cancer survivors: cardiac and pulmonary late effects. J. Clin. Oncol. 2007;25,:3991–4008. doi: 10.1200/JCO.2007.10.9777. 1:CAS:528:DC%2BD2sXhtFanu73F. [DOI] [PubMed] [Google Scholar]
- 209.Vagane R, Bruland OS, Fossa SD, Olsen DR. Radiological and functional assessment of radiation-induced pulmonary damage following breast irradiation. Acta. Oncol. 2008;47,:248–254. doi: 10.1080/02841860701630267. [DOI] [PubMed] [Google Scholar]
- 210.Ghafoori P, Marks LB, Vujaskovic Z, Kelsey CR. Radiation-induced lung injury. Assessment, management, and prevention. Oncology (Williston Park) 2008;22,:37–47. [PubMed] [Google Scholar]
- 211.Beinert T. Oxidant-induced lung injury in anticancer therapy. Eur. J. Med. Res. 1999;4,:43–53. [PubMed] [Google Scholar]
- 212.Rodningen OK, Borresen-Dale AL, Alsner J, Hastie T, Overgaard J. Radiation-induced gene expression in human subcutaneous fibroblasts is predictive of radiation-induced fibrosis. Radiother Oncol. 2008;86,:314–320. doi: 10.1016/j.radonc.2007.09.013. 1:CAS:528:DC%2BD1cXjtlSkur8%3D. [DOI] [PubMed] [Google Scholar]
- 213.Lemay AM, Haston CK. Radiation-induced lung response of AcB/BcA recombinant congenic mice. Radiat Res. 2008;170,:299–306. doi: 10.1667/RR1319.1. [DOI] [PubMed] [Google Scholar]
- 214.Johnston CJ, Williams JP, Elder A, Hernady E, Finkelstein JN. Inflammatory cell recruitment following thoracic irradiation. Exp. Lung Res. 2004;30,:369–382. doi: 10.1080/01902140490438915. 1:CAS:528:DC%2BD2cXksFShu7Y%3D. [DOI] [PubMed] [Google Scholar]
- 215.Westermann W, Schobl R, Rieber EP, Frank KH. Th2 cells as effectors in postirradiation pulmonary damage preceding fibrosis in the rat. Int. J. Radiat. Biol. 1999;75,:629–638. doi: 10.1080/095530099140276. [DOI] [PubMed] [Google Scholar]
- 216.Barthelemy-Brichant N. Increased IL-6 and TGF-beta1 concentrations in bronchoalveolar lavage fluid associated with thoracic radiotherapy. Int. J. Radiat. Oncol. Biol. Phys. 2004;58,:758–767. doi: 10.1016/S0360-3016(03)01614-6. 1:CAS:528:DC%2BD2cXhtVyhuro%3D. [DOI] [PubMed] [Google Scholar]
- 217.Hill RP. Radiation effects on the respiratory system. BJR Suppl. 2005;27,:75–81. doi: 10.1259/bjr/34124307. [DOI] [Google Scholar]
- 218.Matej R, Housa D, Pouckova P, Zadinova M, Olejar T. Radiation-induced production of PAR-1 and TGF-beta 1 mRNA in lung of C57Bl6 and C3H murine strains and influence of pharmacoprophylaxis by ACE inhibitors. Pathol. Res. Pract. 2007;203,:107–114. doi: 10.1016/j.prp.2006.10.006. 1:CAS:528:DC%2BD2sXjvFKqur0%3D. [DOI] [PubMed] [Google Scholar]
- 219.Molteni A. Effect of an angiotensin II receptor blocker and two angiotensin converting enzyme inhibitors on transforming growth factor-beta (TGF-beta) and alpha-actomyosin (alpha SMA), important mediators of radiation-induced pneumopathy and lung fibrosis. Curr. Pharm. Des. 2007;13,:1307–1316. doi: 10.2174/138161207780618777. [DOI] [PubMed] [Google Scholar]
- 220.Yang K. Matrix-Metallo-Proteinases and their tissue inhibitors in radiation-induced lung injury. Int. J. Radiat. Biol. 2007;83,:665–676. doi: 10.1080/09553000701558977. 1:CAS:528:DC%2BD2sXps1GktbY%3D. [DOI] [PubMed] [Google Scholar]
- 221.Johnston CJ, Williams JP, Okunieff P, Finkelstein JN. Radiation-induced pulmonary fibrosis: examination of chemokine and chemokine receptor families. Radiat. Res. 2002;157,:256–265. doi: 10.1667/0033-7587(2002)157[0256:RIPFEO]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 222.Haase MG, Klawitter A, Geyer P, Baretton GB. Expression of the immunomodulator IL-10 in type I pneumocytes of the rat: alterations of IL-10 expression in radiation-induced lung damage. J. Histochem Cytochem. 2007;55,:1167–1172. doi: 10.1369/jhc.7A7173.2007. 1:CAS:528:DC%2BD2sXht1aju7rN. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Sharplin J, Franko AJ. A quantitative histological study of strain-dependent differences in the effects of irradiation on mouse lung during the intermediate and late phases. Radiat Res. 1989;119,:15–31. doi: 10.2307/3577364. [DOI] [PubMed] [Google Scholar]
- 224.Giotopoulos G. The late radiotherapy normal tissue injury phenotypes of telangiectasia, fibrosis and atrophy in breast cancer patients have distinct genotype-dependent causes. Br. J. Cancer. 2007;96,:1001–1007. doi: 10.1038/sj.bjc.6603637. 1:CAS:528:DC%2BD2sXjt1Srs7Y%3D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Abid SH, Malhotra V, Perry MC. Radiation-induced and chemotherapy-induced pulmonary injury. Curr. Opin. Oncol. 2001;13,:242–248. doi: 10.1097/00001622-200107000-00006. [DOI] [PubMed] [Google Scholar]
- 226.Sleijfer S. Bleomycin-induced pneumonitis. Chest. 2001;120,:617–624. doi: 10.1378/chest.120.2.617. [DOI] [PubMed] [Google Scholar]
- 227.Umezawa H, Ishizuka M, Maeda K, Takeuchi T. Studies on bleomycin. Cancer. 1967;20,:891–895. doi: 10.1002/1097-0142(1967)20:5. [DOI] [PubMed] [Google Scholar]
- 228.Umezawa H. Chemistry and mechanism of action of bleomycin. Fed. Proc. 1974;33,:2296–2302. [PubMed] [Google Scholar]
- 229.Van Barneveld PW. Predictive factors for bleomycin-induced pneumonitis. Am. Rev. Respir. Dis. 1984;130,:1078–1081. doi: 10.1164/arrd.1984.130.6.1078. [DOI] [PubMed] [Google Scholar]
- 230.Onuma T, Holland JF, Masuda H, Waligunda JA, Goldberg GA. Microbiological assay of bleomycin: inactivation, tissue distribution, and clearance. Cancer. 1974;33,:1230–1238. doi: 10.1002/1097-0142(197405)33:5. [DOI] [PubMed] [Google Scholar]
- 231.Doelman CJ, Bast A. Oxygen radicals in lung pathology. Free Radic. Biol. Med. 1990;9,:381–400. doi: 10.1016/0891-5849(90)90015-B. [DOI] [PubMed] [Google Scholar]
- 232.Burger RM, Peisach J, Horwitz SB. Activated bleomycin. A transient complex of drug, iron, and oxygen that degrades DNA. J. Biol. Chem. 1981;256,:11636–11644. [PubMed] [Google Scholar]
- 233.Adamson IY, Bowden DH. The pathogenesis of bloemycin-induced pulmonary fibrosis in mice. Am. J. Pathol. 1974;77,:185–197. [PMC free article] [PubMed] [Google Scholar]
- 234.Piguet PF, Rosen H, Vesin C, Grau GE. Effective treatment of the pulmonary fibrosis elicited in mice by bleomycin or silica with anti-CD-11 antibodies. Am. Rev. Respir. Dis. 1993;147,:435–441. doi: 10.1164/ajrccm/147.2.435. [DOI] [PubMed] [Google Scholar]
- 235.Piguet PF, Collart MA, Grau GE, Kapanci Y, Vassalli P. Tumor necrosis factor/cachectin plays a key role in bleomycin-induced pneumopathy and fibrosis. J. Exp. Med. 1989;170,:655–663. doi: 10.1084/jem.170.3.655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Scheule RK, Perkins RC, Hamilton R, Holian A. Bleomycin stimulation of cytokine secretion by the human alveolar macrophage. Am. J. Physiol. 1992;262(4 Part 1):L386–L391. doi: 10.1152/ajplung.1992.262.4.L386. 1:CAS:528:DyaK38Xkt1Sjurs%3D, 1373569. [DOI] [PubMed] [Google Scholar]
- 237.Smith RE, Strieter RM, Phan SH, Lukacs N, Kunkel SL. TNF and IL-6 mediate MIP-1alpha expression in bleomycin-induced lung injury. J. Leukoc. Biol. 1998;64,:528–536. doi: 10.1002/jlb.64.4.528. [DOI] [PubMed] [Google Scholar]
- 238.Santana A, Saxena B, Noble NA, Gold LI, Marshall BC. Increased expression of transforming growth factor beta isoforms (beta 1, beta 2, beta 3) in bleomycin-induced pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 1995;13,:34–44. doi: 10.1165/ajrcmb.13.1.7541221. [DOI] [PubMed] [Google Scholar]
- 239.Zhang K, Flanders KC, Phan SH. Cellular localization of transforming growth factor-beta expression in bleomycin-induced pulmonary fibrosis. Am. J. Pathol. 1995;147,:352–361. [PMC free article] [PubMed] [Google Scholar]
- 240.Hagimoto N, Kuwano K, Nomoto Y, Kunitake R, Hara N. Apoptosis and expression of Fas/Fas ligand mRNA in bleomycin-induced pulmonary fibrosis in mice. Am. J. Respir. Cell Mol. Biol. 1997;16,:91–101. doi: 10.1165/ajrcmb.16.1.8998084. [DOI] [PubMed] [Google Scholar]
- 241.Giri SN, Hyde DM, Hollinger MA. Effect of antibody to transforming growth factor beta on bleomycin induced accumulation of lung collagen in mice. Thorax. 1993;48,:959–966. doi: 10.1136/thx.48.10.959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Piguet PF, Vesin C, Grau GE, Thompson RC. Interleukin 1 receptor antagonist (IL-1ra) prevents or cures pulmonary fibrosis elicited in mice by bleomycin or silica. Cytokine. 1993;5,:57–61. doi: 10.1016/1043-4666(93)90024-Y. [DOI] [PubMed] [Google Scholar]
- 243.Hao H, Cohen DA, Jennings CD, Bryson JS, Kaplan AM. Bleomycin-induced pulmonary fibrosis is independent of eosinophils. J. Leukoc. Biol. 2000;68,:515–521. [PubMed] [Google Scholar]
- 244.Izbicki G, Breuer R. IL-4 is not a key profibrotic cytokine in bleomycin-induced lung fibrosis model. J. Immunol. 2003;171,:2767–2768. doi: 10.4049/jimmunol.171.6.2767-b. [DOI] [PubMed] [Google Scholar]
- 245.McKay DL, Jr, Fuqua F, Weinberg AG. Balanitis xerotica obliterans in children. J. Urol. 1975;114,:773–775. doi: 10.1016/S0022-5347(17)67141-6. [DOI] [PubMed] [Google Scholar]
- 246.Segel MJ. Role of interferon-gamma in the evolution of murine bleomycin lung fibrosis. Am. J. Physiol. 2003;285,:L1255–L1262. doi: 10.1152/ajpcell.00149.2003. [DOI] [PubMed] [Google Scholar]
- 247.Chen ES, Greenlee BM, Wills-Karp M, Moller DR. Attenuation of lung inflammation and fibrosis in interferon-gamma-deficient mice after intratracheal bleomycin. Am. J. Respir. Cell Mol. Biol. 2001;24,:545–555. doi: 10.1165/ajrcmb.24.5.4064. [DOI] [PubMed] [Google Scholar]
- 248.Sakamoto H, Zhao LH, Jain F, Kradin R. IL-12p40(−/−) mice treated with intratracheal bleomycin exhibit decreased pulmonary inflammation and increased fibrosis. Exp. Mol. Pathol. 2002;72,:1–9. doi: 10.1006/exmp.2001.2409. 1:CAS:528:DC%2BD38XisVygtw%3D%3D. [DOI] [PubMed] [Google Scholar]
- 249.Moseley PL, Hemken C, Hunninghake GW. Augmentation of fibroblast proliferation by bleomycin. J. Clin. Invest. 1986;78,:1150–1154. doi: 10.1172/JCI112695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Phan SH, Gharaee-Kermani M, Wolber F, Ryan US. Stimulation of rat endothelial cell transforming growth factor-beta production by bleomycin. J. Clin. Invest. 1991;87,:148–154. doi: 10.1172/JCI114964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Eder W, Ege MJ, von Mutius E. The asthma epidemic. N. Engl. J. Med. 2006;355,:2226–2235. doi: 10.1056/NEJMra054308. [DOI] [PubMed] [Google Scholar]
- 252.Martinez FD. Gene-environment interactions in asthma and allergies: a new paradigm to understand disease causation. Immunol. Allergy Clin. North Am. 2005;25,:709–721. doi: 10.1016/j.iac.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 253.Cohn L, Elias JA, Chupp GL. Asthma: mechanisms of disease persistence and progression. Annu. Rev. Immunol. 2004;22,:789–815. doi: 10.1146/annurev.immunol.22.012703.104716. 1:CAS:528:DC%2BD2cXktlOgsb0%3D. [DOI] [PubMed] [Google Scholar]
- 254.Boulet LP, Sterk PJ. Airway remodelling: the future. Eur. Respir. J. 2007;30,:831–834. doi: 10.1183/09031936.00110107. [DOI] [PubMed] [Google Scholar]
- 255.Broide DH. Immunologic and inflammatory mechanisms that drive asthma progression to remodeling. J. Allergy Clin. Immunol. 2008;121,:560–570. doi: 10.1016/j.jaci.2008.01.031. 1:CAS:528:DC%2BD1cXjtVSqsr0%3D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Huang J, Olivenstein R, Taha R, Hamid Q, Ludwig M. Enhanced proteoglycan deposition in the airway wall of atopic asthmatics. Am. J. Resp. Crit. Care Med. 1999;160,:725–729. doi: 10.1164/ajrccm.160.2.9809040. [DOI] [PubMed] [Google Scholar]
- 257.Ward C. Airway inflammation, basement membrane thickening and bronchial hyperresponsiveness in asthma. Thorax. 2002;57,:309–316. doi: 10.1136/thorax.57.4.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Angkasekwinai P. Interleukin 25 promotes the initiation of proallergic type 2 responses. J. Exp. Med. 2007;204,:1509–1517. doi: 10.1084/jem.20061675. 1:CAS:528:DC%2BD2sXnslymu7g%3D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Owyang AM. Interleukin 25 regulates type 2 cytokine-dependent immunity and limits chronic inflammation in the gastrointestinal tract. J. Exp. Med. 2006;203,:843–849. doi: 10.1084/jem.20051496. 1:CAS:528:DC%2BD28Xjsl2isrg%3D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Sharkhuu T. Mechanism of interleukin-25 (IL-17E)-induced pulmonary inflammation and airways hyper-reactivity. Clin. Exp. Allergy. 2006;36,:1575–1583. doi: 10.1111/j.1365-2222.2006.02595.x. [DOI] [PubMed] [Google Scholar]
- 261.Min B. Basophils produce IL-4 and accumulate in tissues after infection with a Th2-inducing parasite. J. Exp. Med. 2004;200,:507–517. doi: 10.1084/jem.20040590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Voehringer D, Shinkai K, Locksley RM. Type 2 immunity reflects orchestrated recruitment of cells committed to IL-4 production. Immunity. 2004;20,:267–277. doi: 10.1016/S1074-7613(04)00026-3. [DOI] [PubMed] [Google Scholar]
- 263.Webb DC, Cai Y, Matthaei KI, Foster PS. Comparative roles of IL-4, IL-13, and IL-4Ralpha in dendritic cell maturation and CD4+ Th2 cell function. J. Immunol. 2007;178,:219–227. doi: 10.4049/jimmunol.178.1.219. [DOI] [PubMed] [Google Scholar]
- 264.Hauber HP, Bergeron C, Hamid Q. IL-9 in allergic inflammation. Int. Arch. Allergy Immunol. 2004;134,:79–87. doi: 10.1159/000078384. 1:CAS:528:DC%2BD2cXktFKlsrw%3D. [DOI] [PubMed] [Google Scholar]
- 265.Woodman L. Mast cells promote airway smooth muscle cell differentiation via autocrine up-regulation of TGF-beta1. J. Immunol. 2008;181,:5001–5007. doi: 10.4049/jimmunol.181.7.5001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Coutts A. Release of biologically active TGF-beta from airway smooth muscle cells induces autocrine synthesis of collagen. Am. J. Physiol. 2001;280,:L999–L1008. doi: 10.1152/ajplung.2001.280.5.L999. [DOI] [PubMed] [Google Scholar]
- 267.Balzar S. Increased TGF-beta2 in severe asthma with eosinophilia. J. Allergy Clin. Immunol. 2005;115,:110–117. doi: 10.1016/j.jaci.2004.09.034. 1:CAS:528:DC%2BD2MXks1ek. [DOI] [PubMed] [Google Scholar]
- 268.Batra V. Bronchoalveolar lavage fluid concentrations of transforming growth factor (TGF)-beta1, TGF-beta2, interleukin (IL)-4 and IL-13 after segmental allergen challenge and their effects on alpha-smooth muscle actin and collagen III synthesis by primary human lung fibroblasts. Clin. Exp. Allergy. 2004;34,:437–444. doi: 10.1111/j.1365-2222.2004.01885.x. [DOI] [PubMed] [Google Scholar]
- 269.Levi-Schaffer F. Human eosinophils regulate human lung- and skin-derived fibroblast properties in vitro: a role for transforming growth factor beta (TGF-beta) Proc. Natl. Acad. Sci. USA. 1999;96,:9660–9665. doi: 10.1073/pnas.96.17.9660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Sagara H. Activation of TGF-beta/Smad2 signaling is associated with airway remodeling in asthma. J. Allergy Clin. Immunol. 2002;110,:249–254. doi: 10.1067/mai.2002.126078. 1:CAS:528:DC%2BD38XntFSlsLo%3D. [DOI] [PubMed] [Google Scholar]
- 271.Flood-Page P. Anti-IL-5 treatment reduces deposition of ECM proteins in the bronchial subepithelial basement membrane of mild atopic asthmatics. J. Clin. Invest. 2003;112,:1029–1036. doi: 10.1172/JCI17974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Kang HR, Cho SJ, Lee CG, Homer RJ, Elias JA. Transforming growth factor (TGF)-beta1 stimulates pulmonary fibrosis and inflammation via a Bax-dependent, bid-activated pathway that involves matrix metalloproteinase-12. J. Biol. Chem. 2007;282,:7723–7732. doi: 10.1074/jbc.M610764200. 1:CAS:528:DC%2BD2sXitlSru74%3D. [DOI] [PubMed] [Google Scholar]
- 273.Phipps S, Flood-Page P, Menzies-Gow A, Ong YE, Kay AB. Intravenous anti-IL-5 monoclonal antibody reduces eosinophils and tenascin deposition in allergen-challenged human atopic skin. J. Invest Dermatol. 2004;122,:1406–1412. doi: 10.1111/j.0022-202X.2004.22619.x. 1:CAS:528:DC%2BD2cXkvFCqtLs%3D. [DOI] [PubMed] [Google Scholar]
- 274.Menzies-Gow A. Anti-IL-5 (mepolizumab) therapy induces bone marrow eosinophil maturational arrest and decreases eosinophil progenitors in the bronchial mucosa of atopic asthmatics. J. Allergy Clin. Immunol. 2003;111,:714–719. doi: 10.1067/mai.2003.1382. 1:CAS:528:DC%2BD3sXjs1equ7o%3D. [DOI] [PubMed] [Google Scholar]
- 275.Flood-Page PT, Menzies-Gow AN, Kay AB, Robinson DS. Eosinophil??s role remains uncertain as anti-interleukin-5 only partially depletes numbers in asthmatic airway. Am. J. Resp. Crit. Care Med. 2003;167,:199–204. doi: 10.1164/rccm.200208-789OC. [DOI] [PubMed] [Google Scholar]
- 276.O'Byrne PM. The demise of anti IL-5 for asthma, or not. Am. J. Resp. Crit. Care Med. 2007;176,:1059–1060. doi: 10.1164/rccm.200708-1264ED. [DOI] [PubMed] [Google Scholar]
- 277.Cho JY. Inhibition of airway remodeling in IL-5-deficient mice. J. Clin. Invest. 2004;113,:551–560. doi: 10.1172/JCI19133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Humbles AA. A critical role for eosinophils in allergic airways remodeling. Science. 2004;305,:1776–1779. doi: 10.1126/science.1100283. 1:CAS:528:DC%2BD2cXnsFajtbY%3D. [DOI] [PubMed] [Google Scholar]
- 279.Lee JJ. Defining a link with asthma in mice congenitally deficient in eosinophils. Science. 2004;305,:1773–1776. doi: 10.1126/science.1099472. 1:CAS:528:DC%2BD2cXnsFajtbk%3D. [DOI] [PubMed] [Google Scholar]
- 280.McMillan SJ, Xanthou G, Lloyd CM. Manipulation of allergen-induced airway remodeling by treatment with anti-TGF-beta antibody: effect on the Smad signaling pathway. J. Immunol. 2005;174,:5774–5780. doi: 10.4049/jimmunol.174.9.5774. [DOI] [PubMed] [Google Scholar]
- 281.Le AV, Cho JY, Miller M, McElwain S, Golgotiu K, Broide DH. Inhibition of allergen-induced airway remodeling in Smad 3-deficient mice. J. Immunol. 2007;178,:7310–7316. doi: 10.4049/jimmunol.178.11.7310. [DOI] [PubMed] [Google Scholar]
- 282.Kondo M. Elimination of IL-13 reverses established goblet cell metaplasia into ciliated epithelia in airway epithelial cell culture. Allergol. Int. 2006;55,:329–336. doi: 10.2332/allergolint.55.329. [DOI] [PubMed] [Google Scholar]
- 283.Fanta CH. Clinical aspects of mucus and mucous plugging in asthma. J. Asthma. 1985;22,:295–301. doi: 10.3109/02770908509087113. [DOI] [PubMed] [Google Scholar]
- 284.Ramalingam TR. Unique functions of the type II interleukin 4 receptor identified in mice lacking the interleukin 13 receptor alpha1 chain. Nat. Immunol. 2008;9,:25–33. doi: 10.1038/ni1544. 1:CAS:528:DC%2BD2sXhsVegtLjL. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Allahverdian S, Harada N, Singhera GK, Knight DA, Dorscheid DR. Secretion of IL-13 by airway epithelial cells enhances epithelial repair via HB-EGF. Am. J. Respir. Cell Mol. Biol. 2008;38,:153–160. doi: 10.1165/rcmb.2007-0173OC. 1:CAS:528:DC%2BD1cXhsVagsr0%3D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Booth BW, Adler KB, Bonner JC, Tournier F, Martin LD. Interleukin-13 induces proliferation of human airway epithelial cells in vitro via a mechanism mediated by transforming growth factor-alpha. Am. J. Respir. Cell Mol. Biol. 2001;25,:739–743. doi: 10.1165/ajrcmb.25.6.4659. [DOI] [PubMed] [Google Scholar]
- 287.Saito A, Okazaki H, Sugawara I, Yamamoto K, Takizawa H. Potential action of IL-4 and IL-13 as fibrogenic factors on lung fibroblasts in vitro. Int. Arch. Allergy Immunol. 2003;132,:168–176. doi: 10.1159/000073718. 1:CAS:528:DC%2BD3sXpvVGlt7o%3D. [DOI] [PubMed] [Google Scholar]
- 288.Richter A. The contribution of interleukin (IL)-4 and IL-13 to the epithelial-mesenchymal trophic unit in asthma. Am. J. Respir. Cell Mol. Biol. 2001;25,:385–391. doi: 10.1165/ajrcmb.25.3.4437. [DOI] [PubMed] [Google Scholar]
- 289.Chiba, Y., Nakazawa, S., Todoroki, M., Shinozaki, K., Sakai, H. & Misawa, M. Interleukin-13 Augments Bronchial Smooth Muscle Contractility with an Upregulation of RhoA Protein. Am. J. Respir. Cell Mol. Biol. (2008). (E-pub ahead of print) [DOI] [PubMed]
- 290.Wen FQ. Interleukin-4- and interleukin-13-enhanced transforming growth factor-beta2 production in cultured human bronchial epithelial cells is attenuated by interferon-gamma. Am. J. Respir. Cell Mol. Biol. 2002;26,:484–490. doi: 10.1165/ajrcmb.26.4.4784. [DOI] [PubMed] [Google Scholar]
- 291.Zhou X. Interleukin-13 augments transforming growth factor-beta1-induced tissue inhibitor of metalloproteinase-1 expression in primary human airway fibroblasts. Am. J. Physiol. Cell Physiol. 2005;288,:C435–C442. doi: 10.1152/ajpcell.00035.2004. 1:CAS:528:DC%2BD2MXhsVCksbg%3D. [DOI] [PubMed] [Google Scholar]
- 292.Zhou X. Mechanisms of tissue inhibitor of metalloproteinase 1 augmentation by IL-13 on TGF-beta 1-stimulated primary human fibroblasts. J. Allergy Clin. Immunol. 2007;119,:1388–1397. doi: 10.1016/j.jaci.2007.02.011. 1:CAS:528:DC%2BD2sXmtlaisb4%3D. [DOI] [PubMed] [Google Scholar]
- 293.Paris C. Factors associated with early-stage pulmonary fibrosis as determined by high-resolution computed tomography among persons occupationally exposed to asbestos. Scand J. Work Environ. Health. 2004;30,:206–214. doi: 10.5271/sjweh.781. [DOI] [PubMed] [Google Scholar]
- 294.Schenker M. Exposures and health effects from inorganic agricultural dusts. Environ. Health Perspect. 2000;108(Suppl 4):661–664. doi: 10.1289/ehp.00108s4661. 1637665, 1:CAS:528:DC%2BD3cXms1yls7g%3D, 10931784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Von Essen S, Robbins RA, Thompson AB, Rennard SI. Organic dust toxic syndrome: an acute febrile reaction to organic dust exposure distinct from hypersensitivity pneumonitis. J. Toxicol. Clin. Toxicol. 1990;28,:389–420. doi: 10.3109/15563659009038584. [DOI] [PubMed] [Google Scholar]
- 296.Buerke U, Schneider J, Rosler J, Woitowitz HJ. Interstitial pulmonary fibrosis after severe exposure to welding fumes. Am. J. Ind. Med. 2002;41,:259–268. doi: 10.1002/ajim.10055. [DOI] [PubMed] [Google Scholar]
- 297.Che DY, Liu SC, Huang XZ. Pathogenesis of extrinsic allergic alveolitis and pulmonary fibrosis induced by streptomyces thermohygroscopicus. Chin. Med. J. (Engl.) 1989;102,:563–567. [PubMed] [Google Scholar]
- 298.Cormier Y, Brown M, Worthy S, Racine G, Muller NL. High-resolution computed tomographic characteristics in acute farmer??s lung and in its follow-up. Eur. Respir. J. 2000;16,:56–60. doi: 10.1034/j.1399-3003.2000.16a10.x. [DOI] [PubMed] [Google Scholar]
- 299.Lalancette M. Farmer??s lung. Long-term outcome and lack of predictive value of bronchoalveolar lavage fibrosing factors. Am. Rev. Respir. Dis. 1993;148,:216–221. doi: 10.1164/ajrccm/148.1.216. [DOI] [PubMed] [Google Scholar]
- 300.Toubas D, Prevost A, Deschamps F, Pinon JM. Extrinsic allergic alveolitis of occupational origin] Presse Med. 1995;24,:1391–1396. [PubMed] [Google Scholar]
- 301.Hillerdal G, Nou E, Osterman K, Schmekel B. Sarcoidosis: epidemiology and prognosis. A 15-year European study. Am. Rev. Respir. Dis. 1984;130,:29–32. doi: 10.1164/arrd.1984.130.1.29. [DOI] [PubMed] [Google Scholar]
- 302.Nunes H, Bouvry D, Soler P, Valeyre D. Sarcoidosis. Orphanet J. Rare Dis. 2007;2,:46. doi: 10.1186/1750-1172-2-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Gaede KI, Mamat U, Muller-Quernheim J. Differential gene expression pattern in alveolar macrophages of patients with sarcoidosis and tuberculosis. J. Mol. Med. 2004;82,:206–210. doi: 10.1007/s00109-003-0511-2. 1:CAS:528:DC%2BD2cXhsl2hsL0%3D. [DOI] [PubMed] [Google Scholar]
- 304.Abehsera M, Valeyre D, Grenier P, Jaillet H, Battesti JP, Brauner MW. Sarcoidosis with pulmonary fibrosis: CT patterns and correlation with pulmonary function. AJR Am. J. Roentgenol. 2000;174,:1751–1757. doi: 10.2214/ajr.174.6.1741751. [DOI] [PubMed] [Google Scholar]
- 305.Roman J, Jeon YJ, Gal A, Perez RL. Distribution of extracellular matrices, matrix receptors, and transforming growth factor-beta 1 in human and experimental lung granulomatous inflammation. Am. J. Med. Sci. 1995;309,:124–133. doi: 10.1097/00000441-199503000-00002. [DOI] [PubMed] [Google Scholar]
- 306.Hutyrova B. Interleukin-1 gene cluster polymorphisms in sarcoidosis and idiopathic pulmonary fibrosis. Am. J. Resp. Crit. Care Med. 2002;165,:148–151. doi: 10.1164/ajrccm.165.2.2106004. [DOI] [PubMed] [Google Scholar]
- 307.Muller-Quernheim J. Sarcoidosis: immunopathogenetic concepts and their clinical application. Eur. Respir. J. 1998;12,:716–738. doi: 10.1183/09031936.98.12030716. [DOI] [PubMed] [Google Scholar]
- 308.Rottoli P. Cytokine profile and proteome analysis in bronchoalveolar lavage of patients with sarcoidosis, pulmonary fibrosis associated with systemic sclerosis and idiopathic pulmonary fibrosis. Proteomics. 2005;5,:1423–1430. doi: 10.1002/pmic.200301007. 1:CAS:528:DC%2BD2MXjtlSjurg%3D. [DOI] [PubMed] [Google Scholar]
- 309.Ziegenhagen MW, Schrum S, Zissel G, Zipfel PF, Schlaak M, Muller-Quernheim J. Increased expression of proinflammatory chemokines in bronchoalveolar lavage cells of patients with progressing idiopathic pulmonary fibrosis and sarcoidosis. J. Investig Med. 1998;46,:223–231. [PubMed] [Google Scholar]
- 310.Iida K, Kadota J, Kawakami K, Matsubara Y, Shirai R, Kohno S. Analysis of T cell subsets and beta chemokines in patients with pulmonary sarcoidosis. Thorax. 1997;52,:431–437. doi: 10.1136/thx.52.5.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Limper AH, Colby TV, Sanders MS, Asakura S, Roche PC, DeRemee RA. Immunohistochemical localization of transforming growth factor-beta 1 in the nonnecrotizing granulomas of pulmonary sarcoidosis. Am. J. Resp. Crit. Care Med. 1994;149,:197–204. doi: 10.1164/ajrccm.149.1.8111583. [DOI] [PubMed] [Google Scholar]
- 312.Marshall BG, Wangoo A, Cook HT, Shaw RJ. Increased inflammatory cytokines and new collagen formation in cutaneous tuberculosis and sarcoidosis. Thorax. 1996;51,:1253–1261. doi: 10.1136/thx.51.12.1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Salez F, Gosset P, Copin MC, Lamblin Degros C, Tonnel AB, Wallaert B. Transforming growth factor-beta1 in sarcoidosis. Eur. Respir. J. 1998;12,:913–919. doi: 10.1183/09031936.98.12040913. [DOI] [PubMed] [Google Scholar]
- 314.Kline JN, Schwartz DA, Monick MM, Floerchinger CS, Hunninghake GW. Relative release of interleukin-1 beta and interleukin-1 receptor antagonist by alveolar macrophages. A study in asbestos-induced lung disease, sarcoidosis, and idiopathic pulmonary fibrosis. Chest. 1993;104,:47–53. doi: 10.1378/chest.104.1.47. [DOI] [PubMed] [Google Scholar]
- 315.Baumgartner KB, Samet JM, Stidley CA, Colby TV, Waldron JA. Cigarette smoking: a risk factor for idiopathic pulmonary fibrosis. Am. J. Resp. Crit. Care Med. 1997;155,:242–248. doi: 10.1164/ajrccm.155.1.9001319. [DOI] [PubMed] [Google Scholar]
- 316.Baumgartner KB. Occupational and environmental risk factors for idiopathic pulmonary fibrosis: a multicenter case-control study. Collaborating Centers. Am. J. Epidemiol. 2000;152,:307–315. doi: 10.1093/aje/152.4.307. [DOI] [PubMed] [Google Scholar]
- 317.Iwai K, Mori T, Yamada N, Yamaguchi M, Hosoda Y. Idiopathic pulmonary fibrosis. Epidemiologic approaches to occupational exposure. Am. J. Resp. Crit. Care Med. 1994;150,:670–675. doi: 10.1164/ajrccm.150.3.8087336. [DOI] [PubMed] [Google Scholar]
- 318.Jakab GJ. Sequential virus infections, bacterial superinfections, and fibrogenesis. Am. Rev. Respir. Dis. 1990;142,:374–379. doi: 10.1164/ajrccm/142.2.374. [DOI] [PubMed] [Google Scholar]
- 319.Meliconi R. Incidence of hepatitis C virus infection in Italian patients with idiopathic pulmonary fibrosis. Thorax. 1996;51,:315–317. doi: 10.1136/thx.51.3.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Pinsker KL, Schneyer B, Becker N, Kamholz SL. Usual interstitial pneumonia following Texas A2 influenza infection. Chest. 1981;80,:123–126. doi: 10.1378/chest.80.2.123. [DOI] [PubMed] [Google Scholar]
- 321.Qunn L. Hyperplastic epithelial foci in honeycomb lesions in idiopathic pulmonary fibrosis. Virchows Arch. 2002;441,:271–278. doi: 10.1007/s00428-002-0618-9. [DOI] [PubMed] [Google Scholar]
- 322.Bitterman PB, Rennard SI, Keogh BA, Wewers MD, Adelberg S, Crystal RG. Familial idiopathic pulmonary fibrosis. Evidence of lung inflammation in unaffected family members. N. Engl. J. Med. 1986;314,:1343–1347. doi: 10.1056/NEJM198605223142103. [DOI] [PubMed] [Google Scholar]
- 323.Verleden GM. Genetic predisposition and pathogenetic mechanisms of interstitial lung diseases of unknown origin. Eur. Respir. J. Suppl. 2001;32,:17s–29s. [PubMed] [Google Scholar]
- 324.Gribbin J, Hubbard RB, Le Jeune I, Smith CJ, West J, Tata LJ. Incidence and mortality of idiopathic pulmonary fibrosis and sarcoidosis in the UK. Thorax. 2006;61,:980–985. doi: 10.1136/thx.2006.062836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325.American Thoracic Society Idiopathic pulmonary fibrosis: diagnosis and treatment. International consensus statement. American Thoracic Society (ATS), and the European Respiratory Society (ERS) Am. J. Resp. Crit. Care Med. 2000;161(2 Part 1):646–664. doi: 10.1164/ajrccm.161.2.ats3-00. [DOI] [PubMed] [Google Scholar]
- 326.Crystal RG. Future research directions in idiopathic pulmonary fibrosis: summary of a National Heart, Lung, and Blood Institute working group. Am. J. Resp. Crit. Care Med. 2002;166,:236–246. doi: 10.1164/rccm.2201069. [DOI] [PubMed] [Google Scholar]
- 327.Crystal RG, Bitterman PB, Rennard SI, Hance AJ, Keogh BA. Interstitial lung diseases of unknown cause. Disorders characterized by chronic inflammation of the lower respiratory tract (first of two parts) N. Engl. J. Med. 1984;310,:154–166. doi: 10.1056/NEJM198401193100304. [DOI] [PubMed] [Google Scholar]
- 328.Gauldie J, Bonniaud P, Sime P, Ask K, Kolb M. TGF-beta, Smad3 and the process of progressive fibrosis. Biochem. Soc. Trans. 2007;35(Part 4):661–664. doi: 10.1042/BST0350661. 1:CAS:528:DC%2BD2sXhtVKmt7zO, 17635115. [DOI] [PubMed] [Google Scholar]
- 329.Flaherty KR. Steroids in idiopathic pulmonary fibrosis: a prospective assessment of adverse reactions, response to therapy, and survival. Am. J. Med. 2001;110,:278–282. doi: 10.1016/S0002-9343(00)00711-7. [DOI] [PubMed] [Google Scholar]
- 330.Khalil N, O'Connor RN, Flanders KC, Unruh H. TGF-beta 1, but not TGF-beta 2 or TGF-beta 3, is differentially present in epithelial cells of advanced pulmonary fibrosis: an immunohistochemical study. Am. J. Respir. Cell Mol. Biol. 1996;14,:131–138. doi: 10.1165/ajrcmb.14.2.8630262. [DOI] [PubMed] [Google Scholar]
- 331.Xu YD, Hua J, Mui A, O'Connor R, Grotendorst G, Khalil N. Release of biologically active TGF-beta1 by alveolar epithelial cells results in pulmonary fibrosis. Am. J. Physiol. 2003;285,:L527–L539. doi: 10.1152/ajplung.00298.2002. [DOI] [PubMed] [Google Scholar]
- 332.Pan LH, Ohtani H, Yamauchi K, Nagura H. Co-expression of TNF alpha and IL-1 beta in human acute pulmonary fibrotic diseases: an immunohistochemical analysis. Pathol. Int. 1996;46,:91–99. doi: 10.1111/j.1440-1827.1996.tb03584.x. [DOI] [PubMed] [Google Scholar]
- 333.Zhang Y, Lee TC, Guillemin B, Yu MC, Rom WN. Enhanced IL-1 beta and tumor necrosis factor-alpha release and messenger RNA expression in macrophages from idiopathic pulmonary fibrosis or after asbestos exposure. J. Immunol. 1993;150,:4188–4196. [PubMed] [Google Scholar]
- 334.Antoniou KM. Th1 cytokine pattern (IL-12 and IL-18) in bronchoalveolar lavage fluid (BALF) before and after treatment with interferon gamma-1b (IFN-gamma-1b) or colchicine in patients with idiopathic pulmonary fibrosis (IPF/UIP) Sarcoidosis Vasc. Diffuse Lung Dis. 2004;21,:105–110. [PubMed] [Google Scholar]
- 335.Antoniades HN. Platelet-derived growth factor in idiopathic pulmonary fibrosis. J. Clin. Invest. 1990;86,:1055–1064. doi: 10.1172/JCI114808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336.Standiford TJ. Altered production and regulation of monocyte chemoattractant protein-1 from pulmonary fibroblasts isolated from patients with idiopathic pulmonary fibrosis. Chest. 1993;103(2 Suppl):121S. doi: 10.1378/chest.103.2_Supplement.121S. 1:STN:280:DyaK3s7lsVWmuw%3D%3D, 8428531. [DOI] [PubMed] [Google Scholar]
- 337.Furuie H, Yamasaki H, Suga M, Ando M. Altered accessory cell function of alveolar macrophages: a possible mechanism for induction of Th2 secretory profile in idiopathic pulmonary fibrosis. Eur. Respir. J. 1997;10,:787–794. [PubMed] [Google Scholar]
- 338.Jakubzick C. Augmented pulmonary IL-4 and IL-13 receptor subunit expression in idiopathic interstitial pneumonia. J. Clin. Pathol. 2004;57,:477–486. doi: 10.1136/jcp.2003.012799. 1:CAS:528:DC%2BD2cXmtl2qurs%3D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339.Hunninghake GW, Gadek JE, Lawley TJ, Crystal RG. Mechanisms of neutrophil accumulation in the lungs of patients with idiopathic pulmonary fibrosis. J. Clin. Invest. 1981;68,:259–269. doi: 10.1172/JCI110242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 340.Baptista AL, Parra ER, Filho JV, Kairalla RA, de Carvalho CR, Capelozzi VL. Structural features of epithelial remodeling in usual interstitial pneumonia histologic pattern. Lung. 2006;184,:239–244. doi: 10.1007/s00408-005-2585-9. [DOI] [PubMed] [Google Scholar]
- 341.Agusti AG, Roca J, Gea J, Wagner PD, Xaubet A, Rodriguez-Roisin R. Mechanisms of gas-exchange impairment in idiopathic pulmonary fibrosis. Am. Rev. Respir. Dis. 1991;143,:219–225. doi: 10.1164/ajrccm/143.2.219. [DOI] [PubMed] [Google Scholar]
- 342.Holsti MA. Regulation of postsurgical fibrosis by the programmed death-1 inhibitory pathway. J. Immunol. 2004;172,:5774–5781. doi: 10.4049/jimmunol.172.9.5774. [DOI] [PubMed] [Google Scholar]
- 343.Moore KW, de Waal Malefyt R, Coffman RL, O'Garra A. Interleukin-10 and the interleukin-10 receptor. Annu. Rev. Immunol. 2001;19,:683–765. doi: 10.1146/annurev.immunol.19.1.683. [DOI] [PubMed] [Google Scholar]
- 344.Garantziotis S. Leukocyte-derived IL-10 reduces subepithelial fibrosis associated with chronically inhaled endotoxin. Am. J. Respir. Cell Mol. Biol. 2006;35,:662–667. doi: 10.1165/rcmb.2006-0055OC. 1:CAS:528:DC%2BD28XhtlSmtbvL. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345.Arai T. Introduction of the interleukin-10 gene into mice inhibited bleomycin-induced lung injury in vivo. Am. J. Physiol. 2000;278,:L914–L922. doi: 10.1152/ajpcell.2000.278.5.C914. [DOI] [PubMed] [Google Scholar]
- 346.Dosanjh AK, Elashoff D, Robbins RC. The bronchoalveolar lavage fluid of cystic fibrosis lung transplant recipients demonstrates increased interleukin-8 and elastase and decreased IL-10. J. Interferon Cytokine Res. 1998;18,:851–854. doi: 10.1089/jir.1998.18.851. [DOI] [PubMed] [Google Scholar]
- 347.Millar AB. IL-10: another therapeutic target in idiopathic pulmonary fibrosis? Thorax. 2006;61,:835–836. doi: 10.1136/thx.2006.060772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 348.Mu W. IL-10 suppresses chemokines, inflammation, and fibrosis in a model of chronic renal disease. J. Am. Soc. Nephrol. 2005;16,:3651–3660. doi: 10.1681/ASN.2005030297. 1:CAS:528:DC%2BD28XhvFajtw%3D%3D. [DOI] [PubMed] [Google Scholar]
- 349.Wynn TA. IL-10 regulates liver pathology in acute murine Schistosomiasis mansoni but is not required for immune down-modulation of chronic disease. J. Immunol. 1998;160,:4473–4480. [PubMed] [Google Scholar]
- 350.Hoffmann KF, Cheever AW, Wynn TA. IL-10 and the dangers of immune polarization: excessive type 1 and type 2 cytokine responses induce distinct forms of lethal immunopathology in murine schistosomiasis. J. Immunol. 2000;164,:6406–6416. doi: 10.4049/jimmunol.164.12.6406. [DOI] [PubMed] [Google Scholar]
- 351.Wilson MS, Pesce JT, Ramalingam TR, Thompson RW, Cheever A, Wynn TA. Suppression of murine allergic airway disease by IL-2:anti-IL-2 monoclonal antibody-induced regulatory T cells. J. Immunol. 2008;181,:6942–6954. doi: 10.4049/jimmunol.181.10.6942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352.Kasaian MT, Miller DK. IL-13 as a therapeutic target for respiratory disease. Biochem Pharmacol. 2008;76,:147–155. doi: 10.1016/j.bcp.2008.04.002. 1:CAS:528:DC%2BD1cXnvVGmtL0%3D. [DOI] [PubMed] [Google Scholar]
- 353.Zhu Z. IL-13-induced chemokine responses in the lung: role of CCR2 in the pathogenesis of IL-13-induced inflammation and remodeling. J. Immunol. 2002;168,:2953–2962. doi: 10.4049/jimmunol.168.6.2953. [DOI] [PubMed] [Google Scholar]
- 354.Bottinger EP. The recombinant proregion of transforming growth factor beta1 (latency-associated peptide) inhibits active transforming growth factor beta1 in transgenic mice. Proc. Natl. Acad. Sci. USA. 1996;93,:5877–5882. doi: 10.1073/pnas.93.12.5877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355.Rodriguez-Pena A. Up-regulation of endoglin, a TGF-beta-binding protein, in rats with experimental renal fibrosis induced by renal mass reduction. Nephrol. Dial. Transplant. 2001;16(Suppl 1):34–39. doi: 10.1093/ndt/16.suppl_1.34. 1:CAS:528:DC%2BD3MXltVOntrk%3D, 11369818. [DOI] [PubMed] [Google Scholar]
- 356.Gurujeyalakshmi G, Hollinger MA, Giri SN. Regulation of transforming growth factor-beta1 mRNA expression by taurine and niacin in the bleomycin hamster model of lung fibrosis. Am. J. Respir. Cell Mol. Biol. 1998;18,:334–342. doi: 10.1165/ajrcmb.18.3.2867. [DOI] [PubMed] [Google Scholar]
- 357.Yang KL, Chang WT, Chuang CC, Hung KC, Li EI. Antagonizing TGF-beta induced liver fibrosis by a retinoic acid derivative through regulation of ROS and calcium influx. Biochem. Biophys. Res. Commun. 2008;365,:484–489. doi: 10.1016/j.bbrc.2007.10.203. 1:CAS:528:DC%2BD2sXhtlyrsbrM. [DOI] [PubMed] [Google Scholar]
- 358.Cutroneo KR, White SL, Phan SH, Ehrlich HP. Therapies for bleomycin induced lung fibrosis through regulation of TGF-beta1 induced collagen gene expression. J. Cell Physiol. 2007;211,:585–589. doi: 10.1002/jcp.20972. 1:CAS:528:DC%2BD2sXkvFCnsrk%3D. [DOI] [PubMed] [Google Scholar]
- 359.Chiaramonte MG, Donaldson DD, Cheever AW, Wynn TA. An IL-13 inhibitor blocks the development of hepatic fibrosis during a T-helper type 2-dominated inflammatory response. J. Clin. Invest. 1999;104,:777–785. doi: 10.1172/JCI7325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 360.Fallon PG, Richardson EJ, McKenzie GJ, McKenzie AN. Schistosome infection of transgenic mice defines distinct and contrasting pathogenic roles for IL-4 and IL-13: IL-13 is a profibrotic agent. J. Immunol. 2000;164,:2585–2591. doi: 10.4049/jimmunol.164.5.2585. [DOI] [PubMed] [Google Scholar]
- 361.Zheng T. IL-13 receptor alpha2 selectively inhibits IL-13-induced responses in the murine lung. J. Immunol. 2008;180,:522–529. doi: 10.4049/jimmunol.180.1.522. [DOI] [PubMed] [Google Scholar]
- 362.Wills-Karp M. Interleukin-13: central mediator of allergic asthma. Science. 1998;282,:2258–2261. doi: 10.1126/science.282.5397.2258. [DOI] [PubMed] [Google Scholar]
- 363.Mentink-Kane MM, Wynn TA. Opposing roles for IL-13 and IL-13 receptor alpha 2 in health and disease. Immunol. Rev. 2004;202,:191–202. doi: 10.1111/j.0105-2896.2004.00210.x. [DOI] [PubMed] [Google Scholar]
- 364.Mentink-Kane MM. IL-13 receptor alpha 2 down-modulates granulomatous inflammation and prolongs host survival in schistosomiasis. Proc. Natl. Acad. Sci. USA. 2004;101,:586–590. doi: 10.1073/pnas.0305064101. 1:CAS:528:DC%2BD2cXmsFGmsg%3D%3D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 365.Mu D. The integrin alpha(v)beta8 mediates epithelial homeostasis through MT1-MMP-dependent activation of TGF-beta1. J. Cell Biol. 2002;157,:493–507. doi: 10.1083/jcb.200109100. 1:CAS:528:DC%2BD38XjtleltLk%3D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 366.Maeda S, Dean DD, Gomez R, Schwartz Z, Boyan BD. The first stage of transforming growth factor beta1 activation is release of the large latent complex from the extracellular matrix of growth plate chondrocytes by matrix vesicle stromelysin-1 (MMP-3) Calcif Tissue Int. 2002;70,:54–65. doi: 10.1007/s002230010032. 1:CAS:528:DC%2BD38XjvFSjurw%3D. [DOI] [PubMed] [Google Scholar]
- 367.Alfranca A. PGE2 induces angiogenesis via MT1-MMP-mediated activation of the TGFbeta/Alk5 signaling pathway. Blood. 2008;112,:1120–1128. doi: 10.1182/blood-2007-09-112268. 1:CAS:528:DC%2BD1cXpvVOksbY%3D. [DOI] [PubMed] [Google Scholar]
- 368.Kim HS, Luo L, Pflugfelder SC, Li DQ. Doxycycline inhibits TGF-beta1-induced MMP-9 via Smad and MAPK pathways in human corneal epithelial cells. Invest Ophthalmol Vis Sci. 2005;46,:840–848. doi: 10.1167/iovs.04-0929. [DOI] [PubMed] [Google Scholar]
- 369.Agren MS. Matrix metalloproteinases (MMPs) are required for re-epithelialization of cutaneous wounds. Arch. Dermatol. Res. 1999;291,:583–590. doi: 10.1007/s004030050459. [DOI] [PubMed] [Google Scholar]
- 370.Pourgholami MH, Morris DL. Inhibitors of vascular endothelial growth factor in cancer. Cardiovasc Hematol. Agents Med. Chem. 2008;6,:343–347. doi: 10.2174/187152508785909528. [DOI] [PubMed] [Google Scholar]
- 371.Yoshiji H, Kuriyama S, Fukui H. Angiotensin-I-converting enzyme inhibitors may be an alternative anti-angiogenic strategy in the treatment of liver fibrosis and hepatocellular carcinoma. Possible role of vascular endothelial growth factor. Tumour Biol. 2002;23,:348–356. doi: 10.1159/000069792. 1:CAS:528:DC%2BD3sXis1Cju74%3D. [DOI] [PubMed] [Google Scholar]
- 372.Bizzarri C, Beccari AR, Bertini R, Cavicchia MR, Giorgini S, Allegretti M. ELR+ CXC chemokines and their receptors (CXC chemokine receptor 1 and CXC chemokine receptor 2) as new therapeutic targets. Pharmacol Ther. 2006;112,:139–149. doi: 10.1016/j.pharmthera.2006.04.002. 1:CAS:528:DC%2BD28XptFCitro%3D. [DOI] [PubMed] [Google Scholar]
- 373.Jiang D. Regulation of pulmonary fibrosis by chemokine receptor CXCR3. J. Clin. Invest. 2004;114,:291–299. doi: 10.1172/JCI16861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 374.Pochetuhen K, Luzina IG, Lockatell V, Choi J, Todd NW, Atamas SP. Complex regulation of pulmonary inflammation and fibrosis by CCL18. Am. J. Pathol. 2007;171,:428–437. doi: 10.2353/ajpath.2007.061167. 1:CAS:528:DC%2BD2sXpsFaitbg%3D. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PowerPoint slide for Fig. 1
PowerPoint slide for Fig. 2