

Abstract
In addition to producing analgesia, opioids have also been proposed to regulate glucose homeostasis by altering insulin secretion. A considerable controversy exists, however, regarding the contribution of the μ-opioid receptor (MOR-1) to insulin secretion dynamics. We employed congenic C57BL/6J MOR-1 knockout (KO) mice to clarify the role of MOR in glucose homeostasis. We first found that both sexes of MOR-1 KO mice weigh more than wild-type mice throughout postnatal life and that this increase includes preferentially increased fat deposition. We also found that MOR-1 KO mice exhibit enhanced glucose tolerance that results from insulin hypersecretion that reflects increased β-cell mass and increased secretory dynamics in the MOR-1 mutant mice compared with wild type. Analysis of the isolated islets indicated that islet insulin hypersecretion is mediated directly by MOR expressed on islet cells via a mechanism downstream of ATP-sensitive K+ channel activation by glucose. These findings indicate that MOR-1 regulates body weight by a mechanism that involves insulin secretion and thus may represent a novel target for new diabetes therapies.
MOR-1 KO mice exhibit increased body mass compared to WT mice and also show enhanced glucose tolerance and insulin hypersecretion both in vivo and in vitro.
Although multiple treatments can improve blood glucose control in type II diabetes, enhancing insulin production from the β-cell still remains an important therapeutic approach (1). Recent research has been focused on identifying novel molecular regulators of β-cell insulin secretion, not only to acquire a better understanding of the coupling between glucose sensing and insulin secretion but also to identify and develop novel β-cell-targeted diabetes therapies.
The endogenous opioid system, including its three receptors and endogenous ligands, is known to modulate several physiological systems in vivo in addition to its well-documented contribution to analgesia. For example, pharmacological evidence indicates that the endogenous opioid system negatively regulates insulin release from isolated Langerhans islets in vitro (2,3,4). Although opioid receptor mRNA and protein have not previously been identified in the islet, evidence for both opioid binding sites and endogenous opioid ligands in the endocrine pancreas have been reported (5,6,7), suggesting potential peripheral opioid action in insulin secretion and glucose homeostasis.
Although numerous in vitro and in vivo studies have demonstrated an effect of endogenous opioids and certain opioid agonists on insulin release, considerable controversy has arisen regarding the contribution of the μ-opioid receptor (MOR-1) system to insulin secretion dynamics. In vitro, β-endorphin (primarily MOR targeting) inhibits insulin release from isolated islets (3,4). In vivo, β-endorphin also attenuates insulin release when administered by iv infusion (8,9), although in contrast, a potentiating effect was found after iv infusion and bolus injection (10,11,12). In addition, some groups also reported dual stimulatory/inhibitory action of β-endorphin on insulin release depending on dose, obesity state, or circulating glucose concentration (13,14). These pharmacological studies have thus yielded conflicting results so that the specific role of the MOR system in glucose homeostasis remains unclear.
Therefore, the aim of this study was to employ MOR-1 knockout (KO) mice to determine genetically the role of MOR in glucose homeostasis in vivo and in vitro. One recent study using MOR-1 KO mice had shown that body weight is increased in these mice without a change in glucose tolerance (15). Here we demonstrate that mice lacking MOR-1 indeed have a heavier body weight after standard diet feeding, which is due to a preferential increase in fat mass as well as an increase in lean mass compared with WT mice. In addition, this strain of MOR-1 KO mice display enhanced glucose tolerance, which is accompanied by an enhanced insulin secretion upon glucose exposure both in vivo and in vitro. This process is regulated by peripheral islet MOR at a step after KATP channel activation. In addition, MOR deletion leads to an increase in β-cell mass shown by greater islet size. Thus, our studies have identified a novel mechanism contributing to glucose metabolism that is a potential target for novel anti-type II diabetes therapy.
Results
C57BL/6J MOR-1 KO mice show enhanced body weight and a preferential increase in fat deposition
Because it had been suggested that MOR is involved in some metabolic parameters such as insulin secretion (8) and feeding behavior (16), we first wanted to determine whether the body weight, as a final outcome, is altered between wild-type (WT) and MOR-1 KO mice. We measured body weights of animals fed on standard diet from the day of birth to d 180. Male MOR-1 KO mice weigh significantly more than WT control mice throughout the 180-d measuring span except on d 0 (day of birth) and during the weaning period (wk 2–4) when daily weights are more variable. Female MOR-1 KO mice also showed the same pattern (Fig. 1A).
Figure 1.

The body weight and composition of WT and MOR-1 KO mice. A, From the day of birth, animals were weighed periodically to build the body weight chart until d 180. MOR-1 KO mice weigh significantly more than WT mice throughout the measured time span independent of genders. Data are represented as means ± sem. Male WT vs. MOR-1 KO mice: *, P < 0.05; **, P < 0.01; ***, P < 0.001. Female WT vs. MOR-1 KO mice: †, P < 0.05; ††, P < 0.01; †††, P < 0.001. B, Male WT and MOR-1 KO were fed on standard diet for 9 wk and subjected to a body composition analysis. Fat and lean tissues were graphed by absolute value in grams and percent body weight. An increase in both fat and lean tissues is observed in MOR-1 KO mice compared with WT control. From a percentage view, the MOR KO mice have more fat tissue percentage and less lean tissue percentage compared with WT. Data are represented as means ± sem. **, P < 0.01; ***, P < 0.001.
To determine which tissue type accounts for the increased body weight, male WT and MOR-1 KO were maintained on standard chow until 9 wk of age and then body composition was analyzed by quantitative nuclear magnetic resonance (qNMR). A significant increase in both fat tissue and lean tissue absolute weight between WT and MOR-1 mutant mice was seen, although the increase in fat tissue (73% increase) was substantially more pronounced than the increase in lean tissue increase (11%) in the MOR KO mice (Fig. 1B, left). In whole body composition percentage, there is thus a significant 5% increase in fat after MOR deletion, accompanied by a 5% decrease in lean tissue (Fig. 1B, right). These body composition studies indicate that the higher body weight in MOR-1 mutant mice can be attributed to an increase in both fat and lean tissue, although MOR deletion preferentially promotes an increase in fat tissue compared with lean tissue. We have also analyzed 4-month-old male WT and MOR KO mice using the same protocol and observed identical results (data not shown).
MOR-1 KO mice display enhanced glucose tolerance due to insulin hypersecretion
Because insulin promotes adipogenesis and thus obesogenesis (17), the enhanced fat deposition in MOR-1 KO mice could result from a chronic alteration in glucose homeostasis/insulin dynamics. We first found that basal glucose levels (after overnight fasting) between MOR-1 KO and WT mice were comparable. Then, to determine whether glucose tolerance in MOR-1 KO mice is altered in vivo, we measured whole-blood glucose levels after a 2 g/kg ip glucose injection after overnight (1800–1000 h) fasting. MOR-1 KO mice showed a significantly enhanced glucose tolerance, which persists throughout all time points after injection (Fig. 2A). We also tested other opioid receptor KO mice that we previously produced and now maintain on C57BL/6J background, including DOR-1 (18), KOR-1 (19), ORL1 (20), and enkephalin (21) KO mice; no effect of these genes on glucose tolerance was observed (data not shown).
Figure 2.

The effect of MOR on glucose homeostasis. A, A 2 g/kg glucose bolus was administered to WT and MOR-1 KO mice fasted overnight. The blood glucose level was measured via tail vein at the different time points. As shown, MOR mice had significantly enhanced glucose tolerance at indicated time points. B, Animals were fasted for 6 h followed by a 0.75 U/kg ip insulin injection. Blood glucose level was monitored at different time points. No statistical significance was found at any time point. C, Animals were fasted overnight followed by an ip 2 g/kg glucose injection. Plasma was taken from the tail vein at indicated time points and assayed for insulin concentration by ELISA. MOR-1 KO mice displayed insulin hypersecretion compared with WT C57BL/6J mice at the indicated time points. Data are represented as means ± sem. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
To determine whether the enhanced glucose tolerance in the MOR-1 KO is a pre-insulin receptor or postreceptor activity, we carried out an insulin tolerance test on MOR-1 KO and WT control mice. Mice were fasted for 6 h and given 0.75 U/kg ip insulin. Serum glucose levels were monitored at times 0, 15, 30, and 60 min after insulin injection. The insulin tolerance curves of WT and MOR-1 KO mice were indistinguishable (Fig. 2B), indicating that the enhanced glucose tolerance in the mutant strain is due to a mechanism arising before insulin receptor activation.
Because our data indicated the enhanced glucose tolerance in MOR-1 KO mice is due to a pre-insulin receptor mechanism, we next wanted to determine whether circulating insulin secretion dynamics were altered after in vivo glucose exposure. Mice were fasted overnight (16 h) and given a 2 g/kg glucose challenge on the next morning. Plasma was collected at 0, 2, 5, 10, 15, and 30 min after glucose ip injection and then analyzed using a mouse insulin ELISA. MOR-1 KO mice generally secreted more insulin than the WT controls, with the early-phase insulin surge significantly potentiated (Fig. 2C). Therefore, these data strongly suggest that increased insulin secretion leads to the enhanced glucose uptake exhibited by MOR-1 KO mice.
Aging does not modify the enhanced glucose tolerance of MOR-1 KO mice
To determine whether the insulin hypersecretion in MOR-1 KO mice might represent predisposition to type II diabetes, we performed a glucose tolerance test on older WT and MOR KO mice. After a 2 g/kg ip glucose challenge, MOR KO mice at 7 months of age again exhibited enhanced tolerance (Fig. 3A). We also maintained these mice until 12 months of age, when they were retested. The fasting blood glucose levels for both genotypes were below 150 mg/dl at both ages. The result from glucose tolerance was the same as in younger mice without a trend of diabetes development in the mutant animals (Fig. 3B). Moreover, the insulin sensitivity of these 1-yr-old WT and MOR KO mice exhibits no difference after 0.75 U/kg ip insulin injection (Fig. 3C). The fasting glucose levels and the glucose tolerance data in the aged mice indicate that the higher insulin levels in the MOR-1 KO mice after glucose exposure do not correlate with any propensity to develop type II diabetes, in which anomalous glucose control may be preceded with an insulin hypersecretion to compensate for peripheral insulin resistance (22).
Figure 3.

The enhanced glucose tolerance remains in aged MOR KO mice. To determine whether type II diabetes propensity accompanies insulin hypersecretion, several tests were performed in aged WT and MOR KO mice. A, After a 2 g/kg glucose ip challenge, the enhanced glucose tolerance remained in MOR KO at 7 months old after overnight fasting. B, When these mice reached 1 yr old, a 1 g/kg glucose ip challenge was performed. The MOR KO mice showed the same trend of enhanced tolerance. C, A 0.75 U/kg insulin ip injection was given to 1-yr-old WT and MOR KO mice fasted for 6 h. There is no change in insulin sensitivity. Data are represented as means ± sem. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
MOR KO mice have larger islets and higher pancreas insulin content
To investigate the cause for the insulin hypersecretion from the perspective of insulin reserve, the pancreatic insulin content was assessed by ELISA after homogenization. MOR-1 KO pancreata showed both increased weight and increased insulin content (Fig. 4, A and B). Although the mutant mice are heavier and their pancreata are proportionally bigger, relative insulin content is still increased after the content is normalized to pancreas weight (Fig. 4C). The insulin content data suggest that the MOR-1 KO mice have either higher β-cell mass (islet population or volume) or greater β-cell insulin concentration. To distinguish these possibilities, the planar secretion area was measured from a pool of WT and mutant enzyme-isolated islets. We found that MOR KO mice have significantly larger islets compared with WT. Measurements were carried out in three independent experiments with each determination comprised of islets isolated from three WT and three MOR KO mice [set 1 (μm2± sem: 15,150 ± 841.2 (n = 135) vs. 19,320 ± 749.6 (n = 279); set 2: 14,880 ± 462.5 (n = 322) vs. 23,320 ± 741.2 (n = 382); set 3: 16,490 ± 435.0 (n = 365) vs. 21,580 ± 582.2 (n = 371)]. Figure 4D presents the average of all three experiments and represents data from 822 and 1032 isolated islets from 3-month-old male WT and MOR KO, respectively. We also carried out the histological comparison of the islet morphology between genotypes. After hematoxylin and eosin staining, the islet sections from MOR KO mice do not exhibit any discernable anatomical difference to WT except for their larger dimension (Fig. 4E). Therefore, the insulin content and histological analysis together demonstrate that higher Langerhans islet mass in the MOR KO mice leads to the increased pancreatic insulin content, which may at least partly contribute to the enhanced insulin secretion and glucose tolerance in the mutant mice.
Figure 4.

MOR KO mice exhibit higher insulin content and larger islet dimension. A, The whole pancreas of 2-month-old WT and MOR KO male mice was carefully dissected and weighed. The MOR-1 KO pancreas is significantly heavier than WT. B, Whole pancreas insulin content of the identical mice was measured by ELISA. MOR KO pancreas processes about 50% more insulin than WT pancreas. C, Because MOR mice are heavier and have larger pancreata, the absolute value of pancreatic insulin content was normalized to pancreatic weight. The mutant pancreas still contains significantly more insulin. D, The islet planar secretion area was calculated by measuring islet diameter of the isolated islet ex vivo. As shown, the mutant islets have significantly larger size (>30%). E, Islet sections (10 μm) from WT and MOR KO pancreas were hematoxylin and eosin stained. Figures represent the size difference between genotypes, with a 100-μm bar superimposed. Data are represented as means ± sem. **, P < 0.01; ***, P < 0.001.
MOR is expressed in Langerhans islets
Because there is controversy regarding islet MOR expression, we next reinvestigated the presence of MOR on the Langerhans islet. Islet cDNA was reverse-transcribed from deoxyribonuclease (DNase)-treated RNA samples extracted from isolated islets. As shown in Fig. 5, a pair of primers spanning the deleted exon 1 (23) of MOR-1 gene detected the 434-bp transcript segment in the two independent WT samples but not in the two mutant samples. These data provide the first direct evidence for the presence of the MOR1 mRNA transcript in the Langerhans islet and suggested a potential role for islet MOR to affect insulin secretion dynamics.
Figure 5.
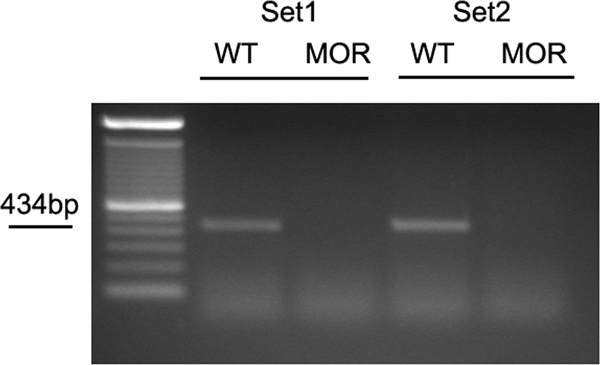
MOR mRNA transcript is detected in Langerhans islet. Islet cDNA was reverse transcribed from RNA extracted from isolated WT and mutant islets as described in Materials and Methods, followed by DNase treatment. Islet cDNA was then subject to a 35-cycle PCR amplification with a pair of primers that detects exon1 (the KO exon) on the mouse oprm1 (MOR) gene. A predicted 434-bp amplicon was present only in WT islet samples in two independent experiments. Each lane (WT or mutant) represents RNA extracted from three independent animals.
MOR regulates β-cell insulin secretion in situ by a post-KATP mechanism
Because MOR mRNA was demonstrated in Langerhans islets, we wanted to know whether MOR-1 is functionally involved in insulin secretion dynamics in situ upon glucose exposure. To evaluate this possibility, an in vitro glucose challenge system was employed with insulin release assessed in isolated WT and MOR islets by ELISA. Because there is an islet size difference between genotypes, five size-controlled islets from both genotypes were challenged with different stimuli in a 250-μl reaction system. Duplicate samples were analyzed for baseline glucose (2.8 mm), whereas at least triplicate samples were analyzed after tolbutamide (which stimulates insulin release by blocking the KATP channel on pancreatic β-cells) and high glucose (16.0 mm) exposure. As shown in Fig. 6, the baseline insulin secretion stimulated by 2.8 mm glucose does not differ between WT and MOR KO islets. In contrast, the mutant islets secreted 2-fold higher insulin after the 16.0 mm high glucose challenge compared with WT islets. Moreover, it is important to note that tolbutamide (275 μm) induced significantly more insulin secretion in the mutant islets at the baseline glucose concentration. Collectively, these in vitro data indicate that MOR inhibits insulin release in situ upon glucose exposure by a post-KATP channel mechanism and that a neuronal mechanism is ruled out because the isolated islets are devoid of any neuronal innervations.
Figure 6.
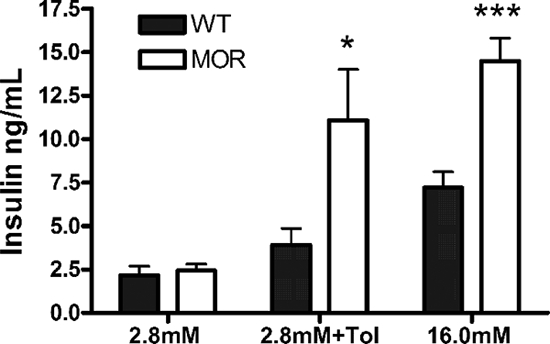
Glucose- and tolbutamide-mediated insulin secretion in isolated islets is enhanced in MOR KO mice. Islets isolated from age-matched (2–3 months old) male WT and MOR KO mice were challenged with 2.8 mm glucose Krebs-Ringer buffer, 2.8 mm Krebs-Ringer buffer plus 275 μm tolbutamide, and 16.0 mm glucose Krebs-Ringer buffer for 60 min, as described in Materials and Methods. The value indicates cumulative insulin secretion from batches of five islets in a 250-μl in vitro system within 1 h. After tolbutamide and high glucose (16.0 mm) challenge, the mutant islets secreted significantly more insulin than WT islets, although the basal insulin secretion (at 2.8 mm glucose) did not differ. Data are represented as means ± sem. *, P < 0.05; ***, P < 0.001.
Discussion
In this study, we first examined the in vivo phenotypes (body weight and glucose tolerance) that might be altered by an insulin secretion change accompanying MOR-1 mutation. We observed a consistent, significant, and early-onset approximately 10% body weight gain in both sexes of MOR-1 KO mice, which is contributed by a large increase (73% by weight) in fat tissue and a relatively small increase in lean tissue (11%). We suggest that the pronounced body weight gain in MOR KO mice may be related to the general growth-stimulatory effect of insulin. Fat tissue takes up glucose for lipid production, a process mediated by the insulin receptor-glucose transporter 4 pathway. Moreover, insulin stimulates hepatic lipogenesis as well as fat cell lipid absorption, which in turn also boosts adipose tissue formation. Thus, a higher insulin level could result in greater glucose uptake by fat tissue, which would alter the lipid metabolism and adipogenesis (17). Muscle, which comprises much of the lean tissue mass, is another major insulin target for peripheral glucose uptake. In both human and bovine models, it has been shown that increased insulin sensitivity could lead to enhanced muscle mass (24,25). It is thus conceivable that higher insulin secretion in MOR KO mice could directly induce greater lean tissue gain as well as fat tissue gain. It should again be noted that MOR deletion causes a significant percent increase in whole-body fat composition and a significant decrease in lean composition. Therefore, the promoting effect of MOR mutation is preferentially on fat rather than lean tissue. Consistent with this observation, the growth-retarded insulin receptor KO mice also exhibit decreased adipose tissue before neonatal lethality (26). In addition, higher insulin secretion dynamics are positively correlated with obesity in human (27), rodent (28), and a nonmammalian avian model (29). The early postnatal effect of MOR mutation on body weight demonstrated here is also consistent with the early postnatal ages in which insulin system disruption has effects in mice (30). Taken together, these comparisons all suggest that the higher body weight gain in MOR-1 KO mice is, at least in part, a consequence of insulin hypersecretion, which then alters adipogenesis in fat cells but also leads to a more limited gain in lean tissue. Because MOR-1 is expressed in the hypothalamus, the regulating center for feeding and metabolism, as well as multiple other brain regions, a centrally mediated alteration affecting metabolism and body composition cannot be ruled out.
Consistent with the enhanced glucose uptake and the prior indications that the opioid system regulates the endocrine function of β-cells (2,3,4), we found that MOR-1 KO congenic mice displayed significantly higher plasma insulin levels than WT mice within the first 10 min after a bolus glucose injection, which corresponds to the first phase of insulin release (31). This increase was substantially greater than the relatively mild elevation of serum insulin in C57BL/6J WT mice after glucose administration (32), which we also observed. The unaltered baseline insulin level between WT and MOR-1 KO mice is consistent with the indistinguishable baseline glucose level seen in WT and MOR-1 KO mice. One question that arises from the insulin hypersecretion in the MOR KO mice is whether it reflects a predisease sign/predisposition to type II diabetes. Various studies have shown that enhanced insulin secretion could be a secondary adjustment to decreased peripheral insulin sensitivity, which eventually leads to islet failure that further exacerbates this disease (22). However, because we did not detect a difference in insulin sensitivity, the insulin hypersecretion appears less likely to be a compensatory response to impaired insulin sensitivity. Moreover, compared with age-matched WT mice, the mutant mice are euglycemic even up to 1 yr of age. Therefore, unlike onset of some forms of type II diabetes, insulin hypersecretion in MOR-1 KO mice does not appear to be a sign of diabetes predisposition.
Detection of endogenous opioid peptide expression in the Langerhans islet and peptide release after glucose stimulation (7,33,34) render the endocrine pancreas a potential candidate for MOR regulation. However, despite the indirect evidence of opioid binding sites (5,7), there is no prior study providing direct evidence of MOR presence in Langerhans islets at either the mRNA or protein level. We here showed that MOR is indeed expressed in the islet although at a relatively low level, which requires islets to be isolated before RNA isolation and 35 cycles of PCR. Higher PCR cycle numbers beyond 30 have been required to detect low abundance but physiologically relevant expression (35,36). The in vivo phenotypes and islet MOR expression led us to propose that MOR plays a role in insulin secretion in situ. This possibility was confirmed by the in vitro 16.0 mm glucose challenge, which demonstrated that the mutant islets secrete twice the amount of insulin compared with WT. Therefore, the in situ regulation by islet-derived MOR on β-cell insulin secretion is more likely to be independent of nerve innervations and global endocrine parameters. Notably, the unaltered in vitro baseline secretion at 2.8 mm glucose is again compatible with the similar baseline glucose and insulin levels between MOR-1 KO and WT mice in the in vivo studies, which indicates that contribution of MOR to insulin secretion is glucose dependent.
In a parallel in vitro challenge experiment, we observed that tolbutamide, an ATP-sensitive K+ channel inhibitor, also enhances insulin secretion in the mutant islets compared with WT. The KATP channel is an intermediate component connecting glucose metabolism and electrical activity in the β-cell stimulus-secretion pathway (37). As observed in vivo, insulin hypersecretion in MOR-1 KO mice was a rapid and early-onset process, corresponding to the first phase of glucose-induced insulin release (31), which is thought to be KATP channel dependent (37). Interestingly, in the central nervous system, the MOR-1 receptor is known to be directly coupled to KATP channel in nociceptor neurons, activation of which produces hyperpolarization (38). Indeed, analgesic responses of certain opioids are mediated by KATP channel activity (39). It is therefore reasonable to predict that opioids and the KATP channel could interact in other electronically excitable tissues in which both are present. Thus, the KATP channel could be a candidate target of MOR in the islet. On the other hand, unlike the KATP channel (whose action inhibits the action potential of β-cells), activity of the voltage-dependent K+ channel (Kv family) potentiates the duration of the action potential (37) by delaying repolarization. It has previously been shown that Kv channel activity and insulin secretion by β-cells can be regulated by another G protein-coupled receptor system, the glucagon-like peptide 1 (GLP-1) (40). Therefore, it is also possible that MOR regulates this process at the repolarization stage.
In addition to the changes in islet insulin secretion dynamics, we also demonstrated higher insulin content in the whole pancreas of MOR-1 KO mice. This increase could not be attributed to the larger body/pancreas size because the difference is still pronounced after normalization. Subsequently, we showed that this difference may be at least in part due to an increase in islet mass. The MOR system has been reported to inhibit neurogenesis in the central nervous system (41) and suppress immunocyte proliferation (42). Importantly, several lines of evidence demonstrated that β-cell progenitors exist in adult pancreas (43). Together, these findings suggest a novel proliferation-inhibiting role of MOR in the endocrine pancreas, which could occur in either the developing embryo or at a later stage to affect the adult β-cell turnover. Therefore, we believe that the insulin hypersecretion seen in MOR-1 KO mice in vivo reflects combined contribution from 1) enhanced insulin secretion dynamics and 2) increased β-cell mass due to larger islets.
The enhanced adult male body weight (significant from postnatal wk 16–50) in an independent strain of MOR-1 KO mice has been reported (15), which is also accompanied by comparable food intake (which we have also observed in this MOR-1 KO strain; Ansonoff, M. A., and J. E. Pintar, in preparation) and unaltered glucose tolerance. Here we have shown that the enhanced body weight in both genders of the mutant mice was an early event observed even immediately after birth, with subsequent development of a highly reproducible, significantly enhanced glucose tolerance. Several possible explanations could account for the different phenotypes, including the slightly different strategy used in initial KO production, differences in the experimental design (16 h fast used here vs. an apparently more modest at least 8 h fast), and the animal environment. Interestingly, quantitative PCR experiments of hypothalamus expression of NPY did not mimic the up-regulation in NPY seen in the other strain. The reason for these phenotypic differences could be interesting and important.
In conclusion, in these in vivo and ex vivo studies, we show that MOR is involved in glucose homeostasis in C57BL/6J mice by negatively regulating glucose tolerance, which is achieved by inhibiting insulin secretion from the β-cell in situ and reducing β-cell mass. The body weight gain of MOR-1 KO mice is significantly higher than WT mice, which is consistent with the stimulatory effect of insulin hypersecretion. These endocrine-related phenotypes of MOR-1 KO mice support the notion that the opioid system mediates specific endocrine functions. Thus, from a clinical perspective, understanding the relationship more clearly between opioid administration and glucose homeostasis should be beneficial to the clinical management of drug abusers and diabetic patients on analgesic therapy. In addition, the potential to interrupt opioid receptor regulation of insulin secretion may provide a new opportunity to develop a novel tissue-specific therapy for type II diabetes.
Materials and Methods
Mice
To generate MOR-1 KO mice on a C57BL/6J congenic background, WT and MOR-1 KO mice on the 129S6/C57BL/6J mixed background (23) were backcrossed onto C57BL/6J mice for at least 15 generations by heterozygous mating with C57BL/6J WT mice. Animals were maintained in the accredited University of Medicine and Dentistry of New Jersey vivarium facility with 25 C temperature control and a 12-h light, 12-h dark cycle (lights on at 0700 h). Two- to three-month-old age-matched male WT and MOR-1 KO mice were used for all of the studies except for the body weight measurements, in which both genders were used.
Measurement of postnatal weights and body composition
C57BL/6J congenic WT and MOR-1 KO mice were obtained from homozygous mating to determine any effect of MOR on body weight gain. Litters from both genotypes were weighed from d 0 (day of birth) to d 180. All animals were maintained on a normal diet and group housed with fewer than five mice per cage. Age-matched 9-wk-old male WT and MOR KO mice were used for body weight composition studies, determined by qNMR (44). The qNMR study was carried out at the Genome Research Institute, University of Cincinnati using an EchoMRI (QMR; EchoMRI, Houston, TX).
Glucose tolerance test
Mice were fasted for 16 h (1800–1000 h) followed by an ip injection of 2 g/kg d-glucose (Dextrose; Fisher Chemical, Fair Lawn, NJ). Whole blood was obtained from the tail vein at the indicated time points (0, 15, 30, 60, 90, and 120 min) after the glucose injection. Blood glucose levels were measured using an Accu-Chek blood glucose monitor (Roche, Indianapolis, IN).
Insulin tolerance test
Mice were fasted for 6 h (1000–1600 h) followed by an ip injection of 0.75 U/kg bovine insulin (Sigma, St. Louis, MO). Whole blood was obtained from the tail vein at the indicated time points (0, 15, 30, and 60 min) after the glucose injection. Blood glucose levels were measured using an Accu-Chek blood glucose monitor (Roche).
Plasma insulin level measurements
To measure insulin levels during glucose tolerance test, mice were fasted for 16 h (1800–1000 h) followed by an ip injection of glucose (2 g/kg body weight). Blood was collected in EDTA-containing tubes and then centrifuged to obtain plasma. Insulin levels were measured at 0, 2, 5, 10, 15, and 30 min using an insulin ELISA kit (Linco, St. Charles, MO), following the protocol provided by the manufacturer. The same kit was used for insulin measurement for the following studies.
Islet isolation and in vitro insulin secretion assay
Islets were isolated from cervically dislocated mice by an initial injection of 2 ml 0.5 mg/ml type XI collagenase (C9407; Sigma) dissolved in Hanks’ balanced salt solution (HBSS) (1×; Mediatech, Herndon, VA) into the pancreatic duct followed by a digestion with the same enzyme solution at 37 C for 15–20 min. The digested tissue was then washed twice with ice-cold HBSS and passed through an 850-μm sieve and redistributed in 5 ml HBSS. Islets were purified by adding 5 ml Ficoll-Paque PLUS solution (Amersham, Uppsala, Sweden) to the bottom of the tube and then centrifuged at level 7 of an IEC clinical centrifuge for 10 min. The isolated islets were washed again with ice-cold HBSS and transferred to RPMI 1640 medium supplemented with 10% fetal bovine serum, 100 U/ml penicillin, and 100 μg/ml streptomycin for overnight recover culture in a 37 C/5% CO2 incubator. In the next morning, islets were transferred to Krebs-Ringer bicarbonate buffer [111 mm NaCl, 4.8 mm KCl, 2.3 mm CaCl2, 1.2 mm MgSO4, 1.2 mm KH2PO4, 25 mm NaHCO3 (pH 7.4)] supplemented with 10 mm HEPES, 2.8 mm glucose, 0.2% BSA fraction V (Sigma) and preincubated for 90 min at 37 C with 5% CO2. Triplicate batches (duplicate for 2.8 mm) of five size-matched islets were transferred to a 96-well plate, with each well containing 250 μl experimental medium (Krebs-Ringer bicarbonate buffer with HEPES and BSA) supplemented with different secretagogues, including 2.8 mm glucose alone, 2.8 mm glucose with 275 μm tolbutamide (Sigma), or 16.0 mm glucose alone, and incubated at 37 C/5% CO2 for 1 h. The supernatant medium was collected after the incubation and stored at −80 C for insulin measurement.
Whole pancreas insulin content
The pancreas was carefully dissected from cervically dislocated mice, measured for weight, snipped into small pieces, and homogenized in 25 ml acid-ethanol (0.18 n HCl-70% ethanol) with a polytron probe. The samples were then sonicated for 2 min and centrifuged at 10,000 × g for 10 min. The supernatant was diluted 100×, and 10 μl was used for the insulin ELISA.
Langerhans islet planar area measurement and histology
Islets from WT and MOR KO mice were isolated into HBSS as described above. A picture for each group of islets isolated from a single animal was photographed using a Polaroid DMC Ie stereoscope-mounted digital camera at 1600 × 1200 resolution with a 1000-μm bar superimposed. The diameter of islets with normal morphology was measured using the ImageJ software (National Institutes of Health, Bethesda, MD), and the planar secretion area of each islet was calculated by the formula S = πd2/4. For histology studies, 10-μm sections were acquired after paraffin embedding and hematoxylin and eosin staining.
Islet cDNA RT-PCR
Islets from WT and MOR KO mice were isolated into HBSS as described above. RNA for one determination was extracted from islets pooled from three animals using the QIAGEN RNeasy kit and simultaneously on-column DNase treated by the QIAGEN ribonuclease-free DNase set (QIAGEN, Valencia, CA). For cDNA synthesis, 2 μg RNA was reverse transcribed with random hexamer primer and 200 U Superscript II enzyme (Invitrogen, Carlsbad, CA) in a 100-μl reaction system. Five microliters of the cDNA product was amplified by conventional PCR (94 C for 30 sec, 58 for C 45 sec, and 72 C for 60 sec for 35 cycles; primers for MOR-1 exon1: 5′-ACGCTCAGACGTTCCATTCT-3′ and 5′-TCCAAAGAGGCCCACTACAC-3′) with a PCR MasterAmp premix (vial H; EPICENTRE Biotechnologies, Madison, WI).
Statistics
The unpaired two-tailed t test embedded in the Microsoft Excel software was used for statistical analysis for all of the studies presented. A P value <0.05 was considered as statistically significant with other levels of significance noted as follows. Data are presented as mean ± sem.
Acknowledgments
We thank the Cincinnati Mouse Metabolic Phoenotyping Center (MMPC) DK59630 for providing the qNMR service for the body composition studies.
Footnotes
This work was supported by National Institutes of Health Grants DA-09040 and DA-15237.
Disclosure Summary: The authors have nothing to disclose.
First Published Online February 12, 2009
Abbreviations: DNase, Deoxyribonuclease; HBSS, Hanks’ balanced salt solution; KO, knockout; MOR-1, μ-opioid receptor; qNMR, quantitative nuclear magnetic resonance; WT, wild type.
References
- Krentz AJ, Bailey CJ 2005 Oral antidiabetic agents: current role in type 2 diabetes mellitus. Drugs 65:385–411 [DOI] [PubMed] [Google Scholar]
- Giugliano D, Torella R, Lefèbvre PJ, D'Onofrio F 1988 Opioid peptides and metabolic regulation. Diabetologia 31:3–15 [DOI] [PubMed] [Google Scholar]
- Schleicher RL 1989 β-Endorphin inhibits insulin secretion from isolated pancreatic islets. Endocrinology 124:1254–1258 [DOI] [PubMed] [Google Scholar]
- García-Barrado MJ, Iglesias-Osma MC, Rodríguez R, Martín M, Moratinos J 2002 Role of μ-opioid receptors in insulin release in the presence of inhibitory and excitatory secretagogues. Eur J Pharmacol 448:95–104 [DOI] [PubMed] [Google Scholar]
- Zhang M, Zheng M, Schleicher RL 1994 Autoradiographic localization of β-endorphin binding in the pancreas. Mol Cell Neurosci 5:684–690 [DOI] [PubMed] [Google Scholar]
- von Dorsche HH, Fält K, Zühlke H 1989 Immunohistochemical investigations of β-endorphin in human pancreatic islets. Acta Histochem 85:131–134 [DOI] [PubMed] [Google Scholar]
- Khawaja XZ, Green IC, Thorpe JR, Titheradge MA 1990 The occurrence and receptor specificity of endogenous opioid peptides within the pancreas and liver of the rat. Comparison with brain. Biochem J 267:233–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatouros IG, Goldfarb AH, Jamurtas AZ, Angelopoulos TJ, Gao J 1997 β-Endorphin infusion alters pancreatic hormone and glucose levels during exercise in rats. Eur J Appl Physiol Occup Physiol 76:203–208 [DOI] [PubMed] [Google Scholar]
- Giugliano D, Cozzolino D, Ceriello A, Salvatore T, Paolisso G, Torella R 1989 β-Endorphin and islet hormone release in humans: evidence for interference with cAMP. Am J Physiol 257:E361–E366 [DOI] [PubMed] [Google Scholar]
- Reid RL, Yen SS 1981 β-Endorphin stimulates the secretion of insulin and glucagon in humans. J Clin Endocrinol Metab 52:592–594 [DOI] [PubMed] [Google Scholar]
- Reid RL, Sandler JA, Yen SS 1984 β-Endorphin stimulates the secretion of insulin and glucagon in diabetes mellitus. Metabolism 33:197–199 [DOI] [PubMed] [Google Scholar]
- Giugliano D, Cozzolino D, Salvatore T, Ceriello A, Giunta R, Torella R, D'Onofrio F 1987 β-Endorphin and islet hormone release in type-2 diabetes mellitus the effects of normoglycemia, enkephalin, naloxone and somatostatin. Diabetes Metab 13:618–624 [PubMed] [Google Scholar]
- Khawaja XZ, Green IC 1991 Dual action of β-endorphin on insulin release in genetically obese and lean mice. Peptides 12:227–233 [DOI] [PubMed] [Google Scholar]
- Ahrén B 1989 Effects of β-endorphin, met-enkephalin, and dynorphin A on basal and stimulated insulin secretion in the mouse. Int J Pancreatol 5:165–178 [DOI] [PubMed] [Google Scholar]
- Han W, Hata H, Imbe H, Liu QR, Takamatsu Y, Koizumi M, Murphy NP, Senba E, Uhl GR, Sora I, Ikeda K 2006 Increased body weight in mice lacking μ-opioid receptors. Neuroreport 17:941–944 [DOI] [PubMed] [Google Scholar]
- Hadjimarkou MM, Singh A, Kandov Y, Israel Y, Pan YX, Rossi GC, Pasternak GW, Bodnar RJ 2004 Opioid receptor involvement in food deprivation-induced feeding: evaluation of selective antagonist and antisense oligodeoxynucleotide probe effects in mice and rats. J Pharmacol Exp Ther 311:1188–1202 [DOI] [PubMed] [Google Scholar]
- Rosen ED, Spiegelman BM 2000 Molecular regulation of adipogenesis. Annu Rev Cell Dev Biol 16:145–171 [DOI] [PubMed] [Google Scholar]
- Zhu Y, King MA, Schuller AG, Nitsche JF, Reidl M, Elde RP, Unterwald E, Pasternak GW, Pintar JE 1999 Retention of supraspinal δ-like analgesia and loss of morphine tolerance in δ-opioid receptor knockout mice. Neuron 24:243–252 [DOI] [PubMed] [Google Scholar]
- Ansonoff MA, Zhang J, Czyzyk T, Rothman RB, Stewart J, Xu H, Zjwiony J, Siebert DJ, Yang F, Roth BL, Pintar JE 2006 Antinociceptive and hypothermic effects of Salvinorin A are abolished in a novel strain of κ-opioid receptor-1 knockout mice. J Pharmacol Exp Ther 318:641–648 [DOI] [PubMed] [Google Scholar]
- Clarke S, Chen Z, Hsu MS, Pintar J, Hill R, Kitchen I 2001 Quantitative autoradiographic mapping of the ORL1, μ-, δ- and κ-receptors in the brains of knockout mice lacking the ORL1 receptor gene. Brain Res 906:13–24 [DOI] [PubMed] [Google Scholar]
- Nitsche JF, Schuller AG, King MA, Zengh M, Pasternak GW, Pintar JE 2002 Genetic dissociation of opiate tolerance and physical dependence in δ-opioid receptor-1 and preproenkephalin knock-out mice. J Neurosci 22:10906–10913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerich JE 1998 The genetic basis of type 2 diabetes mellitus: impaired insulin secretion versus impaired insulin sensitivity. Endocr Rev 19:491–503 [DOI] [PubMed] [Google Scholar]
- Schuller AG, King MA, Zhang J, Bolan E, Pan YX, Morgan DJ, Chang A, Czick ME, Unterwald EM, Pasternak GW, Pintar JE 1999 Retention of heroin and morphine-6 β-glucuronide analgesia in a new line of mice lacking exon 1 of MOR-1. Nat Neurosci 2:151–156 [DOI] [PubMed] [Google Scholar]
- Grobet L, Martin LJ, Poncelet D, Pirottin D, Brouwers B, Riquet J, Schoeberlein A, Dunner S, Ménissier F, Massabanda J, Fries R, Hanset R, Georges M 1997 A deletion in the bovine myostatin gene causes the double-muscled phenotype in cattle. Nat Genet 17:71–74 [DOI] [PubMed] [Google Scholar]
- Schuelke M, Wagner KR, Stolz LE, Hubner C, Riebel T, Kömen W, Braun T, Tobin JF, Lee SJ 2004 Myostatin mutation associated with gross muscle hypertrophy in a child. N Engl J Med 350:2682–2688 [DOI] [PubMed] [Google Scholar]
- Cinti S, Eberbach S, Castellucci M, Accili D 1998 Lack of insulin receptors affects the formation of white adipose tissue in mice. A morphometric and ultrastructural analysis. Diabetologia 41:171–177 [DOI] [PubMed] [Google Scholar]
- Lustig RH, Sen S, Soberman JE, Velasquez-Mieyer PA 2004 Obesity, leptin resistance, and the effects of insulin reduction. Int J Obes Relat Metab Disord 28:1344–1348 [DOI] [PubMed] [Google Scholar]
- Rohner-Jeanrenaud F, Jeanrenaud B 1985 Involvement of the cholinergic system in insulin and glucagon oversecretion of genetic preobesity. Endocrinology 116:830–834 [DOI] [PubMed] [Google Scholar]
- Simon J, Leclercq B 1985 Fat and lean chickens: prefattening period and in vivo sensitivity to insulin, atropine, and propranolol. Am J Physiol 249:R393–R401 [DOI] [PubMed] [Google Scholar]
- Accili D 1997 Insulin receptor knock-out mice. Trends Endocrinol Metab 8:101–104 [DOI] [PubMed] [Google Scholar]
- Zawalich WS, Yamazaki H, Zawalich KC 2008 Biphasic insulin secretion from freshly isolated or cultured, perifused rodent islets: comparative studies with rats and mice. Metabolism 57:30–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kooptiwut S, Zraika S, Thorburn AW, Dunlop ME, Darwiche R, Kay TW, Proietto J, Andrikopoulos S 2002 Comparison of insulin secretory function in two mouse models with different susceptibility to β-cell failure. Endocrinology 143:2085–2092 [DOI] [PubMed] [Google Scholar]
- Cetin Y 1990 Immunohistochemistry of opioid peptides in the guinea pig endocrine pancreas. Cell Tissue Res 259:313–319 [DOI] [PubMed] [Google Scholar]
- Josefsen K, Buschard K, Sorensen LR, Wøllike M, Ekman R, Birkenbach M 1998 Glucose stimulation of pancreatic β-cell lines induces expression and secretion of dynorphin. Endocrinology 139:4329–4336 [DOI] [PubMed] [Google Scholar]
- Agirregoitia E, Valdivia A, Carracedo A, Casis L, Gil J, Subiran N, Ochoa C, Irazusta J 2006 Expression and localization of δ-, κ-, and μ-opioid receptors in human spermatozoa and implications for sperm motility. J Clin Endocrinol Metab 91:4969–4975 [DOI] [PubMed] [Google Scholar]
- Springer J, McGregor GP, Fink L, Fischer A 2003 Alternative splicing in single cells dissected from complex tissues: separate expression of prepro-tachykinin A mRNA splice variants in sensory neurones. J Neurochem 85:882–888 [DOI] [PubMed] [Google Scholar]
- MacDonald PE, Wheeler MB 2003 Voltage-dependent K+ channels in pancreatic β-cells: role, regulation and potential as therapeutic targets. Diabetologia 46:1046–1062 [DOI] [PubMed] [Google Scholar]
- Christie MJ 1991 Mechanisms of opioid actions on neurons of the locus coeruleus. Prog Brain Res 88:197–205 [DOI] [PubMed] [Google Scholar]
- Pacheco DF, Duarte ID 2005 δ-Opioid receptor agonist SNC80 induces peripheral antinociception via activation of ATP-sensitive K+ channels. Eur J Pharmacol 512:23–28 [DOI] [PubMed] [Google Scholar]
- MacDonald PE, Salapatek AM, Wheeler MB 2002 Glucagon-like peptide-1 receptor activation antagonizes voltage-dependent repolarizing K+ currents in β-cells: a possible glucose-dependent insulinotropic mechanism. Diabetes 51(Suppl 3):S443–S447 [DOI] [PubMed] [Google Scholar]
- Eisch AJ, Barrot M, Schad CA, Self DW, Nestler EJ 2000 Opiates inhibit neurogenesis in the adult rat hippocampus. Proc Natl Acad Sci USA 97:7579–7584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenstein TK, Hilburger ME 1998 Opioid modulation of immune responses: effects on phagocyte and lymphoid cell populations. J Neuroimmunol 83:36–44 [DOI] [PubMed] [Google Scholar]
- Xu X, D'Hoker J, Stangé G, Bonné S, De Leu N, Xiao X, Van de Casteele M, Mellitzer G, Ling Z, Pipeleers D, Bouwens L, Scharfmann R, Gradwohl G, Heimberg H 2008 β-Cells can be generated from endogenous progenitors in injured adult mouse pancreas. Cell 132:197–207 [DOI] [PubMed] [Google Scholar]
- Tinsley FC, Taicher GZ, Heiman ML 2004 Evaluation of a quantitative magnetic resonance method for mouse whole body composition analysis. Obes Res 12:150–160 [DOI] [PubMed] [Google Scholar]