

Abstract
In order to provide new insight into the determining factors of membrane-bound peptide conformation which might play an important role in peptide-receptor docking and further biological behaviors, the dodecylphosphocholine (DPC) micelle-bound conformations of bifunctional peptide derivatives of δ-preferring opioid agonists and NK1 antagonists (1: Tyr-D-Ala-Gly-Phe-Met-Pro-Leu-Trp-O-3,5-Bzl(CF3)2; 2: Tyr-D-Ala-Gly-Phe-Met-Pro-Leu-Trp-NH-3,5-Bzl(CF3)2; 3: Tyr-D-Ala-Gly-Phe-Met-Pro-Leu-Trp-NH-Bzl) were determined based on 2D NMR studies. Although the differences in the primary sequence were limited to the C-terminus, the obtained NMR conformations were unexpectedly different for each compound. Moreover, their biological activities showed different trends in direct relation to the compound-specific conformations in DPC micelles. The important result is that not only were the NK1 antagonist activities different (the pharmacophore located at the C-terminus), but the opioid agonist activities (this pharmacophore was at the structurally preserved N-terminus) also were shifted, suggesting that a general conformational change in the bioactive state was induced due to relatively small and limited structural modifications.
Keywords: bifunctional peptides, analgesics, opioid induced tolerance, opioid receptor agonists, neurokinin-1 receptor antagonists, structure-activity relationships, NMR structure, DPC micelles, conformation, fluorine
Introduction
The importance of peptide and protein conformation has become increasingly appreciated recently, as their biological importance has been widely recognized. One notable example is protein misfolding which has been implicated in a large number of diseases such as Protein Folding Disorders (PFD).1–3 In the diseases known as amyloidoses,3 large quantities of misfolded proteins or peptides undergo aggregation resulting in destroying brain cells and other tissues. These diseases, such as Alzheimer’s disease and Parkinson’s disease, have high rates of pathogenesis and give rise to the huge social problems all over the world. The Alzheimer’s β-amyloid protein (AβP) is the well-known offending substance whose conformational change to the β-sheet oligomer and subsequent aggregation was shown to enhance its neurotoxicity.3–5 Another important misfolded protein is the cellular prion protein PrPC whose secondary structure was changed into a β-rich conformation (PrPSc) to cause the prion diseases.6 Thus, many scientific efforts have been made to seek the origin of such misfoldings.7–10 However, the full picture of their mechanisms is still largely unknown.
The conformation of small and linear peptides, such as the endogenous opioid peptide enkephalin11 and its analogues,12, 13 have been thought to have “random” conformations in aqueous solution due to their high flexibility.14 However, in the presence of trifluoroethanol (TFE) or membrane-mimicking surroundings, they could have structured conformations, since their secondary structural elements can be stabilized by the environmental effects.12, 13, 15, 16 Therefore, the circumstances surrounding such a peptide should be important especially for their conformational definitions.17
Many G-protein coupled receptors (GPCRs), which are the typical membrane-bound proteins, have their ligand binding sites in or near the lipophilic trans-membrane (TM) domains.18–21 Since the docking event of such a receptor and a ligand must take place near the membrane, ligand-membrane interactions should be important, and this research topic has been occasionally explored for decades.22, 23 In fact, it has been pointed out that the membrane promotes ligand-receptor docking,24–27 and that the ligand adoption into membrane followed by membrane-catalyzed 2D search should be more efficient than the three dimensional ligand-receptor binding through solvent space.12,13,23,24,28 Hence, understanding ligand-membrane interactions and membrane-bound structures of ligands is indispensable for further insight into their diverse biological behaviors.12, 13, 28
The opioid and NK1 receptors are both GPCRs which have significant importance in pain signal transmission, and their ligand binding sites are found in or near the TM domains.20, 21 The opioid receptors are classified into three subtypes (μ, δ and κ) and have endogenous peptides as their ligands such as endomorphin, enkephalin and dynorphin.20, 29, 30 It is well known that the 11-amino acid peptide substance P acts as an excitatory and pronociceptive neurotransmitter of pain signaling through the neurokinin-1 (NK1) receptor.31–37 Recently, combining the agonist effect at the opioid receptors together with the blocking of signals through NK1 receptors has been reported to have several merits for the analgesic effects, such as enhanced potency and the prevention of opioid-induced tolerance.31–37 Thus, we have been developing in one molecule, bifunctional peptide derivatives which act as opioid agonists and NK1 antagonists, with extensive discussions on their structure-activity relationships (SAR), which were based on their primary sequences.38–40 However, their three-dimensional conformations in membrane and compound-membrane interactions should also be important for further understanding the biological activities of ligands both in vitro and in vivo.
In the present article, we report our new findings, that small and local structural modifications induced large conformational changes in membrane-mimicking dodecylphosphocholine (DPC) micelles,41–46 together with a discussion of their relevance to the biological activities. The starting point of our bifunctional design was the successful C-terminal modifications on TY001 (Tyr1-D-Ala2-Gly3-Phe4-Pro5-Leu6-Trp7-O-3′,5′-Bzl(CF3)2) to yield several potent bifunctional derivatives with better affinities at the μ opioid receptors rather than at the δ receptor.40 Because the selective agonists at the δ opioid receptor have analgesic activity with fewer adverse effects, but less potency than μ-selective drugs,47–49 a δ-selective opioid agonist with enhanced analgesic activity which also has NK1 receptor antagonist activity, provides a novel way to find a potent drug candidate for prolonged pain control. Thus, 1 (TY005: Tyr1-D-Ala2-Gly3-Phe4-Met5-Pro6-Leu7-Trp8-O- 3′,5′-Bzl(CF3)2), which has a Met5 as the key amino acid residue for the δ-opioid selectivity and has been shown to have potent analgesic activities in vivo,38–40 was chosen as the current lead compound to find novel δ-preferring bifunctional ligands with strong analgesic potency. The C-terminus of 1 was modified to give two novel derivatives 2 (TY027: Tyr1-D-Ala2-Gly3-Phe4-Met5-Pro6-Leu7-Trp8-NH-3′,5′- Bzl(CF3)2) and 3 (TY025: Tyr1-D-Ala2-Gly3-Phe4-Met5-Pro6-Leu7-Trp8-NH-Bzl) (Figure 1).
Figure 1.

Sequence of the bifunctional ligands synthesized and examined.
Our main intention for these structural modifications was to seek induced conformational changes in the presence of lipid media, and to examine the influences of such changes on manipulating bioactivities. Therefore, the NMR conformations of 1-3 were determined in aqueous solution with membrane-mimicking perdeuterated dodecylphosphocholine (DPC) micelles. Fluorescence experiments as well as additional NMR studies using paramagnetic agents also were performed to determine the difference in membrane-compound interactions. Moreover, the biological activities of 2 and 3 were extensively evaluated using radioligand binding assays, GTPγS binding assays and tissue-based functional experiments using guinea pig ileum (GPI) and mouse isolated vas deferens (MVD) tissues.38, 40, 50, 51
Results and Discussion
Peptide Synthesis
The peptide derivatives 1-3 (Figure 1) were synthesized using previously published methods.40 Briefly, Boc-Tyr(tBu)-D-Ala-Gly-Phe-Met-Pro-Leu-Trp(Boc)-OH was synthesized using Nα-Fmoc chemistry with HBTU as the coupling reagent on a 2-chlorotrityl resin, followed by esterification or amidation of the protected C-terminal intermediate. The esterification was performed employing cesium carbonate to form the cesium salt of the protected peptide to react with 3′,5′-bis(trifluoromethyl)benzyl bromide, and standard EDC/Cl-HOBt coupling chemistry with two equivalents of reactant amine was used for the amidation. Final deprotection with the cleavage cocktail (82.5% v/v TFA, 5% water, 5% thioanisole, 2.5% 1,2-ethanedithiol and 5% phenol to quench the highly stabilized carbocations released from permanent protecting groups), gave the final crude peptides which were purified by RP-HPLC (>99 %) and characterized by analytical HPLC, 1H-NMR, HRMS and TLC. The assignments of NMR resonances are available in the Supporting Information.
Secondary structure analysis based on assigned 1H NMR
NMR experiments were performed to obtain structural information in DPC micelles as previously reported.45, 46 Two-dimensional NMR studies including TOCSY, DQF-COSY and NOESY were performed on all three bifunctional peptide derivatives 1-3 in pH 4.5 buffer (45 mM CD3CO2Na/HCl, 1 mM NaN3, 90% H2O/10% D2O) with 40-fold perdeuterated DPC micelles. At concentrations above the critical micelle point, DPC forms micelles with an aggregate number of 50 to 90, corresponding to one or two peptide molecules per micelle.52
The obtained NOESY data showed the reasonably good quality as seen in Figure 2. A total of 136, 155 and 184 non-redundant NOE restraints were used for 1, 2 and 3, respectively, based on the NOESY cross-peak volumes including sequential (50, 63 and 72, respectively), medium-range (2-4 residues; 31, 36, and 46, respectively) and long-range (1, 0 and 3, respectively) restraints. The distribution of these restraints along the peptide chain is shown in Figure 3D. Only one dihedral angle restraint was used: the Leu ϕ angle in 3. The total numbers of restraints were 136, 155 and 185, respectively (15.1, 17.2 and 20.4 per residue). The large numbers of NOEs per residue for small and linear peptides suggest that the peptide derivatives exist in well-defined conformations in the DPC micelles.
Figure 2.

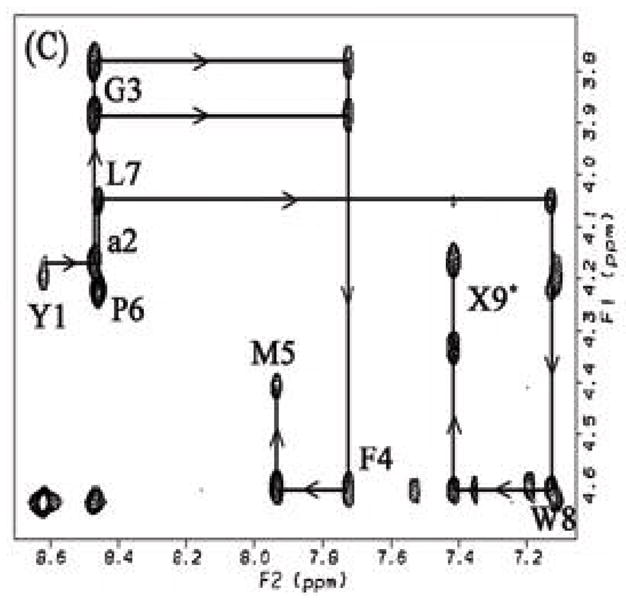
Fingerprint (HN-Hα) region of the NOESY spectrum of (A) 1, (B) 2 and (C) 3 in DPC micelles. Intraresidue HN-HαNOE cross-peaks are labeled with residue number, and arrows indicate the connectivity path from N-terminal to C-terminal. X9 represents the cross-peaks derived from the corresponding C-terminal HN and benzyl protons.
Figure 3.

Diagram of HN-Hα coupling constants, NOE connectivities, and Hα chemical shift index (CSI) for (A) 1, (B) 2 and (C) 3. The Hα CSI was calculated using the random-coil values reported by Andersen et al.86, 87 The residual interresidue NOE distance restraints of 1 (left), 2 (middle) and 3 (right) (D). Each column shows the sequential (i, i + 1; open), medium-range (i, i + 2–4; hatched) and long-range restraints (i, i + >4; filled), respectively. The residue Bzl or 9 stands for the respective C-terminal moieties.
The interresidual NOE connectivities and the 3JHN-Hα coupling constants of all of the peptide derivatives are illustrated in Figure 3. The C-terminal benzyl moiety of 2 and 3 are represented as residue 9. The 3JHN-Hα coupling constants for all residues in all three peptide derivatives were within the range of 6–8 Hz except for Leu7 in 3. This is most likely due to conformational averaging of the peptides in solution.53 The observed NOE patterns, including dNN(i, i + 1), dαN(i, i + 1) and some medium-range (i, i + 2 or 3) connectivities, suggest the possibility of β-turn structures around residues 1-4 in all three peptide derivatives as well as around residues 5-8 in 2.54 A few longer-range dαN(i, i + 3) and dαN(i, i + 4) connectivities found in 3 indicate the existence of a helical structure in this molecule, consistent with its Hα CSI pattern (Figure 3C).54
Structural calculations
The 20 structures with the lowest total energies after rMD refinement were used to represent the structure of the peptide derivatives in DPC micelles. Throughout the 1H-NMR studies, only one major rotamer was found for peptides 1-3 and the populations of minor rotamers were all negligible. The Met5-Pro6 bond of the major rotamers were fixed in the trans configuration based on the observations of 5Hα to Pro6 Hα sequential NOEs together with the absence of sequential 5Hα-6Hα NOEs in the structural calculations of 1-3. Based on the observations, cis-isomers of peptides 1-3 have negligible population in the lipid-mimicking DPC micelles, and thus considered to have negligible effects on the biological behaviors. Since the experimental NOEs and 3JHN-Hα were the averaged values in the NMR experiment time course, the obtained low-energy structures didn’t correspond to a single conformation under these experimental conditions. However, the ensemble of an appropriate number of obtained structures provides sufficient structural figures based on the “averaged” or “dynamic” view of the conformations. Thus, statistics for the 20 best structures were performed as shown in Table 1. The average restraint violation energies were low (2.48, 2.95 and 1.13 kcal mol−1 for 1, 2 and 3, respectively), with average maximum NOE distance violations of 0.17, 0.11 and 0.11 Å with no dihedral angle violations. The 19 structures were aligned with the most stable structure using all backbone atoms (Figure 4A), only the backbone atoms of residues 1-4 (Figure 4B) or 5-8 (Figure 4C). The backbone rmsd’s of the 19 structures with respect to the most stable structure were 1.80, 1.14 and 0.19 Å for 1, 2 and 3, respectively, for all residues. The rmsd values are significantly decreased if alignment is carried out only on the backbone atoms of residues 5-8 (1: 0.75; 2: 0.45; 3: 0.04), indicating that the C-terminal half is much better defined by the NMR restraints than the N-terminal half (residues 1-4). This may be due to greater flexibility in the N-terminal portion. The decrease in rmsd going from a flexible ester (1) to a more rigid amide (2) linkage at the C-terminus was expected, but the much larger decrease resulting from removal of two trifluoromethyl groups of 2 (3) was surprising.
Table 1.
Structural statistics
| Compound | 1 | 2 | 3 | |||
|---|---|---|---|---|---|---|
| final 20 structs | most stable structure | final 20 structs | most stable structure | final 20 structs | most stable structure | |
| rmsd from NOE dist restraints (Å)a | 0.025 ± 0.004 | 0.028 | 0.027 ± 0.004 | 0.016 | 0.016 ± 0.001 | 0.016 |
| rmsd from backbone ϕ angle restraints (deg)b | -c | -c | -c | -c | 0.0 ± 0.0 | 0.0 |
| NOE dist restraints violations | ||||||
| > 0.01 Å | 13.9 ± 2.5 | 11 | 14.6 ± 1.5 | 14 | 14.2 ± 1.1 | 15 |
| > 0.1 Å | 2.5 ± 1.4 | 3 | 3.9 ± 1.2 | 3 | 1.0 ± 0.0 | 1 |
| max dist violations (Å) | 0.17 ± 0.04 | 0.22 | 0.16 ± 0.02 | 0.13 | 0.11 ± 0.00 | 0.12 |
| dihedral backbone angle violations | ||||||
| > 0.1° | -c | -c | -c | -c | 0 ± 0 | 0 |
| > 1° | -c | -c | -c | -c | 0 ± 0 | 0 |
| max dihdral violations (deg) | -c | -c | -c | -c | 0 ± 0 | 0 |
| rms deviation from ideal geometryd | ||||||
| bond length (Å)e | 0.0061 ± 0.0004 | 0.0063 | 0.0052 ±0.0002 | 0.0052 | 0.0035 ± 0.00004 | 0.0035 |
| bond valence angles (deg)f | 2.14 ± 0.11 | 2.16 | 1.78 ± 0.05 | 1.72 | 1.25 ± 0.003 | 1.25 |
| out-of-plane angles (deg)g | 3.57 ± 0.63 | 3.25 | 2.73 ± 0.40 | 2.90 | 1.54 ± 0.09 | 1.45 |
| AMBER energies (kcal mol−1) | 9.08 | |||||
| restrainth | 2.48 ± 0.67 | 2.78 | 2.95 ± 0.58 | 2.41 | 1.13 ± 0.07 | 1.20 |
| bond stretching | 2.07 ± 0.22 | 2.20 | 1.42 ± 0.08 | 1.40 | 1.68 ± 0.02 | 1.70 |
| bond angles | 19.28 ± 1.84 | 19.49 | 12.8 ± 0.8 | 11.89 | 14.28 ± 0.22 | 14.00 |
| dihedral angles | 12.24 ± 1.9 | 9.54 | 9.57 ± 1.61 | 7.99 | 14.52 ± 0.38 | 14.53 |
| planarity | 1.63 ± 1.11 | 1.15 | 0.74 ± 0.33 | 0.61 | 0.19 ± 0.03 | 0.17 |
| van der Waalsi | −11.65 ± 3.1 | −12.61 | −12.23 ± 1.4 | −13.80 | −17.4 ± 0.75 | −18.41 |
| electrostaticj | −9.6 ± 0.91 | −39.93 | −11.5 ± 0.68 | −11.84 | −9.59 ± 0.41 | −9.82 |
| Total | 13.25 ± 2.12 | 9.08 | −0.01 ± 2.25 | −4.44 | −.14 ± 0.78 | 1.61 |
| atomic rmsd (Å): final 19 structures v.s. most stable structure | ||||||
|
|
||||||
| 1 | 2 | 3 | ||||
|
| ||||||
| backbone atoms (N, Cα, C′) | all non-hydrogen atoms | backbone atoms (N, Cα, C′) | all non-hydrogen atoms | backbone atoms (N, Cα, C′) | all non-hydrogen atoms | |
|
| ||||||
| Calculated on whole molecule | 1.80 ± 0.47 | 2.72 ± 0.92 | 1.14 ± 0.43 | 2.09 ± 0.64 | 0.19 ± 0.20 | 0.84 ± 0.28 |
| Calculated only on 1–4 res. | 1.11 ± 0.54 | 2.49 ± 1.12 | 1.05 ± 0.63 | 2.16 ± 0.98 | 0.14 ± 0.30 | 0.32 ± 0.66 |
| Calculated only on 5–8 res. and C-terminus | 0.75 ± 0.26 | 1.82 ± 0.90 | 0.45 ± 0.38 | 1.02 ± 0.25 | 0.04 ± 0.01 | 0.76 ± 0.42 |
The total number of NOE restraints were 136 for 1, 155 for 2 and 184 for 3, respectively.
Two backbone ϕ angle restraints were applied only on 3.
no restraints used.
Derived from the rMD calculations using the AMBER force field in DISCOVER.
The number of bond length were 160 for 1, 161 for 2 and 155 for 3, respectively.
The number of bond valence angles were 285 for 1, 287 for 2 and 275 for 3, respectively.
The number of out-of-plane angles were 36 for 1, 36 for 2 and 37 for 3, respectively.
Calculated with force constants of 25 kcal mol−1 Å−2 and 100 kcal mol−1 rad−2 for the NOE distance and dihedral angle restraints, respectively.
Calculated with the Lennard-Jones potential using the AMBER force field and a 12 Å cutoff.
Calculated with a distance-dependent dielectric constant (ε= 4r).
Figure 4.

Ensembles of the best 20 calculated structures in 40-fold DPC micelle/pH 4.5 buffer for (A) 1, (B) 2 and (C) 3 with the lowest restraint energy, aligned on backbone atoms of residues (a) 1-8, (b) 1-4 and (c) 5-8, from N-terminal (up in the left image) to C-terminal (down). Only backbone atoms were illustrated in (a) and (b) for easier comparison, and the most stable conformers (c) are shown with all non-hydrogen atoms.
In Met-Enkephalin (Tyr1-Gly2-Gly3-Phe4-Met5-OH) and Leu-Enkephalin (Tyr1-Gly2-Gly3-Phe4-Leu5-OH), which form the basis for the design of the N-terminal portion of peptide sequence of 1-3, a β-turn structure was often found between Tyr1 and Phe4 by several methods including X-ray crystallography and NMR spectroscopy in environments which mimic the membrane bilayer.55, 56 In the case of peptide derivatives 1 and 2, a distance of less than 7 Å between the Cα of D-Ala2 and the Cα of Met5 was observed in 15 of the best 20 structures for 1, and in all 20 structures for 2, whereas the β-turn between Tyr1 and Phe4 were found only 5 and 3 structures in the best 20 structures of 1 and 2, respectively (Table 2).57 This result implied the existence of an alternative β-turn in the residues 2 to 5 for both 1 and 2, and all of them were classified as Type IV (“distorted”) β-turn by their backbone dihedral angles. A second β-turn structure was found from Pro6 to the C-terminal benzyl moiety (residue 9) in which the distance between the Cα (Pro6) and the benzylic carbon (CH2) of the C-terminus was less than 7 Å in 17 and 19 of the best 20 structures for 1 and 2, respectively (Table 2). For peptide derivative 2, 6 of 19 turns found in 2 were classified as Type I β-turns, and a hydrogen bond between the HN of residue 9 and the carbonyl oxygen atom in Pro6 was observed in 9 of the 19 (Table 3), The C-terminal ester (1) showed only distorted β-turns in this region, with no hydrogen bonds, consistent with the larger backbone rmsd observed for C-terminal half (0.75 Å vs 0.45 Å for 2). These implied that the secondary structure elements were consistent with the observed NOE connectivities (Figure 3).
Table 2.
Number of structures with less than 7 Å distance between alpha carbons of i th and (i + 3) th residues.a
| Residues | Tyr1-Phe4 | DAla2-Met5 | Gly3-Pro6 | Met5-Trp8 | Pro6-Bzl9 |
|---|---|---|---|---|---|
| 1 | 5 | 15 | 0 | 0 | 17 |
| 2 | 3 | 20 | 0 | 0 | 19 |
| 3b | 0 | 2 | 20 | 20 | 18 |
Out of the best 20 calculated structures.
Helical structure was found, in which no β-turn structures should not be defined according to the original definition.57 Bzl stands for the cross-peaks derived from the corresponding aromatic protons of benzyl moiety (residue 9).
Table 3.
Observed hydrogen bondsa
| Molecule | No.b | Donor | Acceptor | Distance (Å)c | Angle (deg)d |
|---|---|---|---|---|---|
| 1 | 14 | Leu7 HN | Met5 O | 1.91 ± 0.07 | 141.1 ± 6.0 |
|
| |||||
| 9 | Bzl9 HNe | Pro6 O | 2.16 ± 0.11 | 158.5 ± 1.9 | |
| 2 | 7 | Gly3 HN | Tyr1 O | 2.05 ± 0.11 | 137.8 ± 8.1 |
| 5 | Trp8 HN | Met5 O | 2.04 ± 0.02 | 132.3 ± 1.1 | |
|
| |||||
| 3 | No hydrogen bond observed | ||||
The hydrogen bonds which were observed in more than five structures were listed.
The number of structures of the final 20 for which the listed hydrogen bond is observed.
The distance is the mean proton-oxygen distance (± SD) in the structures for which a hydrogen bond is observed.
The angle is the mean N-H- - -O angle (± SD) in the structures for which a hydrogen bond is observed.
Amide proton of C-terminal benzyl moiety.
Comparing the tandem β-turn structures of 1 and 2, the peptide with no trifluoromethyl groups in the C-terminus (3) showed significantly different structural properties. First, the backbone of 3 had a well-defined helical structure, consistent with the NOE connectivities and CSI values (Figure 4). It is noteworthy that not only the C-terminal half of 3, but also its N-terminus was found to be quite structured (Table 1). Thus, removal of two trifluoromethyl groups from the C-terminus led to a drastic conformational change for the whole molecules from tandem β-turn structures to a helical structure (Figure 4).
This well-defined structure of 3 was also confirmed by the Ramachandran plot and angular order parameters (Figure 5).58 In the Ramachandran plot of 3, only seven clear spots, corresponding to the respective residues 2-8, were found. Among them, only Gly3 has positive ϕ angles in all of its 20 best structures. On the other hand, the corresponding Ramachandran plots for 1 and 2 showed more scattered views together with positive ϕ angles for Gly3 (10 structures in 1 and 3 structures in 2), Phe4 (3 structures in 1), Met5 (13 structures in 1 and all 20 structures in 2) and Leu7 (1 structures in 1 and 6 structures in 2) in the seven L-amino acids and some of D-Ala2 (3 structures in 1 and 3 structures in 2) have negative ϕ angles. It is interesting that Met5 of 1 and 2, located between two β-turns, had frequent positive ϕ angles. As for the angular order parameters, both the parameters for ϕ and ψ angles in 3 were close to 1 in all the residues, whereas 1 and 2 had smaller values in some residues, implying a better-defined structure for 3 than for 1 and 2, especially in the N-terminus where no structural modification was made.
Figure 5.

The D-Ala2 (crosses), Gly3 (open circle) and Met5 with positive ϕ angles (circled) were indicated in the Ramachandran ϕ, ψ plots for (A) 1, (B) 2 and (C) 3 for residues 2-7 of 20 final structures. Angular order parameters for ϕ (D) and ψ (E) angles calculated from the 20 final structures for 1 (open circles), 2 (filled squares) and 3 (crosses). For calculating the ψ angles of Trp8, Non-carbonyl oxygen atoms of the C-terminal ester (1) and nitrogen atoms of C-terminal amide (2 and 3) were used instead of N (i + 3), respectively.
Based on the NMR structural analysis, it is clear that the limited modifications at the C-terminal moiety gave rise to several changes in the conformations of the peptide derivatives 1-3 in the presence of membrane-like DPC micelles. The change of ester (1) into amide (2) resulted in a better-defined conformation especially in the C-terminal portion of the peptide, and a more rigid β-turn structural element with intra-molecule hydrogen-bond was found for residues 6-9 of 2. The removal of the trifluoromethyl groups from the C-terminus of (3) induced much larger changes in the three-dimensional structures which changed from a tandem β-turn (1 and 2) to a structured helical conformation, with the smallest rmsd values for alignment at residues 5-8 (Table 1), implying a large influence of the trifluoromethyl groups on the conformations of the entire molecules in the lipid environment. Since the structural modifications were limited only to the C-terminus, it is not clear how the simple electrostatic or steric changes of these modifications affects the conformations of the entire molecule. Because the molar ratios of 1-3 were low compared to DPC, they presumably form complexes with lipid molecules and thus induce perturbations in the interaction between the compound and micelles due to their unique C-terminus. It might be suggested that the highly lipophilic and electronegative trifluoromethyl groups induced such a perturbation, if these trifluoromethyl groups interact with the core of micelles which mostly consists of lipophilic and electrostatically neutral hydrocarbon chains. The ester-to-amide substitution, in which the oxygen atom (hydrogen-bond acceptor) was replaced to the nitrogen with a proton to be a hydrogen-bond donor, also can introduce some changes in the interactive mode between these compounds and membrane-like micelles. Therefore, the interaction of compound with DPC micelles might be compound-specific which is consistent with the different three-dimensional conformations for each compound. In order to confirm these implications, further NMR experiments using paramagnetic agents as well as fluorescence experiments were performed to estimate such differences in the membrane-compound interactions.
Fluorescence study
It is well known that the intrinsic fluorescence spectrum of the indole ring in tryptophan shifts to shorter wavelength (“blue shifted”) as the polarity of the solvent surrounding the tryptophan residue decreases, and this blue shift is a good index to monitor the lipophilicity of the environment close to the tryptophan.59, 60 The fluorescence of Trp8 in 1-3 was measured in the presence and absence of DPC micelles, in order to examine the interaction between the peptides and membrane-like micelles. The fluorescence spectra in DPC micelles were compared to the spectra in the EtOH-buffer solution (EtOH : pH 7.4 HEPES buffer = 1 : 1), which was chosen as the standard since the solubilities of the peptide derivatives 1-3 in aqueous media were too low to run the experiment (Table 4).61 The emission spectra were obtained by excitation at 290 nm to avoid excitement of the tyrosine residue.
Table 4.
. Solubility and lipophilicity of peptide derivatives
Obvious blue shifts of fluorescence spectra from the standard solution were observed in all three peptide derivatives (6 nm, 1; 10 nm, 2; 10 nm, 3, respectively, Figure 6), implying that the indole ring in Trp8 at the C-terminal of all peptides was buried inside the micelles, and that the peptides have strong interactions with the lipophilic core of the micelles, at least the C-terminus.
Figure 6.

Fluorescence spectra of (A) 1 (B) 2 (C) 3: the spectra were recorded at 500 μg/mL at 290 nm excitation in 40-fold DPC micelle/pH 7.4 HEPES buffer (solid line) or EtOH : pH 7.4 HEPES buffer = 1 : 1 solution (broken line).
Because poorly water-soluble peptides 1-3 were easily dissolved at millimolar concentration in the presence of micelles, strong interactions between the peptide derivatives 1-3 and micelles are also suggested (Table 4). Among compounds 1-3, 1 showed a smaller blue-shift than 2 and 3, suggesting that the surroundings near the indole ring of Trp8 in C-terminal ester (1) were less lipophilic than was the case for the C-terminal amides (2 and 3), both of which had better-defined structures in DPC micelles than 1 (Table 1). Therfore, the lipophilicity near Trp8 was mostly influenced by ester-amide substitution rather than the removal of trifluoromethyl groups, and the lipophilic environment at the C-terminus might have some responsibility for the structured conformations of the peptide derivatives.
Paramagnetic Broadening Studies on 1H NMR
To obtain further information about the interaction between peptide derivatives 1-3 and DPC micelles, we used a nitroxyl spin-label, 5-doxylstearic acid, and Mn2+ ions (MnCl2) to induce selective broadening of NMR resonances close to the paramagnetic probes.12, 62 The cross-peaks of protons exposed to an aqueous exterior are broadened or disappear due to the paramagnetic effect of Mn2+, while cross-peaks of protons located inside the micelles and close to the phosphate groups of DPC are broadened by the free radical on the doxyl group, which is bound to carbon 5 of the stearic acid. The paramagnetic effects of these agents on the peptide resonances were studied by comparing TOCSY spectra in the presence and absence of the paramagnetic agents, and all peaks were classified into three categories according to their sensitivities to the paramagnetic agents: missed by 5-DOXYL only (in the micelle, but not deeply buried); missed by both Mn2+ and 5-DOXYL (the proton is at or near the surface of micelles ) and preserved by either agent (deeply buried in the micelle) (Figure 7 and Supporting Information). In fact, none of the 1H resonances were missed by Mn2+ only, indicating a strong association with the micelle for all three derivatives.
Figure 7.

Typical example of the paramagnetic effects on TOCSY Spectra. The aromatic region of 3 with DPC micelles (left column), with 200 μM Mn2+ (middle) and 5-DOXYL stearic acid (right). Preserved resonances (labeled) are in a phase not missed by the phase-specific radical probe (Mn2+ or DOXYL). Spectra were compared from the same noise level. The full spectra for 1-3 are available in the Supporting Information.
For all three peptide derivatives 1-3, nearly all of the HN related cross-peaks were categorized as sensitive to both Mn2+ and 5-DOXYL, implying that the peptide backbones are located at or near the surface of micelles. On the other hand, most of the side-chain resonances were missed only by 5-DOXYL or were non-sensitive to either agent. Thus, generally, the backbones of 1-3 lie close to the surface with their side-chains buried in the micelles. However, there is one notable exception to this general observation: the HN resonances of Met5 in 1-3 were affected only by 5-DOXYL, implying that the protons are not exposed to the surface of the micelles. Moreover, the side-chain cross-peaks of Met5 in only 1 were missed by either paramagnetic agent, indicating that Met5 side-chain of 1 was exposed to the surface of the micelle, whereas those of 2 and 3 were sensitive only to 5-DOXYL, indicating a different orientation in the side-chain of Met5.
The sensitivity of side-chain protons to broadening by 5-DOXYL provides insight into their portioning in depth in the micelles. The cross-peaks of two different aromatic protons (para and ortho) in the C-terminal benzyl moiety were eliminated in 1 and 2 by 5-DOXYL, but the resonance of 3 were preserved in spite of the hydrophobic trifluoromethyl group in 1 and 2. On the other hand, the cross-peaks of the aromatic protons of Trp8 were unaffected in 1, but were sensitive to 5-DOXYL for 2 and 3. Therefore, the C-terminal benzyl moiety (1 and 2) and Trp8 (2 and 3) appear to be located close to the phosphate moiety of DPC micelles, but the Trp8 of 1 and the C-terminus of 3 were rather deeply buried into the micelles.
The important implication from these paramagnetic experiments was that all the compounds (1-3) showed different orientations at each C-terminus in DPC micelles. Thus, both of the modifications, the ester-to-amide substitution and the removal of trifluoromethyl groups, was shown to change the interaction between compound and membrane-like micelles, as suggested from the NMR conformations of 1-3.
Biological activities
Finally, in order to evaluate the biological influences of conformations in the presence of membrane-mimicking micelles and compound-micelles interactions, the bioactivities of peptide derivatives 2 and 3 were evaluated using well-established assay systems (Tables 5–7; the details of assay systems are found in the Experimental Section and references herein).38, 40, 50, 51
Table 5.
Binding affinities of bifunctional peptides at δ/μ opioid receptors and NK1 receptors
| no | hDORa, [3H]DPDPEb | rMORa, [3H]DAMGO c | Ki(μ)/Ki(δ) | hNK1d, [3H]Substance Pe | rNK1d, [3H]Substance Pf | Ki(hNK1)/Ki(rNK1) | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| LogIC50g | Ki (nM) | LogIC50g | Ki (nM) | LogIC50g | Ki (nM) | LogIC50g | Ki (nM) | |||
| 1h | −8.2 ± 0.06 | 2.8 | −7.1 ± 0.11 | 36 | 13 | −9.9 ± 0.25 | 0.084 | −9.0 ± 0.10 | 0.29 | 3.4 |
| 2 | −8.8 ± 0.07 | 0.66 | −7.4 ± 0.05 | 16 | 24 | −10.9 ± 0.10 | 0.0065 | −7.6 ± 0.05 | 7.3 | 1100 |
| 3 | −9.1 ± 0.09 | 0.44 | −8.4 ± 0.03 | 1.8 | 4.1 | −8.4 ± 0.42 | 3.20 | −5.6 ± 0.06 | 700 | 220 |
| Biphalini | 2.6 | 1.4 | 0.54 | |||||||
| 4 | −8.8 ± 0.02 | 0.73 | −6.4 ± 0.12 | 130 | 180 | |||||
Competition analyses were carried out using membrane preparations from transfected HN9.10 cells that constitutively expressed the δ and μ opioid receptors, respectively.
Kd = 0.45 ± 0.1 nM.
Kd = 0.50 ± 0.1 nM.
Competition analyses were carried out using membrane preparations from transfected CHO cells that constitutively expressed rat or human NK1 receptors.
Kd = 0.40 ± 0.17 nM
Kd = 0.16 ± 0.03 nM
The logIC50 ± standard errors are expressed as logarithmic values determined from the non linear regression analysis of data collected from 2 independent experiments performed in duplicate (4 independent experimental values per drug concentration). The Ki values are calculated using the Cheng and Prusoff equation to correct for the concentration of the radioligand used in the assay.
See a reference.38
See a reference.85
Table 7.
Functional assay result for bifunctional peptide ligands at opioid and Substance P receptors
| No | Opioid agonist | Substance P GPI | |
|---|---|---|---|
| MVD (δ), IC50 (nM)a | GPI (μ), IC50 (nM)a | antagonist, Ke (nM)b | |
| 1c | 22 ± 1.2 | 360 ± 130 | 25 ± 8.8 |
| 2 | 15 ± 2.0 | 490 ± 29 | 10 ± 2.1 |
| 3 | 4.8 ± 0.35 | 61 ± 9.6 | 9.9 ± 2.8 |
| Biphalin | 2.7 ± 1.5 | 8.8 ± 0.3 | |
| 4 | 250 ± 87 | ||
Concentration at 50% inhibition of muscle concentration at electrically stimulated isolated tissues.
Inhibitory activity against the Substance P induced muscle contraction in the presence of 1 μM naloxone, Ke: concentration of antagonist needed to inhibit Substance P to half its activity.
See a reference.38
Compared to the subnanomolar-level affinities of the C-terminal ester derivative 1 (Ki = 0.29 nM), the C-terminal amide derivative 2 was 25 times less potent, but still showed binding affinities in the nanomolecular range for the rNK1 receptor (Ki = 7.3 nM) (Table 5). The binding affinity at the rNK1 of 3, which had no trifluoromethyl group in the C-terminal benzyl moiety, was decreased to a Ki = 700 nM. However, in the functional assay using the GPI to examine their antagonist activities, the Ke value for 3 (10 nM) was similar to that for 2 (9.9 nM) and better than that for 1 (25 nM) (Table 7). Several factors, such as pharmacokinetic differences in the different assay systems might be responsible for this inconsistency, but one good explanation should be provided by the known species difference between rat and guinea pig NK1 receptors.63 It is well known that the human NK1 receptor has higher homology to the guinea pig NK1 receptor than to the rat or mouse NK1, and some NK1 antagonists have a large species difference.63 In fact, 3 showed a 220 times better Ki value at the hNK1 receptor (3.2 nM) compared to the value at the rNK1 receptor. A smaller difference was found in 1 whose Ki value for the hNK1 was 0.084 nM (3.4-fold species difference). Surprisingly, 2, which is the C-terminal benzyl amide with two trifluoromethyl groups, showed the largest difference between the affinities at the rNK1 and at the hNK1 (1100-fold), and the Ki value for the hNK1 receptor was 6.5 pM suggesting that this analogue would be very potent in humans.
Therefore, substitution of the C-terminal ester for an amide gave rise to increased species difference at the human and rat NK1 receptors. This substitution changed hydrogen-bonding interactions and planarity of the C-terminus which might induce a conformational change in the lipidic surroundings as found in the NMR conformations (Figure 4). The summation of these changes could explain the activity shifts at the NK1 receptors. The existence of two trifluoromethyl groups at the C-terminus plays an important role in the affinities for both rNK1 and hNK1 receptors. Since the 3′,5′-dimethyl-substituted analogue of Ac-Trp- O-3′,5′-Bzl(CF3)2 (4, L-732,138)21 was reported to have 16-fold reduced affinity for the human NK1 receptor as compared to 4,21 one good explanation of the influence by trifluoromethyl introduction was that a more electronegative phenyl ring at the C-terminus might be preferred by the NK1 receptors.64 Another possibility is that the induced conformational changes to a well-defined helical structure due to the trifluoromethyl groups interfered with the binding of the compound at the NK1 receptor.
For the δ and μ opioid agonist activities, 2 showed 24-fold δ selectivity, as expected from the existence of Met5,38 with four-fold higher affinity at the hDOR (Ki = 0.66 nM) and two times higher affinity at the rMOR (Ki = 16 nM) than 1. It is interesting that a small modification at the C-terminus, which is far from the opioid agonist pharmacophore, can influence the opioid activities so much (Table 5). The elimination of two trifluoromethyl groups led to further increased affinity at both the DOR (Ki = 0.44 nM) and the rMOR (Ki = 1.8 nM) with 4.1-fold δ-selectivity. The binding affinity results correlated well with the in vitro GTPγS binding assays, and the functional assays using GPI and MVD tissues (Table 6 and 7).
Table 6.
Opioid agonist functional activities in [35S]GTPγS binding assays
| No | hDORa | rMORa | ||||
|---|---|---|---|---|---|---|
| LogEC50b | EC50 (nM) | Emax (%)c | LogEC50b | EC50 (nM) | Emax (%)c | |
| 1d | −8.5 ± 0.21 | 2.9 | 48 | −7.5 ± 0.09 | 32 | 46 |
| 2 | −8.1 ± 0.11 | 8.6 | 58 | −8.2 ± 0.17 | 7.0 | 55 |
| 3 | −8.6 ± 0.13 | 2.6 | 52 | −7.7 ± 0.18 | 21 | 47 |
| Biphalin | −9.0 ± 0.17 | 1.1 | 83 | |||
| DPDPE | −8.8 ± 0.25 | 1.6 | 69 | |||
| DAMGO | −7.4 ± 0.19 | 37 | 150 | |||
Expressed from HN9.10 cell.
The log EC50 ± standard error are logarithmic values determined from the non-linear regression analysis of data collected from 2 independent experiments performed in duplicate (4 independent experimental values per drug concentration)
Net total bound/basal binding ×100.
See a reference.38
Here, 2 showed potent activity in the MVD assay with the best δ selectivity (IC50 = 15 nM in MVD and 490 nM in GPI). The IC50 value of 3 in the GPI assay (IC50 = 61 nM) was a large increase from those of 1 and 2, with the best IC50 value in the MVD assay (4.8 nM). Therefore, 3 was found to be a bifunctional peptide derivative possessing potent agonist activities for both δ and μ opioid receptors together with a nanomolar level hNK1 antagonist activity. On the other hand, 2 was characterized as a very potent hNK1 antagonist with potent and δ selective opioid agonist activities, which also has nanomolar level affinity at the rNK1 receptor. Thus, 2 can be considered as a promising candidate for a potent analgesic drug, and it is currently being tested in several animal models using rats and will be published as a separate biological paper. Though both 2 and 3 have different biological profiles, they are expected to be potent analgesics for pain control in humans.
It should be noted that 2, which had the better-defined conformation in DPC micelles than 1, showed increased opioid activities, although the primary sequences for opioid agonist pharmacophore were exactly the same for both of 1 and 2 (Figure 1). Moreover, 3, which also had the same sequence for opioid pharmacophore at the N-terminus, showed much improved opioid activities versus 1 and 2. As discussed above, 3 had a structured helical conformation in DPC micelles, whereas 1 and 2 showed β-rich conformations. Moreover, the interactions between the compounds and the membranes were different for compounds 1-3.
Consequently, these results show that even small and limited structural modification in the ligand could induce relatively large conformational changes from a β-structure to a helical conformation in membrane-mimicking DPC micelles, together with significantly shifted biological activities. In addition, the reported data suggests that the removal of trifluoromethyl groups from the C-terminus induced much larger changes in the three-dimensional conformations than the substitution of the ester to amide, implying a large influence of lipophilic and electronegative trifluoromethyl groups on the conformations of whole molecules in a lipid media. The importance of this article is that such a small and local structural modification in the C-terminus might affect on the bioactive states not only at the C-terminus (NK1 pharmacophore), but also at the N-terminal (opioid pharmacophore which was preserved for all three derivatives), and the observed differences of the conformations in the membrane-like micelles could indicate their relevance to the changes in their bioactive form. These findings provide significant information regarding the nature of peptide drug-membrane interactions as well as the specific driving force to use conformations to manipulate bioactivities, both of which can be a guide to new aspects of modern medicinal chemistry to understand the folding of bioactive peptides or proteins.
Experimental Section
Materials
All amino acid derivatives and coupling reagents were purchased from EMD Biosciences (Madison, WI), Bachem (Torrance, CA), SynPep (Dublin, CA) and Chem Impex International (Wood Dale, IL). 2-Chlorotrityl resin was acquired from Iris Biotech GmbH (Marktredwitz, Germany). Perdeuterated DPC was purchased from C/D/N Isotopes (Quebec, Canada). ACS grade organic solvents were purchased from VWR Scientific (West Chester, PA), and other reagents were obtained from Sigma-Aldrich (St. Louis, MO) and used as obtained. The polypropylene reaction vessels (syringes with frits) were purchased from Torviq (Niles, MI). Myo-[2-3H(N)]-inositol; [Tyrosyl-3,5-3H(N)] D-Ala2-MePhe4-Glyol5-enkephalin (DAMGO); [Tyrosyl-2,6-3H(N)]-[2-D-Penicillamine, 5-D-Penicillamine] enkephalin (DPDPE); [3H]-substance P; and [35S]-guanosine 5′-(γ-thio) triphosphate were purchased from Perkin Elmer (Wellesley, MA). Bovine serum albumin (BSA), protease inhibitors, Tris and other buffer reagents were obtained from Sigma (St. Louis, MO). Culture medium (MEM, DMEM and IMDM), Penicillin/Streptomycin and fetal calf serum (FCS) were purchased from Invitrogen (Carlsbad, CA).
Boc-Tyr(tBu)-D-Ala-Gly-Phe-Met-Pro-Leu-Trp(Boc)-OH
The peptide was synthesized manually by the Nα-Fmoc solid-phase methodology using HBTU as the coupling reagents as previously reported.40 2-Chlorotrityl resin (2.0 g, 1.56 mmol/g) was placed into a 50 mL polypropylene syringe with the frit on the bottom and swollen in DMF (20 mL) for 1 h. The resin was washed with DMF (3 × 15 mL) and then with DCM (3 × 15 mL). Fmoc-Trp(Boc)-OH (1.2 equiv) was dissolved in 30 mL of DCM, and then DIEA (5 equiv) was added. The reaction mixture was transferred into the syringe with the resin then shaken for 2 h. The resin was washed three times with DMF (15 mL) and three times with DCM (15 mL), and then with DMF (3 × 15 mL). The Fmoc protecting group was removed by 20% piperidine in DMF (1 × 2 min and 1 × 20 min). The deprotected resin was washed with DMF (3 × 15 mL), DCM (3 × 15 mL) and then with DMF (3 × 15 mL). Fmoc-Leu-OH (3 equiv) and HBTU (2.9 equiv) were dissolved in 30 mL of DMF, then DIEA (6 equiv) was added. The coupling mixture was transferred into the syringe with the resin, then shaken for 2 h. All other amino acids, Pro, Met, Phe, Gly, D-Ala and Tyr were consecutively coupled using the procedures described above, using the TNBS test (all the amino acids except for Met) or chloranil test (only for Met) to check the extent of coupling. In case of a positive test result, the coupling was repeated until a negative test result was obtained. The resulting batch of the resin-bound protected Boc-Tyr(tBu)-D-Ala-Gly-Phe-Met-Pro-Leu-Trp(Boc) was carefully washed with DMF (3 × 15 mL), DCM (3 × 15 mL), DMF (3 × 15 mL), and DCM (3 × 15 mL), and dried under reduced pressure. The peptide was cleaved off the solid support with 1% v/v TFA in DCM (30 mL) for 30 min, and most of the organic solvent was removed under reduced pressure. The obtained crude peptides were precipitated out by the addition of chilled petroleum ether (45 mL) to give a white precipitate. The suspensions were centrifuged for 20 min at 7000 rpm, then the liquid was decanted off. The crude peptides were washed with petroleum ether (2 × 50 mL), and after the final centrifugation, the peptides were dried under vacuum (2 h) to obtain the title compound (2.89 g, 74.8%). The purity of the final products (93.6%) was checked by analytical RP-HPLC using a Hewlett Packard 1100 system (230 nm) on a reverse phase column (Waters NOVA-Pak C-18 column, 3.9 × 150 mm, 5 μm, 60Å). The peptide was eluted with a linear gradient of aqueous CH3CN/0.1% CF3CO2H (10–90% in 40 minutes) at a flow rate of 1.0 mL/min. The crude peptide was used for next reactions without further purification. 1H- NMR (DMSO-d6): 0.79(3H, d, J=6.6Hz), 0.84(3H, d, J=6.6Hz), 1.10(3H, d, J=6.6Hz), 1.24(9H, s), 1.26(9H, s), 1.38–1.41(2H, m), 1.61(10H, s), 1.70–1.77(3H, m), 1.80–1.94(3H, m), 2.01(3H, s), 2.42–2.47(2H, m), 2.67(1H, d-d, J=3.6, 13.2Hz), 2.75(1H, d-d, J=9.6, 13.8Hz), 2.87(1H, d-d, J=4.2, 13.2Hz), 2.94(1H, d-d, 2.4, 13.2Hz), 3.04(1H, d-d, 8.4, 15.0Hz), 3.14(1H, d-d, 4.8, 15.0Hz), 3.48–3.53(1H, m), 3.54–3.63(2H, m), 3.68(1H, d-d, J=5.4, 16.8Hz), 4.15(1H, d-d, J=6.0, 12.0Hz), 4.20–4.30(2H, m), 4.32(1H, d-d, 3.6, 7.8Hz), 4.48–4.55(2H, m), 4.58(1H, J=7.2, 14.4Hz), 6.83(2H, d, J=7.8Hz), 6.92(1H, d, J=8.4Hz), 7.12(2H, d, J=7.8Hz), 7.14–7.24(7H, m), 7.30(1H, t, J=7.8Hz), 7.47(1H, s), 7.87–7.92(2H, m), 7.98–8.08(4H, m), 8.30(1H, d, J=7.2Hz). MS (ESI): 1262 (M+Na)+.
H-Tyr-D-Ala-Gly-Phe-Met-Pro-Leu-Trp-O-3,5-Bzl(CF3)2·TFA(1)
Boc-Tyr(tBu)-D-Ala-Gly-Phe-Met-Pro-Leu-Trp(Boc)-OH (2.0 g, 1.61 mmol) and 3,5-bis(trifluoromethyl)benzyl bromide (1.24 g, 4.02 mmol) were dissolved in DMF (8 mL). Cesium carbonate (1.05 g, 3.22mmol) was added to the solution at r.t. After stirring for 2 h, saturated aqueous sodium bicarbonate (200 mL) was added to the solution, and extracted with ethyl acetate (200 mL) three times. The combined organic phases were washed with 5% aqueous citrate and saturated aqueous sodium chloride (200 mL each), and dried over sodium sulfate. The solvent was evaporated off and the crude peptide was precipitated in cold petroleum ether (45 mL) and centrifuged two times, and dried under reduced pressure. The obtained protected peptide was treated with 82.5% v/v TFA, 5% water, 5% thioanisole, 2.5% 1,2-ethanedithiol, and 5% phenol (10 mL, 1 h). The crude peptide was precipitated out by the addition of chilled diethyl ether (45 mL) to give white precipitates. The suspension was centrifuged for 20 min at 7000 rpm, then the liquid was decanted. The crude peptides were washed with diethyl ether (2 × 45 mL), and after the final centrifugation, the peptides were dried under vacuum (2 h). The resulting white residues (2.48 g, quantitative) were dissolved in 3: 1 mixture of acetonitrile and distilled water (5 mL), and the insoluble impurities were removed by passing the solutions through syringe filters (Gelman Laboratory, Ann Arbor, MI, Acrodisc 13 mm syringe filter with 0.45 μM PTFE membrane). Final purification was accomplished by preparative RP-HPLC, then lyophilized.
H-Tyr-D-Ala-Gly-Phe-Met-Pro-Leu-Trp-NH-3,5-Bzl(CF3)2·TFA(2)
Boc-Tyr(tBu)-D-Ala- Gly-Phe-Met-Pro-Leu-Trp(Boc)-OH (3.0 g, 2.42 mmol) and Cl-HOBt (428 mg, 2.66 mmol) were dissolved in DMF (10 mL). 3,5-bistrifluoromethylbenzyl amine (1.17 g, 4.84 mmol) and EDC (508 mg, 2.66 mmol) were added to the solution at r.t and stirred until the starting material wasn’t detected in TLC; then saturated aqueous sodium bicarbonate (250 mL) was added. The reaction mixture was extracted with ethyl acetate (250 mL) three times. The combined organic phases were washed with 5% aqueous citrate and saturated aqueous sodium chloride (250 mL each), then dried over sodium sulfate. The solvent was evaporated and the crude peptide was precipitated in cold petroleum ether (45 mL). The product was twice dispersed in cold petroleum ether, centrifuged and decanted, then dried under reduced pressure. The obtained protected peptide was treated with 82.5% v/v TFA, 5% water, 5% thioanisol, 2.5% 1,2-ethanedithiol, and 5% phenol (1.5 mL, 1 h). The crude peptide was precipitated out by the addition of chilled diethyl ether (45 mL) to give a white precipitate. The resulting peptide suspension was centrifuged for 20 min at 7000 rpm, and the liquid was decanted. The crude peptide was washed with diethyl ether (2 × 45 mL), and after a final centrifugation, the peptides were dried under vacuum (2 h). The resulting white residue (3.42 g, quantitative) was dissolved in a 3 : 1 mixture of acetonitrile and distilled water (5 mL), and the insoluble impurities were removed by passing the solutions through syringe filters (Gelman Laboratory, Acrodisc 13 mm syringe filter with 0.45 μM PTFE membrane). Final purification was accomplished by preparative RP-HPLC. The pure title compound was obtained after lyophilization.
H-Tyr-D-Ala-Gly-Phe-Met-Pro-Leu-Trp-NH-Bzl·TFA (3)
The title peptide was prepared using the same method as described for H-Tyr-D-Ala-Gly-Phe-Pro-Leu-Trp-NH-3,5-Bzl(CF3)2·TFA (2). The crude peptide was obtained quantitatively.
Characterization of peptides
Preparative RP-HPLC was performed on Waters Delta Prep 4000 with Waters XTerra C-18 column (19 × 250 mm, 10 μm, a linear gradient of 33–53% or 40–60% acedtonitrile/0.1% TFA at a flow rate of 15.0 mL/min). The purified peptides were characterized by HRMS, TLC, analytical HPLC and 1H-1D-NMR. Sequential assignment of proton resonances was achieved by 2D-TOCSY NMR experiments.65–69 High-resolution MS were taken in the positive ion mode using ESI methods at the University of Arizona Mass Spectrometry Facility. TLC was performed on aluminum sheets coated with a 0.2 mm layer of silica gel 60 F254 Merck using the following solvent systems: (1) CHCl3 : MeOH : AcOH = 90 : 10 : 3; (2) EtOAc : n-BuOH : water : AcOH = 5 : 3 : 1 : 1; and (3) n-BuOH : water : AcOH = 4 : 1 : 1. TLC chromatograms were visualized by UV light and by ninhydrin spray followed by heating (hot plate). Analytical HPLC was performed on a Hewlett Packard 1100 or Hewlett Packard 1090m with Waters NOVA-Pak C-18 column (3.9 × 150 mm, 5 μm, 60Å) or Vydac 218TP104 C-18 column (4.6 × 250 mm, 10 μm, 300 Å). 1H-1D-NMR spectra were obtained on Bruker DRX-500 or DRX-600 spectrometer. 2D-TOCSY NMR spectra were performed on a Bruker DRX-600 spectrometer equipped with a 5mm Nalorac triple-resonance single-axis gradient probe. The NMR experiments were conducted in DMSO-d6 solution at 298K. Spectra were referenced to residual solvent protons as 2.49 ppm. The processing of NMR data was performed with the XwinNmr software (Bruker BioSpin, Fremont, CA). In the TOCSY experiments, the TPPI mode70 with MLEV-17 Mixing Sequence65 were used with a mixing time of 62.2 ms, at a spin-lock field of 8.33 kHz. TOCSY spectra were acquired with 2k complex pairs in t2 and 750 or 1024 FIDs using a 90°-shifted sine-squared window function in both dimensions and zero filling to a final matrix size of 2048 (F2) × 1024(F1) points.
NMR Spectroscopy in DPC amphipathic media. 45, 46
All NMR spectra were recorded on a Bruker DRX600 600 MHz spectrometer with 5mm Nalorac triple-resonance single-axis gradient probe. The peptide concentration for the NMR experiments varied from 3.5 to 4 mM. The samples were prepared by dissolving the peptide in 0.5 mL of 45 mM sodium acetate-d3 buffer (pH 4.5) containing 40 equivalents of dodecylphosphocholine-d38 and 1 mM sodium azide (90% H2O/10% D2O) followed by sonication for 5 min. Two-dimensional double quantum filtered correlation (DQF-COSY), nuclear Overhauser effect71 (NOESY), and total correlation spectra66 (TOCSY) were acquired using standard pulse sequences and processed using XwinNmr and Felix 2000 (Accelrys Inc, San Diego, CA). The mixing time for NOESY spectra was 450 ms. All 2D spectra were acquired in the TPPI mode with 2k or 1k complex data points in t2 and 750 real data points in t1, and the spectral processing used 90°-shifted sine bell window functions in both dimensions. For suppressing the H2O signal, the 3-9-19 WATERGATE pulse sequence was used.72 Experiments were conducted at 310 K, and referenced to the H2O shift (4.631 ppm). Coupling constants (3JNH-Hα) were measured from 2D DQF-COSY spectra by analysis of the fingerprint region. The matrix rows of each of the upper and lower halves of a cross-peak were summed to give an antiphase 1D spectrum, which was fitted using a 5-parameter Levenberg-Marquardt nonlinear least-squares protocol73 to a general antiphase doublet. The analysis yielded two independent determinations of the J coupling and line width for each cross-peak, one from the upper half and one from the lower half, and the one with better fitted curve was used for structure calculations. Cross-peak volumes for determination of distance restraints were measured using the Felix 2000 software. In the radical experiment using Mn2+, a stock solution of 5 mM MnCl2 was prepared and added to the sample to achieve a total concentration of 200 μM in Mn2+. The DPC micelles with 5-DOXYL stearic acid were prepared as the same procedure with about 1 mg mL−1 of 5-DOXYL stearic acid but sonicating for 30 min.
Conformational Structure Determination
The methods used for structure calculations have been described previously.45, 46 Throughout the 1H-NMR studies, only one major rotamer for 1-3 was found and the populations of minor rotamers were all negligible. The Met5-Pro6 bond of the major rotamers were fixed in the trans configuration based on the observations of 5Hα to Pro6 Hα sequential NOEs together with the absence of sequential 5Hα-6Hα NOEs in the structural calculations of 1-3. The volumes of the assigned cross-peaks in the 2D NOESY spectrum were converted into upper distance bounds of 3.0, 3.8, 4.8, or 5.8 Å. For overlapping cross-peaks, the distance categories was increased by one or two levels, depending on the qualitative estimate of the extent of overlap. Pseudoatoms were created for nonstereospecifically assigned methylene protons with a correction of 1.0 Å applied to their upper bound distances. In addition to the distance constraints, ϕ dihedral angle constraints derived from 3JHN-Hα coupling constants were set to between −90 and 40° for 3JHN-Hα< 6 Hz and to between −150 and −90° for 3JHN-Hα> 8 Hz. Dihedral angle constraints of 180° ± 5° for peptide bonds (ω) were also used to maintain the planarity of these bonds.
Simulated annealing molecular dynamics analysis was done for all the peptides to obtain an ensemble of NMR structures using the NOE-derived distance constraints and dihedral angle (ϕ) constraints using the DGII74 program within the software package Insight II 2000 (Accelrys Inc., San Diego, CA). Solvent was not explicitly included in the calculations. All the embedded structures successfully passed the simulated annealing step and were minimized using the consistent valency force field (CVFF) (Accelrys Inc.). The 50 structures with the lowest penalty function were further refined by two rounds of restrained molecular dynamics (rMD) using the all-atom AMBER force field with additional parameters for fluorine atom,75–78 using the standalone DISCOVER ver. 2.98 program (Accelrys Inc.). A 12.0 Å cutoff for nonbonded interactions and a distance-dependent dielectric constant (4r) were used. All amide bonds were constrained to trans conformation by a 100 kcal mol−1 rad−2 energy penalty. The distance constraints and dihedral angles (ϕ) constraints were applied with a force constant of 25 kcal mol−1 Å−2 and 100 kcal mol−1 rad−2 were applied, respectively. After 100 steps of steepest descents minimization and 1000 steps of conjugate gradient minimization on the initial structures, an rMD equilibration at 500 K was performed for 1.5 ps, during which a scale factor of 0.1 was applied to the experimental restraint force constants. During the next 2 ps, full values of the experimental restraint force constants were applied. A further 1 ps rMD simulation was run at 500 K, and the system was then cooled to 0 K over 3 ps. After another 1 ps at 0 K, 100 cycles of steepest descents and 2000 steps of conjugate gradient minimization were performed. The final 20 structures with the lowest energies were used for the analysis. All calculations were performed on a Silicon Graphics Octane computer.
Fluorescence emission spectra
The Fluorescence spectra were recorded on a Cary Eclipse fluorescence spectrometer (Varian, Darmstadt, Germany). Emission spectra were obtained by excitation at 290 nm and recorded in the wavelength range of 310–420 nm with continuous stirring at 25°C. A scan step was 1 nm and scan speed was 120 nm min−1. Excitation and emission bandwidths were set at 5 and 10 nm, respectively. The peptide concentration was 500μM in HEPES buffer (10 mM HEPES, 150 mM NaCl, 1 mN NaN3, 0.1 mM EDTA, pH = 7.40) with 40-fold DPC or standard solution (EtOH : HEPES buffer = 1 : 1).61 At least two scans were accumulated and averaged for each spectrum.
Octanol/saline distribution (logD7.4).38
HEPES buffer (0.05 M HEPES buffer in 0.1 M NaCl, pH 7.4, 500 μL) was added to 2 mg of peptide and mixed with 500 μL of 1-octanol. The sample was shaken at r.t. for 12 h to allow equilibrating. The sample was centrifuged at 6500 rpm in a VanGuard V6500 (GlaxoSmithKline, Research Triangle Park, NC) for 15 min. The layers were separated and each layer was centrifuged once again. The peptide concentrations in the obtained layers were analyzed by HPLC (30–70% of acetonitrile containing 0.1% TFA within 20 min and up to 95% within additional 5 min, 1 mL/min, 230 nm, Vydac 218TP104 C-18 column). The logarithm of 1-octanol/saline distribution (logD7.4) was calculated as the ratio of peptide concentration in the 1-octanol and saline phases.
Solubility
HEPES buffer (0.05 M HEPES buffer in 0.1 M NaCl, pH 7.4, 500 μL) was added to 1 mg of peptide. The sample was vortexed for 30 sec, sonicated for 5 min, shaken at r.t. for 2 h, and then allowed to be stayed overnight to equilibration. The sample was filtrated with an Acrodisc Syringe Filter (13 mm, 0.45 μm pore, PTFE membrane, Pall Life Sciences, East Hills, NY). The peptide concentration of the obtained filtrate was analyzed by HPLC (30–70% of acetonitrile containing 0.1% TFA within 20 min and up to 95% within additional 5 min, 1 mL/min, 230 nm, Vydac 218TP104 C-18 column).
Cell Lines
For opioid receptors, the cDNA for the human δ opioid receptor (DOR) was a gift from Dr. Brigitte Kieffer (IGBMC, Illkirch, France). The cDNA for the rat μ opioid receptor (MOR) was a gift from Dr. Huda Akil (University of Michigan, MI). Stable expression of the rat MOR (pCDNA3) and the human DOR (pCDNA3) in the neuroblastoma cell line, HN9.10 were achieved with the respective cDNAs by calcium phosphate precipitation followed by clonal selection in neomycin. Expression of the respective receptors was initially verified and the level of expression periodically monitored by radioligand saturation analysis (see below). All cells were maintained at 37°C, with a 95% air/5% CO2 humidified atmosphere in a Forma Scientific incubator in Dulbecco’s modified Eagle’s medium (DMEM) with 10% bovine serum albumin (BSA) and 100 U mL penicillin/100 μg mL streptomycin. For NK1 receptor, the hNK1/CHO and rNK1/CHO cell lines were obtained from Dr. James Krause (University of Washington Medical School, St. Louis, MI). Expression of the receptor was verified as previously described by Krause et al.79 All cells were maintained at a 37° C, 95% air and 5% CO2, humidified atmosphere, in a Forma Scientific incubator in Ham’S F12 with 2.5 mM HEPES, 10% fetal bovine serum and 100 U mL penicillin/100 μg mLstreptomycin/500 μg mL Geneticin.
Radioligand Labeled Binding Assays
For opioid receptors, crude membranes were prepared as previously described80 from the transfected cells that express the MOR or the DOR. The protein concentration of the membrane preparations was determined by the Lowry method and the membranes were stored at −80°C until use. Membranes were resuspended in assay buffer (50 mM Tris, pH 7.4, containing 50 μg/mL bacitracin, 30 μM bestatin, 10 μM captopril, 100 μM phenylmethylsulfonylfluoride (PMSF), 1 mg/mL BSA). For saturation analysis, six concentrations of tritiated [D-Ala2, NMePhe4, Gly5-ol]-enkephalin ([3H]DAMGO) (0.02–6 nM, 47.2 Ci/mmol), or six concentrations of tritiated c[D-Pen2, D-Pen5]-enkephalin ([3H]DPDPE) (0.1nM–10nM, 44Ci/mmol) were each mixed with 200 μg of membranes from MOR or DOR expressing cells, respectively, in a final volume of 1 mL. For competition analysis, ten concentrations of a test compound were each incubated with 50 μg of membranes from MOR or DOR expressing cells and the Kd concentration of [3H]DAMGO (1.0 nM, 50 Ci/mmol), or of [3H]DPDPE (1.0 nM, 44 Ci/mmol), respectively. Naloxone at 10 μM was used to define the non-specific binding of the radioligands in all assays. All assays were carried out in duplicates and were repeated. The samples were incubated in a shaking water bath at 25°C for 3 hours, followed by rapid filtration through Whatman Grade GF/B filter paper (Gaithersburg, MD) pre-soaked in 1% polyethyleneimine, washed 4 times each with 2 mL of cold saline, and the radioactivity determined by liquid scintillation counting (Beckman LS5000 TD).
For the NK1 receptors, competition binding assays for the NK1 receptors were carried out on crude membranes prepared from transfected CHO cells expressing the human or rat NK1 receptor, respectively. Ten concentrations of a test compound were each incubated with 50–100 μg of membrane homogenate and 0.3 – 0.4 nM [3H] substance P (135 Ci/mmol, Perkin Elmer) in 1 mL final volume of assay buffer (50 mM Tris, pH 7.4, containing 5 mM MgCl2, 50 μg/mL bacitracin, 30 μM bestatin, 10 μM captopril, 100 μM phenylmethylsulfonylfluoride (PMSF) at 25°C for 20 min. Substance P at 10 μM was used to define the non-specific binding. Membrane concentrations used in the assay were within the tissue linearity range. The [3H] substance P concentration was selected based on the saturation binding experiments which showed a high affinity binding with Kd values of 0.40 ± 0.17 for hNK1 and 0.16 ± 0.03 nM for rNK1, respectively. The incubation times correspond to the binding equilibrium as determined from the kinetics experiments. The incubation was stopped by rapid filtration through a GF/B glass filter (Brandel Inc.) pre-soaked in 0.5% polyethyleneimide, followed by four washes with 2 mL of ice-cold saline, using a Brandel Harvester apparatus. The filter-bound radioactivity was measured by liquid scintillation counting (Beckman LS 6000SC). Log IC50 values were determined with standard errors from non-linear regression analysis of at least two independent experiments performed in duplicates using GraphPad Prizm 4 software (GraphPad, San Diego, CA).
For competition analysis using [3H]DAMGO, [3H]DPDPE or [3H]substance P values were calculated from the IC50 by the Cheng and Prusoff equation.
[35S]GTPγS Binding Assay
The method was carried out as previously described.80 Membrane preparation (10 μg) to a final volume of 1 mL incubation mix (50 mM Hepes, pH 7.4, 1 mM EDTA, 5 mM MgCl2, 30 μM GDP, 1 mM dithiothreitol, 100 mM NaCl, 0.1 mM PMSF, 0.1% BSA, 0.1 nM [35S]GTPγS) was added along with various concentrations, in duplicates or triplicates, of the test drug and incubated for 90 min at 30 °C in a shaking water bath. Reactions were terminated by rapid filtration through Whatman GF/B filters (pre-soaked in water), followed by 4 washes with 4 mL of ice-cold wash buffer (50 mM Tris, 5 mM MgCl2, 100 mM NaCl, pH 7.4). The radioactivity was determined by liquid scintillation counting as above. Basal level of [35S]GTPγS binding was defined as the amount bound in the absence of any test drug. Non-specific binding was determined in the presence of 10 μM unlabeled GTPγS. Total binding was defined as the amount of radioactivity bound in the presence of test drug. The effect of the drug at each concentration on [35S]GTPγS binding was calculated as a percentage by the following equation: [Total bound- Basal]/[Basal - Non-specific]×100. Data were expressed as logEC50 ± standard error from at least two independent experiments performed in duplicate analyzed by non-linear regression analysis using GraphPad Prism4.
Guinea Pig Isolated Ileum Assay
The in vitro tissue bioassays were performed as described previously.38 Male Hartley guinea pigs under ether anesthesia were sacrificed by decapitation and a non-terminal portion of the ileum removed and the longitudinal muscle with myenteric plexus (LMMP) was carefully separated from the circular muscle as described previously.81 These tissues were tied to gold chains with suture silk and mounted between platinum wire electrodes in 20 mL organ baths at a tension of 1 g and bathed in oxygenated (95:5 O 2 :CO 2 ) Kreb’s bicarbonate buffer at 37°C and stimulated electrically (0.1 Hz, 0.4 msec duration) at supramaximal voltage. Following an equilibration period, compounds were added cumulatively to the bath in volumes of 14–60 μL until maximum inhibition was reached. A baseline for PL-017 was constructed to determine tissue integrity and allow calculation of antagonist activity before opioid analogue testing began. If no agonist activity was observed at 1 μM, a repeat PL-017 dose-response curve was constructed to test for antagonist qualities.
All substance P antagonist compound testing was performed in the presence of 1 μM naloxone to block opioid effects on unstimulated tissue. Two minutes after naloxone was added to the bath, the test compound was added. Four minutes after naloxone was added, the test dose of substance P was added to the bath, the peak height was noted and the tissues were washed. Agonist activity of the analog was also observed during this period. Testing stopped at 1 mM concentrations of the test compound.
Mouse Isolated Vas Deferens (MVD) Assay
The in vitro tissue bioassay was performed as described previously.38 Male ICR mice under ether anesthesia were sacrificed by cervical dislocation and the vasa deferentia removed. Tissues were tied to gold chains with suture silk and mounted between platinum wire electrodes in 20 mL organ baths at a tension of 0.5 g and bathed in oxygenated (95:5 O2:CO2) magnesium free Kreb’s buffer at 37°C and stimulated electrically (0.1 Hz, single pulses, 2.0 msec duration) at supramaximal voltage as previously described.82 Following an equilibration period, compounds were added to the bath cumulatively in volumes of 14–60 μL until maximum inhibition was reached. Response to an IC50 dose of DPDPE (10 nM) was measured to determine tissue integrity before compound testing.
Analysis of the GPI and MVD assays
For opioid data analysis, percentage inhibition was calculated using the average tissue contraction height for 1 min preceding the addition of the agonist divided by the contraction height 3 min after exposure to the dose of agonist. IC50 values represent the mean of not less than 4 tissues. IC50 and Emax estimates were determined by computerized nonlinear least-squares analysis (the pharmacological statistics package FlashCalc: Dr. Michael Ossipov, University of Arizona, Tucson, AZ). For substance P data analysis, the height of the maximum peak produced during the control substance P dose-response curve was used as a 100 % response and other values calculated as a percentage. Ke values represent the mean of not less than 4 tissues. Ke estimates were determined by computerized nonlinear least-squares analysis (FlashCalc).
Supplementary Material
Acknowledgments
The work was supported by a grant from the USDHS, National Institute on Drug Abuse, DA-13449. We thank Dr. Guangxin Lin and Dr. Jinfa Ying for kind assistance and advice with the NMR measurements and structure calculations, Dr. Robin Polt for fruitful discussion especially about the paramagnetic NMR experiments, Dr. John J. Osterhout for use of the fluorescence spectrometer, Ms. Magdalena Kaczmarska for culturing cells, and the University of Arizona Mass Spectrometry Facility for the mass spectra measurements. We express appreciation to Ms. Margie Colie and Ms. Brigid Blazek for assistance with the manuscript.
Abbreviations
Abbreviations used for amino acids and designation of peptides follow the rules of the IUPAC-IUB Commission of Biochemical Nomenclature in J. Biol. Chem. 1972, 247, 977-83. The following additional abbreviations are used
- AcOH
acetic acid
- Boc
tert-butyloxycarbonyl
- BuOH
butanol
- Bzl
benzyl
- BSA
bovine serum albumin
- CCK
cholecystokinin
- Cl-HOBt
1-hydroxy-6-chlorobenzotriazole
- CHO
Chinese hamster ovary
- DMEM
Dulbecco’s modified Eagle’s medium
- DCM
dichloromethane
- DIEA
diisopropylethylamine
- DMF
N, N-dimethylformamide
- DMSO
dimethylsulfoxide
- DOR
δ opioid receptor
- DPC
Dodecylphosphocholine
- DPDPE
c[D-Pen2, D-Pen5]-enkephalin
- DQF-COSY
double quantum filtered correlation spectroscopy
- DAMGO
[D-Ala2, NMePhe4, Gly5-ol]-enkephalin
- ESI
Electrospray Ionization
- Fmoc
fluorenylmethoxycarbonyl
- GPI
guinea pig isolated ileum
- HCTU
1H-Benzotriazolium-1-[bis(dimethylamino)methylene]-5-chloro-hexafluorophosphate-(1-),3-oxide
- HEK
human embryonic kidney
- HRMS
high-resolution mass spectroscopy
- LMMP
longitudinal muscle/myenteric plexus
- MOR
μ opioid receptor
- MVD
mouse vas deferens
- NK1
neurokinin-1
- NOESY
nuclear Overhauser enhancement
- rmsd
root mean square deviation
- RP-HPLC
reverse phase high performance liquid chromatography
- SAR
structure-activity relationships
- SEM
mean standard error
- SPPS
solid phase peptide synthesis
- TFA
trifluoroacetic acid
- TOCSY
total correlation spectroscopy
- Trp-NH-3
5-Bzl(CF3)2, 3′,5′-(bistrifluoromethyl)-benzyl amide of tryptophan
- Trp-NH-Bzl
benzyl amide of tryptophan
- Trp-O-3
5-Bzl(CF3)2, 3′,5′-(bistrifluoromethyl)-benzyl ester of tryptophan
Footnotes
Supporting Information Available: The 1H-NMR, HPLC and MS data of the peptide derivatives 1-3 are provided. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Dobson CM. Protein folding and misfolding. Nature. 2003;426:884–890. doi: 10.1038/nature02261. [DOI] [PubMed] [Google Scholar]
- 2.Stefani M, Dobson CM. Protein aggregation and aggregate toxicity: new insights into protein folding, misfolding diseases and biological evolution. J Mol Med. 2003;81:678–699. doi: 10.1007/s00109-003-0464-5. [DOI] [PubMed] [Google Scholar]
- 3.Selkoe DJ. Folding proteins in fatal ways. Nature. 2003;426:900–904. doi: 10.1038/nature02264. [DOI] [PubMed] [Google Scholar]
- 4.Barrow CJ, Zagorski MG. Solution structures of beta peptide and its constituent fragments: relation to amyloid deposition. Science. 1991;253:179–182. doi: 10.1126/science.1853202. [DOI] [PubMed] [Google Scholar]
- 5.Fraser PE, Nguyen JT, Surewicz WK, Kirschner DA. pH-dependent structural transitions of Alzheimer amyloid peptides. Biophys J. 1991;60:1190–1201. doi: 10.1016/S0006-3495(91)82154-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Prusiner SB. Prions. Proc Natl Acad Sci U S A. 1998;95:13363–13383. doi: 10.1073/pnas.95.23.13363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Trzesniewska K, Brzyska M, Elbaum D. Neurodegenerative aspects of protein aggregation. Acta Neurobiol Exp (Wars) 2004;64:41–52. doi: 10.55782/ane-2004-1490. [DOI] [PubMed] [Google Scholar]
- 8.Reiss C, Lesnik T, Parvez H, Parvez S, Ehrlich R. Conformational toxicity and sporadic conformational diseases. Toxicology. 2000;153:115–121. doi: 10.1016/s0300-483x(00)00308-5. [DOI] [PubMed] [Google Scholar]
- 9.Vendruscolo M, Zurdo J, MacPhee CE, Dobson CM. Protein folding and misfolding: a paradigm of self-assembly and regulation in complex biological systems. Philos Transact A Math Phys Eng Sci. 2003;361:1205–1222. doi: 10.1098/rsta.2003.1194. [DOI] [PubMed] [Google Scholar]
- 10.Lopez de la Paz M, de Mori GM, Serrano L, Colombo G. Sequence dependence of amyloid fibril formation: insights from molecular dynamics simulations. J Mol Biol. 2005;349:583–596. doi: 10.1016/j.jmb.2005.03.081. [DOI] [PubMed] [Google Scholar]
- 11.Hughes J, Smith TW, Kosterlitz HW, Fothergill LA, Morgan BA, Morris HR. Identification of two related pentapeptides from the brain with potent opiate agonist activity. Nature. 1975;258:577–580. doi: 10.1038/258577a0. [DOI] [PubMed] [Google Scholar]
- 12.Palian MM, Boguslavsky VI, O’Brien DF, Polt R. Glycopeptide-membrane interactions: glycosyl enkephalin analogues adopt turn conformations by NMR and CD in amphipathic media. J Am Chem Soc. 2003;125:5823–5831. doi: 10.1021/ja0268635. [DOI] [PubMed] [Google Scholar]
- 13.Dhanasekaran M, Palian MM, Alves I, Yeomans L, Keyari CM, Davis P, Bilsky EJ, Egleton RD, Yamamura HI, Jacobsen NE, Tollin G, Hruby VJ, Porreca F, Polt R. Glycopeptides related to beta-endorphin adopt helical amphipathic conformations in the presence of lipid bilayers. J Am Chem Soc. 2005;127:5435–5448. doi: 10.1021/ja0432158. [DOI] [PubMed] [Google Scholar]
- 14.Spadaccini R, Temussi PA. Natural peptide analgesics: the role of solution conformation. Cell Mol Life Sci. 2001;58:1572–1582. doi: 10.1007/PL00000797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rudolph-Bohner S, Quarzago D, Czisch M, Ragnarsson U, Moroder L. Conformational preferences of Leu-enkephalin in reverse micelles as membrane-mimicking environment. Biopolymers. 1997;41:591–606. [Google Scholar]
- 16.Kallick DA, Tessmer MR, Watts CR, Li CY. The use of dodecylphosphocholine micelles in solution NMR. J Magn Reson B. 1995;109:60–65. doi: 10.1006/jmrb.1995.1146. [DOI] [PubMed] [Google Scholar]
- 17.Kameda T, Takada S. Secondary structure provides a template for the folding of nearby polypeptides. Proc Natl Acad Sci U S A. 2006;103:17765–17770. doi: 10.1073/pnas.0602632103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Berthold M, Bartfai T. Modes of peptide binding in G protein-coupled receptors. Neurochem Res. 1997;22:1023–1031. doi: 10.1023/a:1022483027858. [DOI] [PubMed] [Google Scholar]
- 19.Noeskea TGA, Parsonsa CG, Weila T. Allosteric modulation of family 3 GPCRs. QSAR Comb Sci. 2006;25:134–146. [Google Scholar]
- 20.Eguchi M. Recent advances in selective opioid receptor agonists and antagonists. Med Res Rev. 2004;24:182–212. doi: 10.1002/med.10059. [DOI] [PubMed] [Google Scholar]
- 21.Cascieri MA, Macleod AM, Underwood D, Shiao LL, Ber E, Sadowski S, Yu H, Merchant KJ, Swain CJ, Strader CD, Fong TM. Characterization of the interaction of N-acyl-L-tryptophan benzyl ester neurokinin antagonists with the human neurokinin-1 receptor. J Biol Chem. 1994;269:6587–6591. [PubMed] [Google Scholar]
- 22.D’Alagni M, Delfini M, Di Nola A, Eisenberg M, Paci M, Roda LG, Veglia G. Conformational study of [Met5]enkephalin-Arg-Phe in the presence of phosphatidylserine vesicles. Eur J Biochem. 1996;240:540–549. doi: 10.1111/j.1432-1033.1996.0540h.x. [DOI] [PubMed] [Google Scholar]
- 23.Deber CM, Behnam BA. Role of membrane lipids in peptide hormone function: binding of enkephalins to micelles. Proc Natl Acad Sci U S A. 1984;81:61–65. doi: 10.1073/pnas.81.1.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sargent DF, Schwyzer R. Membrane lipid phase as catalyst for peptide-receptor interactions. Proc Natl Acad Sci U S A. 1986;83:5774–5778. doi: 10.1073/pnas.83.16.5774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wienk HL, Wechselberger RW, Czisch M, de Kruijff B. Structure, dynamics, and insertion of a chloroplast targeting peptide in mixed micelles. Biochemistry. 2000;39:8219–27. doi: 10.1021/bi000110i. [DOI] [PubMed] [Google Scholar]
- 26.Wienk HL, Wechselberger RW, Czisch M, de Kruijff B. Structure, dynamics, and insertion of a chloroplast targeting peptide in mixed micelles. Biochemistry. 2000;39:8219–8227. doi: 10.1021/bi000110i. [DOI] [PubMed] [Google Scholar]
- 27.Bryson EA, Rankin SE, Carey M, Watts A, Pinheiro TJ. Folding of apocytochrome c in lipid micelles: formation of alpha-helix precedes membrane insertion. Biochemistry. 1999;38:9758–9767. doi: 10.1021/bi990119o. [DOI] [PubMed] [Google Scholar]
- 28.Saffman PG, Delbruck M. Brownian motion in biological membranes. Proc Natl Acad Sci U S A. 1975;72:3111–3113. doi: 10.1073/pnas.72.8.3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Janecka A, Fichna J, Janecki T. Opioid receptors and their ligands. Curr Top Med Chem. 2004;4:1–17. doi: 10.2174/1568026043451618. [DOI] [PubMed] [Google Scholar]
- 30.Gentilucci L, Tolomelli A. Recent advances in the investigation of the bioactive conformation of peptides active at the micro-opioid receptor. conformational analysis of endomorphins. Curr Top Med Chem. 2004;4:105–121. doi: 10.2174/1568026043451627. [DOI] [PubMed] [Google Scholar]
- 31.King T, Gardell LR, Wang R, Vardanyan A, Ossipov MH, Malan TP, Jr, Vanderah TW, Hunt SP, Hruby VJ, Lai J, Porreca F. Role of NK-1 neurotransmission in opioid-induced hyperalgesia. Pain. 2005;116:276–288. doi: 10.1016/j.pain.2005.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ripley TL, Gadd CA, De Felipe C, Hunt SP, Stephens DN. Lack of self-administration and behavioural sensitisation to morphine, but not cocaine, in mice lacking NK1 receptors. Neuropharmacology. 2002;43:1258–1268. doi: 10.1016/s0028-3908(02)00295-2. [DOI] [PubMed] [Google Scholar]
- 33.King T, Ossipov MH, Vanderah TW, Porreca F, Lai J. Is paradoxical pain induced by sustained opioid exposure an underlying mechanism of opioid antinociceptive tolerance? Neurosignals. 2005;14:194–205. doi: 10.1159/000087658. [DOI] [PubMed] [Google Scholar]
- 34.Ma W, Zheng WH, Kar S, Quirion R. Morphine treatment induced calcitonin gene-related peptide and substance P increases in cultured dorsal root ganglion neurons. Neuroscience. 2000;99:529–539. doi: 10.1016/s0306-4522(00)00226-8. [DOI] [PubMed] [Google Scholar]
- 35.Powell KJ, Quirion R, Jhamandas K. Inhibition of neurokinin-1-substance P receptor and prostanoid activity prevents and reverses the development of morphine tolerance in vivo and the morphine-induced increase in CGRP expression in cultured dorsal root ganglion neurons. Eur J Neurosci. 2003;18:1572–1583. doi: 10.1046/j.1460-9568.2003.02887.x. [DOI] [PubMed] [Google Scholar]
- 36.Misterek K, Maszczynska I, Dorociak A, Gumulka SW, Carr DB, Szyfelbein SK, Lipkowski AW. Spinal co-administration of peptide substance P antagonist increases antinociceptive effect of the opioid peptide biphalin. Life Sci. 1994;54:939–944. doi: 10.1016/0024-3205(94)00494-3. [DOI] [PubMed] [Google Scholar]
- 37.Gu G, Kondo I, Hua XY, Yaksh TL. Resting and evoked spinal substance P release during chronic intrathecal morphine infusion: parallels with tolerance and dependence. J Pharmacol Exp Ther. 2005;314:1362–1369. doi: 10.1124/jpet.105.087718. [DOI] [PubMed] [Google Scholar]
- 38.Yamamoto T, Nair P, Davis P, Ma SW, Navratilova E, Moye M, Tumati S, Vanderah TW, Lai J, Porreca F, Yamamura HI, Hruby VJ. Design, Synthesis and Biological Evaluation of Novel Bifunctional C-terminal Modified Peptides for δ/μ Opioid Receptor Agonists and Neurokinin-1 Receptor Antagonists. J Med Chem. 2007;50:2779 – 2786. doi: 10.1021/jm061369n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yamamoto T, Nair P, Davis P, Ma SW, Moye S, Largent T, Vanderah TW, Lai J, Porreca F, Yamamura HI, Hruby VJ. Design, Structure-Activity Relationships and Biological Evaluation of Novel Bifunctional C-terminal Modified Peptides for δ/μ Opioid Receptor Agonists and Neurokinin-1 Receptor Antagonists. 232nd ACS National Meeting; San Francisco, CA, United States. 2006. MEDI–7. [Google Scholar]
- 40.Yamamoto T, Nair P, Vagner J, Davis P, Ma SW, Navratilova E, Moye M, Tumati S, Vanderah TW, Lai J, Porreca F, Yamamura HI, Hruby VJ. A Structure Activity Relationship Study and Combinatorial Synthetic Approach of C-Terminal Modified Bifunctional Peptides That Are δ/μ Opioid Receptor Agonists and Neurokinin 1 Receptor Antagonists. J Med Chem. 2008;51:1369–1376. doi: 10.1021/jm070332f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Braun W, Wider G, Lee KH, Wuthrich K. Conformation of glucagon in a lipid-water interphase by 1H nuclear magnetic resonance. J Mol Biol. 1983;169:921–948. doi: 10.1016/s0022-2836(83)80143-0. [DOI] [PubMed] [Google Scholar]
- 42.Thornton K, Gorenstein DG. Structure of glucagon-like peptide (7–36) amide in a dodecylphosphocholine micelle as determined by 2D NMR. Biochemistry. 1994;33:3532–3539. doi: 10.1021/bi00178a009. [DOI] [PubMed] [Google Scholar]
- 43.Arora A, Abildgaard F, Bushweller JH, Tamm LK. Structure of outer membrane protein A transmembrane domain by NMR spectroscopy. Nat Struct Biol. 2001;8:334–338. doi: 10.1038/86214. [DOI] [PubMed] [Google Scholar]
- 44.Karslake C, Piotto ME, Pak YK, Weiner H, Gorenstein DG. 2D NMR and structural model for a mitochondrial signal peptide bound to a micelle. Biochemistry. 1990;29:9872–9878. doi: 10.1021/bi00494a017. [DOI] [PubMed] [Google Scholar]
- 45.Ying J, Ahn JM, Jacobsen NE, Brown MF, Hruby VJ. NMR solution structure of the glucagon antagonist [desHis1, desPhe6, Glu9]glucagon amide in the presence of perdeuterated dodecylphosphocholine micelles. Biochemistry. 2003;42:2825–2835. doi: 10.1021/bi026629r. [DOI] [PubMed] [Google Scholar]
- 46.Jacobsen NE, Abadi N, Sliwkowski MX, Reilly D, Skelton NJ, Fairbrother WJ. High-resolution solution structure of the EGF-like domain of heregulin-alpha. Biochemistry. 1996;35:3402–3417. doi: 10.1021/bi952626l. [DOI] [PubMed] [Google Scholar]
- 47.Porreca F, Heyman JS, Mosberg HI, Omnaas JR, Vaught JL. Role of mu and delta receptors in the supraspinal and spinal analgesic effects of [D-Pen2, D-Pen5]enkephalin in the mouse. J Pharmacol Exp Ther. 1987;241:393–400. [PubMed] [Google Scholar]
- 48.Porreca F, Mosberg HI, Hurst R, Hruby VJ, Burks TF. Roles of mu, delta and kappa opioid receptors in spinal and supraspinal mediation of gastrointestinal transit effects and hot-plate analgesia in the mouse. J Pharmacol Exp Ther. 1984;230:341–348. [PubMed] [Google Scholar]
- 49.Audigier Y, Mazarguil H, Gout R, Cros J. Structure-activity relationships of enkephalin analogs at opiate and enkephalin receptors: correlation with analgesia. Eur J Pharmacol. 1980;63:35–46. doi: 10.1016/0014-2999(80)90114-4. [DOI] [PubMed] [Google Scholar]
- 50.Agnes RS, Lee YS, Davis P, Ma SW, Badghisi H, Porreca F, Lai J, Hruby VJ. Structure-activity relationships of bifunctional peptides based on overlapping pharmacophores at opioid and cholecystokinin receptors. J Med Chem. 2006;49:2868–2875. doi: 10.1021/jm050921q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lee YS, Agnes RS, Badghisi H, Davis P, Ma SW, Lai J, Porreca F, Hruby VJ. Design and synthesis of novel hydrazide-linked bifunctional peptides as delta/mu opioid receptor agonists and CCK-1/CCK-2 receptor antagonists. J Med Chem. 2006;49:1773–1780. doi: 10.1021/jm05085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lazaridis T, Mallik B, Chen Y. Implicit solvent simulations of DPC micelle formation. J Phys Chem B. 2005;109:15098–15106. doi: 10.1021/jp0516801. [DOI] [PubMed] [Google Scholar]
- 53.Smith LJ, Bolin KA, Schwalbe H, MacArthur MW, Thornton JM, Dobson CM. Analysis of main chain torsion angles in proteins: prediction of NMR coupling constants for native and random coil conformations. J Mol Biol. 1996;255:494–506. doi: 10.1006/jmbi.1996.0041. [DOI] [PubMed] [Google Scholar]
- 54.Wagner G, Neuhaus D, Worgotter E, Vasak M, Kagi JH, Wuthrich K. Nuclear magnetic resonance identification of “half-turn” and 3(10)-helix secondary structure in rabbit liver metallothionein-2. J Mol Biol. 1986;187:131–135. doi: 10.1016/0022-2836(86)90413-4. [DOI] [PubMed] [Google Scholar]
- 55.Smith D, Griffin JF. Conformation of [Leu5]enkephalin from X-ray diffraction: features important for recognition at opiate receptor. Science. 1978;199:1214–1216. doi: 10.1126/science.204006. [DOI] [PubMed] [Google Scholar]
- 56.Graham WH, Carter ES, 2nd, Hicks RP. Conformational analysis of Met-enkephalin in both aqueous solution and in the presence of sodium dodecyl sulfate micelles using multidimensional NMR and molecular modeling. Biopolymers. 1992;32:1755–1764. doi: 10.1002/bip.360321216. [DOI] [PubMed] [Google Scholar]
- 57.Wilmot CM, Thornton JM. Analysis and prediction of the different types of beta-turn in proteins. J Mol Biol. 1988;203:221–232. doi: 10.1016/0022-2836(88)90103-9. [DOI] [PubMed] [Google Scholar]
- 58.Hyberts SG, Goldberg MS, Havel TF, Wagner G. The solution structure of eglin c based on measurements of many NOEs and coupling constants and its comparison with X-ray structures. Protein Sci. 1992;1:736–751. doi: 10.1002/pro.5560010606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Beechem JM, Brand L. Time-resolved fluorescence of proteins. Annu Rev Biochem. 1985;54:43–71. doi: 10.1146/annurev.bi.54.070185.000355. [DOI] [PubMed] [Google Scholar]
- 60.Vivian JT, Callis PR. Mechanisms of tryptophan fluorescence shifts in proteins. Biophys J. 2001;80:2093–2109. doi: 10.1016/S0006-3495(01)76183-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Raguz M, Brnjas-Kraljevic J. Resolved fluorescence emission spectra of PRODAN in ethanol/buffer solvents. J Chem Inf Model. 2005;45:1636–1340. doi: 10.1021/ci050166p. [DOI] [PubMed] [Google Scholar]
- 62.Neidigh JW, Fesinmeyer RM, Prickett KS, Andersen NH. Exendin-4 and glucagon-like-peptide-1: NMR structural comparisons in the solution and micelle-associated states. Biochemistry. 2001;40:13188–13200. doi: 10.1021/bi010902s. [DOI] [PubMed] [Google Scholar]
- 63.Datar P, Srivastava S, Coutinho E, Govil G. Substance P: structure, function, and therapeutics. Curr Top Med Chem. 2004;4:75–103. doi: 10.2174/1568026043451636. [DOI] [PubMed] [Google Scholar]
- 64.Lewis RT, Macleod AM, Merchant KJ, Kelleher F, Sanderson I, Herbert RH, Cascieri MA, Sadowski S, Ball RG, Hoogsteen K. Tryptophan-derived NK1 antagonists: conformationally constrained heterocyclic bioisosteres of the ester linkage. J Med Chem. 1995;38:923–933. doi: 10.1021/jm00006a011. [DOI] [PubMed] [Google Scholar]
- 65.Braunschweiler L, Ernst RR. Coherence Transfer by Isotropic Mixing: Application of Proton Correlation Spectroscopy. J Magn Reson. 1983;53:521–528. [Google Scholar]
- 66.Davis DG, Bax A. Assignment of complex proton NMR spectra via two-dimensional homonuclear Hartmann-Hahn spectroscopy. J Am Chem Soc. 1985;107:2820–2821. [Google Scholar]
- 67.Subramanian S, Bax A. Generation of pure phase NMR subspectra for measurement of homonuclear coupling constants. J Magn Reson. 1987;71:325–330. [Google Scholar]
- 68.Rance M. Improved techniques for homonuclear rotating-frame and isotropic mixing experiments. J Magn Reson. 1987;74:557–564. [Google Scholar]
- 69.Bax A, Davis DG. MLEV-17-based two-dimensional homonuclear. magnetization transfer spectroscopy. J Magn Reson. 1985;65:355–360. [Google Scholar]
- 70.Marion DW, Wüthrich K. Application of phase sensitive two-dimensional correlated spectroscopy (COSY) for measurements of 1H-1H spin-spin coupling constants in proteins. Biochem Biophys Res Commun. 1983;113:967–974. doi: 10.1016/0006-291x(83)91093-8. [DOI] [PubMed] [Google Scholar]
- 71.Kumar A, Ernst RR, Wüthrich K. A two-dimensional nuclear Overhauser enhancement (2D NOE) experiment for the elucidation of complete proton-proton cross-relaxation networks in biological macromolecules. Biochem Biophys Res Commun. 1980;95:1–6. doi: 10.1016/0006-291x(80)90695-6. [DOI] [PubMed] [Google Scholar]
- 72.Piotto M, Saudek V, Sklenář V. Gradient-tailored excitation for single-quantum NMR spectroscopy of aqueous solutions. J Biomol NMR. 1992;2:661–665. doi: 10.1007/BF02192855. [DOI] [PubMed] [Google Scholar]
- 73.Press WH, Vetterling WT, Teukolsky SA. The Art of Scientific Computing. Cambridge University Press; New York: 1988. Numerical Recipes in C. [Google Scholar]
- 74.Havel TF. An evaluation of computational strategies for use in the determination of protein structure from distance constraints obtained by nuclear magnetic resonance. Prog Biophys Mol Biol. 1991;56:43–78. doi: 10.1016/0079-6107(91)90007-f. [DOI] [PubMed] [Google Scholar]
- 75.Weiner SJ, Kollman PA, Case DA, Singh UC, Ghio C, Alagona GS, Profeta J, Weiner P. A New Force Field for Molecular Mechanical Simulation of Nucleic Acids and Proteins. J Am Chem Soc. 1984;106:765–784. [Google Scholar]
- 76.Weiner SJ, Kollman PA, Case DA. An all atom force field for simulations of proteins and nucleic acids. J Comput Chem. 1986;7:230–252. doi: 10.1002/jcc.540070216. [DOI] [PubMed] [Google Scholar]
- 77.Gough CA, DeBolt SE, Kollman PA. Derivation of fluorine and hydrogen atom parameters using liquid simulations. J Comp Chem. 1992;13:963–970. [Google Scholar]
- 78.http://amber.scripps.edu/Questions/fluorine.html.
- 79.Boyd ND, Kage R, Dumas JJ, Krause JE, Leeman SE. The peptide binding site of the substance P (NK-1) receptor localized by a photoreactive analogue of substance P: presence of a disulfide bond. Proc Natl Acad Sci U S A. 1996;93:433–737. doi: 10.1073/pnas.93.1.433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lorenzen A, Fuss M, Vogt H, Schwabe U. Measurement of guanine nucleotide-binding protein activation by A1 adenosine receptor agonists in bovine brain membranes: stimulation of guanosine-5′-O-(3-[35S]thio) triphosphate binding. Mol Pharmacol. 1993;44:115–123. [PubMed] [Google Scholar]
- 81.Porreca F, Burks TF. Affinity of normorphine for its pharmacologic receptor in the naive and morphine-tolerant guinea-pig isolated ileum. J Pharmacol Exp Ther. 1983;225:688–693. [PubMed] [Google Scholar]
- 82.Porreca F, LoPresti D, Ward SJ. Opioid agonist affinity in the guinea-pig ileum and mouse vas deferens. Eur J Pharmacol. 1990;179:129–139. doi: 10.1016/0014-2999(90)90410-8. [DOI] [PubMed] [Google Scholar]
- 83.Tetko IV, Gasteiger J, Todeschini R, Mauri A, Livingstone D, Ertl P, Palyulin VA, Radchenko EV, Zefirov NS, Makarenko AS, Tanchuk VY, Prokopenko VV. Virtual computational chemistry laboratory - design and description. J Comput Aid Mol Des. 2005;19:453–63. doi: 10.1007/s10822-005-8694-y. [DOI] [PubMed] [Google Scholar]
- 84.VCCLAB. http://www.vcclab.org.
- 85.Lipkowski AW, Misicka A, Davis P, Stropova D, Janders J, Lachwa M, Porreca F, Yamamura HI, Hruby VJ. Biological activity of fragments and analogues of the potent dimeric opioid peptide, biphalin. Bioorg Med Chem Lett. 1999;9:2763–2766. doi: 10.1016/s0960-894x(99)00464-3. [DOI] [PubMed] [Google Scholar]
- 86.Wishart DS, Sykes BD, Richards FM. The Chemical Shift Index: A Fast and Simple Method for the Assignment of Protein Secondary Structure through NMR Spectroscopy. Biochemistry. 1992;31:1647–1651. doi: 10.1021/bi00121a010. [DOI] [PubMed] [Google Scholar]
- 87.Andersen NH, Neidigh JW, Harris SW, Lee GM, Liu ZH, Tong H. Extracting Information from the Temperature Gradients of Polypeptide NH Chemical Shifts. 1. The Importance of Conformational Averaging. J Am Chem Soc. 1997;119:8547–8561. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.