

Abstract
As genomic sequences become easier to acquire, shotgun proteomics will play an increasingly important role in genome annotation. With proteomics, researchers can confirm and revise existing genome annotations and discover completely new genes. Proteomic-based de novo gene discovery should be especially useful for sets of genes with characteristics that make them difficult to predict with gene-finding algorithms. Here, we report the proteomic discovery of 19 previously unannotated genes encoding seminal fluid proteins (Sfps) that are transferred from males to females during mating in Drosophila. Using bioinformatics, we detected putative orthologs of these genes, as well as 19 others detected by the same method in a previous study, across several related species. Gene expression analysis revealed that nearly all predicted orthologs are transcribed and that most are expressed in a male-specific or male-biased manner. We suggest several reasons why these genes escaped computational prediction. Like annotated Sfps, many of these new proteins show a pattern of adaptive evolution, consistent with their potential role in influencing male sperm competitive ability. However, in contrast to annotated Sfps, these new genes are shorter, have a higher rate of nonsynonymous substitution, and have a markedly lower GC content in coding regions. Our data demonstrate the utility of applying proteomic gene discovery methods to a specific biological process and provide a more complete picture of the molecules that are critical to reproductive success in Drosophila.
Advances in DNA sequencing technology have made it cheaper and easier to determine the complete genome sequences of a variety of organisms. However, a fully sequenced genome is only a starting point for understanding an organism's biology. One critical, subsequent step is to annotate the complete sets of proteins used by the organism in specific biological processes. The first pass at genome annotation often comes from gene prediction algorithms, which scan DNA sequences for features of genes (such as open-reading frames and GC content) and examine cross-species conservation to infer functionally important regions (Burge and Karlin 1997; Brent and Guigo 2004). These computational methods have identified many new genes, but they remain imperfect and cannot provide experimental validation of their predicted gene models. Mass spectrometry (MS)-based proteomic methods can be used to refine computational gene annotations and identify novel genes (Ansong et al. 2008; Gupta et al. 2008). Mass spectra are typically searched against a database of predicted proteins; the peptides that are identified confirm and refine gene models derived from computational work. When these searches are expanded to an entire translated genome, identified peptides can reveal novel splice variants, unexpected shifts in a transcript's reading frame, or genes that were completely unknown. Such methods have improved existing gene annotations and discovered new genes in a range of organisms, including humans, plants, flies, nematodes, and algae (McGowan et al. 2004; Brunner et al. 2007; Tanner et al. 2007; Baerenfaller et al. 2008; May et al. 2008; Merrihew et al. 2008).
In addition to improving gene annotations on the whole-organism level, MS can also be used to identify new genes in a specific tissue or biological process. One such class of proteins that is evolutionarily important and should particularly benefit from de novo gene discovery is the seminal fluid proteins (Sfps) of Drosophila. Sfps are soluble proteins that are secreted into seminal fluid from specialized organs in the male reproductive tract (primarily the accessory glands) and transferred with sperm to females during mating. Evolutionarily, Sfps are important because they are a key factor in male reproductive success. Genetic knockout studies show that specific Sfps influence mating behaviors, such as the storage of sperm inside the female and the propensity of a female to remate with subsequent suitors (for review, see Ravi Ram and Wolfner 2007a). Sfps also evolve under positive selection across several species (Swanson et al. 2001; Mueller et al. 2005; Findlay et al. 2008; Wong et al. 2008b) and show patterns of polymorphism within populations that are consistent with recent selective sweeps (Begun et al. 2000; Begun and Lindfors 2005; Wagstaff and Begun 2005b, 2007). When outbred, polymorphic populations of Drosophila melanogaster males are allowed to evolve against a static female genotype, males show significant fitness increases relative to the initial population within 30–40 generations (Rice 1996). Furthermore, coding sequence variants of specific Sfps are associated with different sperm competitive abilities (Fiumera et al. 2005, 2007). Thus, Sfps play important reproductive roles. Additionally, while the specific genes encoding Drosophila Sfps are not conserved outside of insects, the classes of proteins found in Drosophila seminal fluid are the same as those found in diverse animal taxa, including rodents and primates (Swanson and Vacquier 2002; Clark and Swanson 2005; Clark et al. 2006; M.D. Dean, N.L. Clark, G.D. Findlay, R.C. Karn, X. Yi, W.J. Swanson, M.J. MacCoss, and M.W. Nachman, in prep.).
Proteomic discovery of novel Sfps is a useful technique precisely because of the evolutionary dynamics of these proteins. Indeed, a limitation to the comparative genomic approach to gene identification is that those genes that are not predicted are the most likely to have interesting evolutionary histories. As described above, many Sfps evolve rapidly between species, making them harder to detect by conservation. Furthermore, Sfps have a history of lineage-specific gene gains and losses (Holloway and Begun 2004; Wagstaff and Begun 2005a; Begun et al. 2006; Findlay et al. 2008), making it difficult or impossible to identify orthologs across species. Many Sfps are short and show less codon bias than other Drosophila genes (Begun et al. 2000; Swanson et al. 2001; Mueller et al. 2005), which could render de novo prediction more difficult. For these reasons, experimental identification of Sfps has been important. Previous studies predicted Sfps by sequencing expressed sequence tags (ESTs) from male accessory glands, which are the main secretory organs of the male reproductive tract (Wolfner et al. 1997; Swanson et al. 2001; Begun et al. 2006), or by performing MS on proteins isolated from male reproductive tracts (Walker et al. 2006). However, neither approach was able to determine with certainty which proteins are transferred by the male to the female. Transferred Sfps are the most likely to be important for male reproductive success and for modification of female post-mating behavior. We developed a proteomic method to identify transferred male Sfps in mated female Drosophila (Findlay et al. 2008). In addition to confirming the transfer of many predicted Sfps and identifying over 60 annotated proteins that were unknown to function in reproduction, we identified 19 previously unannotated genes by searching our MS data against a translation of the entire D. melanogaster genome. These newly identified genes were confirmed with rapid amplification of cDNA ends (RACE) and RT-PCR. Uncovering so many unannotated Sfps in a genome that has been extensively curated and for which many related species' genomes are available for comparative analysis (Drosophila 12 Genomes Consortium 2007; Stark et al. 2007) confirmed that the evolutionary dynamics of Sfps make this class of proteins difficult to predict computationally.
Here, we further explore the utility of proteomics to identify unannotated Sfps in the genomes of three Drosophila species and investigate why these proteins were not already annotated. We show that searching mass spectra against an entire translated genome is a reliable, robust method of identifying potential new genes by identifying and experimentally validating 19 additional Sfp genes across the three species. We then consider the larger set of 38 novel Sfps and use bioinformatics and expression analysis to show that, rather than being restricted to a single species, these genes are present and expressed across several species. These proteins show similar evolutionary dynamics to the annotated Sfps, including signatures of adaptive evolution and instances of gene duplication and divergence. However, the unannotated proteins differ in important ways from the annotated Sfps: On average they are shorter, have a higher rate of evolution, and show reduced GC content in coding regions. By identifying new genes and investigating the reasons for their lack of prior identification, we describe both general and gene-specific reasons why many Sfps have gone unannotated. Our work demonstrates how proteomics can improve genome annotations and provides a more complete, thoroughly validated set of transferred Sfps for which future functional studies should yield much insight.
Results
Proteomics reveals unannotated genes encoding transferred seminal fluid proteins
Unannotated Sfps comprise a substantial fraction of the transferred Sfps in D. melanogaster: In addition to detecting 138 annotated Sfps that were transferred at mating, an additional search based on just one experiment revealed 19 transferred proteins that were not previously known to exist and that lacked annotations in the genome (Findlay et al. 2008). We used the same proteomics strategy to identify additional unannotated proteins in D. melanogaster and in two related species, Drosophila simulans and Drosophila yakuba. We first performed mating experiments in which 15N-labeled females of each species were mated to unlabeled males. Soluble proteins were isolated from the reproductive tracts of mated females, digested with trypsin, and analyzed with MS. Spectra obtained for each species were filtered in two ways. First, we used a standard Sequest database search to identify those peptides that matched an annotated protein from the relevant species' genome, based on annotations available from FlyBase. (Because 15N labeling increases the masses of female-derived peptides, only male peptides are able to match the database.) Second, we used the Hardklör algorithm (Hoopmann et al. 2007) to predict which MS2 spectra were likely to have been derived from MS1 spectra that showed isotope distributions characteristic of 15N labeling (i.e., those likely to have come from female peptides). These likely female spectra (∼85% of all spectra) were removed from the data sets, and the remaining spectra were then searched against a six-reading-frame translation of the entire genome of the relevant species.
These six-frame searches identified dozens of open reading frames (ORFs) across the three species that represented candidate pieces of possible new Sfp genes (for summary statistics, see Table 1; for a complete list of all male-derived peptides, see Supplemental Table S1). For each species examined, more than half of the identified ORFs were verified to be pieces of new Sfps. This process resulted in identifying several completely new Sfps in each species (eight in D. melanogaster, three in D. simulans, and eight in D. yakuba). It also allowed substantial cross-species validation of the new genes, since in many instances a peptide identified in one species was found to correspond to a predicted ortholog of a new protein found in another species. These results suggest that searching mass spectra against a database consisting of a translated genome is a robust way to identify new genes, particularly in the context of Drosophila Sfps. The results further show that while these new genes have to date escaped annotation by the various Drosophila genome projects, many of these new genes have readily detectable orthologs in additional species (also see below).
Table 1.
Verification of ORFs identified by six-frame translation searches in three species

aVerification was occasionally not attempted if the peptide used to identify the ORF was short, repetitive, flanked closely by in-frame stop codons with no apparent intervening splicing sites, or mapped back to many regions of the genome. Verification was judged unsuccessful if two rounds of RACE primer design (in each direction) failed to produce a complete transcript.
Supplemental Tables S2–S4 show the new genes that were discovered in each species, as well as sequence- and structure-based efforts to determine the potential functional classes of the new proteins these genes encode. Most of the new Sfps show no significant identity to other known proteins and could not be assigned to any functional class. However, several Sfps share identity with protease inhibitors, a class of proteins commonly found in seminal fluid in drosophilids and other taxa (Clark and Swanson 2005; Findlay et al. 2008; M.D. Dean, N.L. Clark, G.D. Findlay, R.C. Karn, X. Yi, W.J. Swanson, M.J. MacCoss, and M.W. Nachman, in prep.). The D. simulans-specific SFP51D showed weak structural similarity to odorant binding proteins (Obps) in other insects. Also, one protein identified in D. melanogaster and predicted to be present in additional species, SFP24F, was predicted to be a C-type lectin. Lectins have been repeatedly detected as transferred Sfps and are known to influence the storage of sperm in females (see below). We also noted that several of the proteins identified in D. melanogaster were located in regions of the genome harboring other, annotated Sfps (Supplemental Table S2), consistent with previous findings (Findlay et al. 2008).
We used RACE and RT-PCR to validate ORFs and to discriminate between MS identifications that represented actual peptides and those that were false positives. To further support these new gene identifications, we also assessed the degree to which the MS experiments overlapped in the specific Sfps they identified. More than half of the Sfps (23/38) were identified in more than one biological replicate (out of six total: the five experiments across three species represented in Table 1, plus the previous experiment in D. melanogaster reported in Findlay et al. 2008), providing strong evidence that these Sfp genes encode translated products that are transferred at mating (Supplemental Table S5). Furthermore, seven of the 15 Sfps identified by only one MS experiment were predicted bioinformatically to be lineage specific or lineage restricted. Within each species, considerable overlap was observed in the proteins identified between the two biological replicates performed (Supplemental Fig. S1). Although many ORFs were detected with only one matching peptide (Table 1; Supplemental Table S5), this result is intuitive given the short lengths of the Sfps (see below), the complexity of the peptide mixture being analyzed (∼85% of which was derived from female proteins), and the fact that the ORF sequences used for peptide identification in many cases were imperfect matches to the mature gene structure (e.g., peptides encoded by intron-containing sequences could not be identified). Regardless of the number of peptides used for identification, however, the RACE validation step allowed for sensitive discrimination between accurate and incorrect peptide identifications.
Unannotated Sfps are conserved across several species
To investigate the evolutionary conservation of the Sfps, we used bioinformatic tools to search for orthologs in the five sequenced species of the melanogaster subgroup: D. melanogaster, D. simulans, Drosophila sechellia, D. yakuba, and Drosophila erecta. We confined our searches to these species because previous experience suggested that many Sfps are not readily found outside of this clade, due to their rapid evolution. Using BLAT and TBLASTN searches, we found that many unannotated Sfps had identifiable orthologs in other species. All 27 Sfps initially identified in D. melanogaster had an ortholog in D. sechellia, and 26 had an ortholog in D. simulans. (The one gene that was not found, Sfp78E, shows >93% identity to an unassembled region from D. simulans chromosome 3L, suggesting that this gene may be present; however, the 5′ end of the coding sequence could not be aligned, preventing positive identification of an ortholog.) Nearly three-fourths (20/27) of the D. melanogaster Sfps had an ortholog in D. yakuba, and nearly half (13/27) had an ortholog in D. erecta. Thus, although these genes were unpredicted, it was straightforward to identify orthologs in additional species, including D. erecta, which is estimated to have diverged from D. melanogaster ∼10 Mya. Through proteomics, we also discovered three new Sfps in D. simulans and eight new Sfps in D. yakuba. One of the three D. simulans Sfps had identifiable orthologs in all four other species, while four of the eight D. yakuba Sfps had identifiable orthologs in the other species. The results of the bioinformatic identification of orthologs for the 38 new Sfp genes discovered here and by Findlay et al. (2008) are shown in Supplemental Table S6.
Because most Sfp genes show male-specific expression patterns, we used RT-PCR to assay for sex-specific expression for each predicted ortholog in D. melanogaster, D. simulans, and D. yakuba. With one exception, all 38 of the genes were expressed in males of all species in which they are predicted (Sfp51E, discovered in D. melanogaster, was not detected in D. simulans). Nearly all the genes showed male-specific or male-biased expression, though expression levels of orthologs between species sometimes appeared variable, which plausibly could contribute to differences in seminal fluid composition between species (Fig. 1; additional examples of RT-PCR gels are shown in Supplemental Fig. S2; complete results are given in Supplemental Table S7). In nearly all cases in which a gene was expressed in all three species, its pattern of sex-specificity was consistent across the species. However, several Sfp genes in D. melanogaster show robust expression in both sexes (Sfp33A3, Sfp53D, and Sfp65A). Such proteins could have been missed by previous attempts to annotate Sfps, which required an Sfp gene to be expressed exclusively in male reproductive organs (Wolfner et al. 1997; Swanson et al. 2001). Therefore, one benefit to our method of gene discovery and annotation is that it relies on the transfer of a protein during mating, rather than expression pattern, to detect Sfps, ensuring that any protein detected may play a role in reproduction regardless of any other pleiotropic function(s).
Figure 1.

Expression analysis for new Sfps and their bioinformatically identified orthologs. RT-PCR was used to assay for expression of two Sfp genes, Sfp53E (top) and Sfp26Ad (middle), as well as a housekeeping control gene, RpL32 (bottom), which is expressed ubiquitously. Sfp53E is expressed exclusively in males of all three species assayed; female D. melanogaster and D. simulans show amplification of a larger than expected product, which could represent unspliced gene product. Sfp26Ad expression levels appeared variable between males of each species, and the pattern of expression is male-specific in D. simulans and D. yakuba and male-biased in D. melanogaster. Note that the D. yakuba Sfp53E and the D. melanogaster Sfp26Ad products are larger than the products from their orthologs in other species. This difference was caused by the use of distinct primer binding sites in each species; comparisons to the expected product sizes confirmed that proper splicing occurred in each of these two cases. Each image contains two ladder lanes with a 100-bp ladder; the smallest band in each lane is 100 bp. For additional RT-PCR data, see Supplemental Figure S2 and Supplemental Table S7.
We also examined the gene structure of each of the 38 new Sfp genes. Each gene was predicted to have the same structure of introns and exons across each species. (These predictions were upheld by the RT-PCR data, which showed spliced products in males in every case where expected.) The most common gene structure (found in 22/38 genes) was two coding exons and one intron, while 11 proteins were encoded by a single exon and five were encoded by three exons. As previously observed in D. melanogaster (Hong et al. 2006), intron lengths were short and tightly distributed (across-species mean ± SD: 58.4 ± 5.6 nucleotides) and did not differ significantly between the three species (one-way ANOVA, F(2,78) = 0.21, P = 0.81). Strikingly, nine of the 38 genes had first exons with fewer than 50 coding nucleotides. Indeed, two genes (Sfp56D and Sfp84E) had first exons whose coding region contained only the ATG initiation codon. These short first exons may make Sfp gene prediction especially complicated (see Discussion).
To investigate why individual Sfps may have escaped computational prediction, we focused on the 11 genes that were initially detected in D. simulans and D. yakuba, since these species' genomes are at earlier stages of annotation that are based more heavily on computational predictions. As shown in Supplemental Tables S3 and S4, there appear to be many reasons why Sfps might have gone unannotated, including short protein lengths, lineage specificity, locations in unassembled genomic regions, and short exon lengths. For example, Sfp56D is present in five species but was correctly predicted only in D. erecta. Through RACE, we identified the first exon of this gene in D. simulans as containing the 5′ UTR and the start codon. The coding sequence is then interrupted by an intron before continuing for 140 codons in the second exon. Comparisons of the D. simulans protein sequence with the putative orthologs in the four other species showed that the protein coding sequence is conserved, with 76% protein identity between D. simulans and D. erecta. However, because the start codon was separated from the rest of the coding sequence, this conservation was insufficient to consider the region a coding sequence. Another new protein discovered in D. simulans, SFP24B3, appears to be a tandem duplicate of the documented Sfp, Acp24A4. After using RACE to determine the gene structure of Sfp24B3, we judged the existing D. simulans annotation of the Acp24A4 gene likely to be incorrect (for the corrected annotation, see Supplemental Fig. S3).
Unannotated D. melanogaster Sfps show canonical signs of rapid evolution and gene duplication
Many reproductive proteins in Drosophila have evolved under positive Darwinian selection (Swanson et al. 2001, 2004; Mueller et al. 2005; Clark et al. 2006; Findlay et al. 2008). We thus examined the unannotated Sfps to determine whether these proteins have experienced similar selective pressures. For these analyses, we focused on the 27 unannotated Sfps that were discovered by MS in D. melanogaster. We first estimated the pairwise dN/dS ratio (ω) between orthologs of D. melanogaster and D. simulans (or D. sechellia for Sfp78E) as a conservative measure of the rate of evolution. Briefly, dN/dS measures the rate of protein coding gene evolution by comparing the rate at which nonsynonymous mutations occur at nonsynonymous sites to the rate at which synonymous mutations (which are assumed to be selectively neutral) occur at synonymous sites. As shown in Figure 2 , many new Sfps have elevated rates of evolution. Five of the 27 proteins have a pairwise estimate of ω > 1, and another five have ω > 0.5. In this conservative test, ω > 1 indicates positive selection, but previous work has shown that proteins with lower pairwise ω values can have specific sites under positive selection when additional sequences are analyzed with more sensitive methods (Yang et al. 2000; Swanson et al. 2003, 2004; Clark and Swanson 2005; Findlay et al. 2008). To this end, we tested each of the 27 proteins for specific sites under selection using all of the species (up to five) for which an ortholog was detected (see above). These methods detected positive selection on specific sites in 10 of the 27 proteins, seven of which remained significant after a strict Bonferroni correction for multiple statistical tests (Supplemental Table S8).
Figure 2.

Whole-gene, pairwise estimates of dN and dS values for 27 Sfps discovered in D. melanogaster and compared with orthologs in D. simulans. The solid line indicates ω = 1; the dashed line indicates ω = 0.5. All genes were also tested across additional species for specific sites evolving under positive selection (Supplemental Table S8).
One of the new proteins found to be under selection, SFP24F, was predicted to be a C-type lectin based on its sequence and predicted structural similarity to other lectin proteins. In D. melanogaster, four annotated lectins were identified previously as transferred Sfps, and genetic studies have shown that when males fail to produce any of three of these proteins, their sperm are stored less efficiently in female sperm storage organs after mating (Ravi Ram and Wolfner 2007b; Findlay et al. 2008; Wong et al. 2008a). More broadly, lectins are known to play an important role in reproduction and are often thought to mediate gametic interactions (Springer and Crespi 2007; Moy et al. 2008). We used homology modeling to predict the three-dimensional structure of the D. melanogaster SFP24F and to map the variable sites predicted from our evolutionary analysis to be under positive selection onto the structure (Fig. 3). Strikingly, some of the sites that are most confidently predicted to be under selection are located next to a region that in other C-type lectins has been implicated in determining the specificity of carbohydrate interactions (Iobst and Drickamer 1994). Given the general importance of lectins in reproduction and the finding that other D. melanogaster seminal fluid lectins play a role in sperm storage, it is possible that the positive selection observed for SFP24F has been driven by the pressure to improve the ability of male sperm to be stored in mated females.
Figure 3.
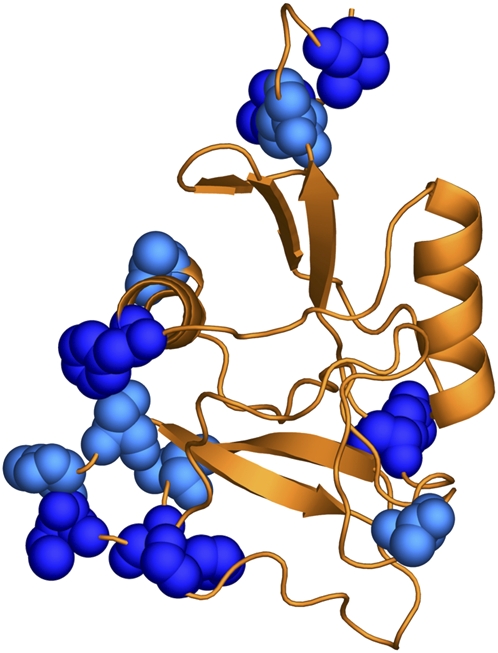
Structural model of the predicted C-type lectin SFP24F. The D. melanogaster SFP24F protein structure was predicted by PHYRE by threading the sequence onto a C-type lectin from mouse (PDB ID no. 2ox9). Sites indicated by space-filled molecules were predicted to have evolved under positive selection (dark blue sites, codeml model M8 BEB P > 0.95, light blue sites, P > 0.90). As oriented in the figure, the carbohydrate recognition domain is located at the bottom of the protein.
Gene duplication can play an important role in the evolution of new genes. We thus examined whether any of the new Sfps appeared to be gene duplicates, either of each other or of one of the annotated Sfps. Previously, we noted that SFP24BA and SFP24BB appeared to be ancient tandem duplicates (Findlay et al. 2008), showing a moderate level of sequence similarity (34% identical, 48% similar based on BLASTP) over most of the length of the protein. One new D. melanogaster protein, SFP24BC, lies just downstream from SFP24BA and SFP24BB. SFP24BC has the same gene structure (one short exon followed by a longer exon) and shows a similar degree of identity to its two adjacent proteins (45% identity to SFP24BA, 30% identity to SFP24BB). All three proteins show identity with Kunitz-type protease inhibitors through BLASTP and PHYRE (Bennett-Lovsey et al. 2008) searches. These data suggest that these three proteins are encoded by a gene cluster that most likely arose anciently, given the high degree of divergence between the three paralogs.
Two Sfps, CG17472 and CG31680, are tandem duplicates on chromosome 2R and are both transferred to females during mating (Findlay et al. 2008). CG17472 has evolved under positive selection and was duplicated in the lineage leading to D. simulans and D. sechellia. After the ancestral duplication of CG17472 and CG31680, there was a burst of positive selection on the lineage leading to the extant orthologs of CG31680. Our six-frame searches identified a third copy of this protein, SFP38D. The Sfp38D gene lies directly downstream from its paralogs, and orthologs were identified in D. simulans, D. sechellia, and D. yakuba. While the gene itself does not appear to have evolved under positive selection in these four species (Supplemental Table S8), a model of evolution that estimated a value of ω for each branch on a phylogeny of the three paralogs was a significantly better fit to the data than a model with a uniform ω across the phylogeny, suggesting rate heterogeneity across the evolutionary histories of these three proteins (χ2 = 53.34, 22 df, P = 0.0002; Fig. 4). In this heterogeneous model, the branch leading to the D. melanogaster, D. simulans, and D. sechellia orthologs of SFP38D had an ω estimate of 1.33, suggesting that a burst of diversifying selection could have acted on the protein after the ancestor to D. yakuba diverged from the ancestor to these three species. Notably, the rest of the phylogeny was consistent with that previously presented for the CG17472 and CG31680 genes by Findlay et al. (2008) and revealed other bursts of positive selection after other duplication events (Fig. 4). In the future, functional analyses and studies of polymorphism in natural populations should shed light on the role of this set of rapidly evolving, repeatedly duplicated genes.
Figure 4.

Phylogenetic analysis of SFP38D and its tandem duplicates. Phylogenetic tree of coding DNA sequences for SFP38D, its orthologs, and its paralogs from D. melanogaster (CG17472 and CG31680) and additional species. Branch color indicates the estimated ω rate for each branch. Red color indicates branches that are predicted to have experienced positive selection. Values of ω for red branches are, from top to bottom: 1.33, ∞, 2.02, ∞. Numbers under each node indicate percentage of bootstrap support for the phylogeny based on 1000 replicates. Sequences for SFP38D and its orthologs were obtained from BLAST and BLAT searches; other sequences represent gene models (numbered as indicated for each species) available from FlyBase. Note that though branches are colored so as to indicate which range of ω values they fell into, the model estimated a precise value for each branch.
Evolutionary proteomic studies of the D. melanogaster sperm proteome have found that several sperm proteins arose through retrotransposition-mediated gene duplication (Dorus et al. 2006, 2008). In searching for duplicates among the 38 new Sfps, we did not find evidence for this sort of duplication: All single-exon Sfps in this study did not have multiple-exon paralogs elsewhere in the genome, and paralogs of multi-exon Sfps shared intron structures (S. Schneider and W. Swanson, unpubl.). The region surrounding Sfp38D and its paralogs contains the remnants of many transposons. While the Sfp genes share a conserved intron and are thus unlikely to be retrogenes, it is possible that their duplication was caused by unequal crossing over mediated by the repetitive sequences.
Unannotated D. melanogaster Sfps are shorter, more rapidly evolving, and less GC-rich than annotated Sfps
The above results demonstrate that novel Sfps can be found by proteomics in several species and that for each new gene identified, one of several features might have contributed to the missing or incorrect annotation. We next considered whether there were features of the broader set of unannotated Sfps that made these proteins different from Sfps that had been annotated. We compared the set of 27 new Sfps detected by MS in D. melanogaster to the set of already-annotated, transferred D. melanogaster Sfps that was previously described (Findlay et al. 2008) across several categories. First, we compared the average lengths of the 27 new Sfps from this study and the previous work to the lengths of the 133 annotated Sfps (a set that excludes five sperm proteins). The unannotated proteins were significantly shorter than the annotated Sfps (median unannotated length, 87 residues; median annotated length, 263 residues; two-sample t-test on log-transformed lengths, t = 8.66, 57.1 df, P < 0.0001). Because of this difference, in subsequent analyses we compared the unannotated Sfps with only those annotated Sfps that were comparably short (200 residues or less).
Previous analyses (see above and Findlay et al. 2008) have shown that both sets of Sfps contain many proteins that have evolved under positive selection. We thus asked whether one set had experienced stronger positive selection by comparing the average dS and dN values between those proteins in each set for which whole-gene pairwise estimates of ω were made from a D. melanogaster–D. simulans comparison (26 unannotated proteins versus 41 annotated proteins with length <200). The sets were not significantly different in their rates of dS (annotated Sfps dS mean ± standard error, 0.149 ± 0.011; unannotated Sfps, 0.164 ± 0.013; t = 1.21, 54 df, P = 0.23). In contrast, the unannotated set of Sfps had a significantly higher mean dN value (annotated Sfps dN mean ± standard error, 0.054 ± 0.007; unannotated Sfps, 0.086 ± 0.013; t = 2.17, 54 df, P = 0.036; Fig. 5A), suggesting that nonsynonymous mutations are more frequently retained in the unannotated set of proteins. Thus, while the annotated set of Sfps has an average dN that is more than twice the average reported for a sample of nonaccessory gland proteins (Acps; 0.024 ± 0.002) (Swanson et al. 2001), the unannotated set of Sfps has a mean dN that is more than three times as high.
Figure 5.

Comparisons of annotated and unannotated transferred Sfps in D. melanogaster. (A) Mean values for annotated and unannotated genes for whole-gene, pairwise estimates of dN. Unannotated genes had a significantly higher rate of nonsynonymous substitution. Error bars, 1 SEM. (B) GC content for annotated and unannotated genes. G+Cc indicates overall GC content in coding regions; G+C2, GC content in second codon positions; and G+C3, GC content in third codon positions. For both panels, gray bars indicate annotated Sfps, and black bars indicate unannotated Sfps.
Short proteins and proteins that are rapidly evolving and/or poorly conserved across species are two classes of proteins that are less accurately predicted by gene prediction algorithms. Another feature of DNA sequences that is used by such programs is the GC content (e.g., Burge and Karlin 1997), with higher GC content expected in protein-coding regions. We investigated whether GC content in the unannotated (unpredicted) Sfps differed from GC content in the annotated set. We concatenated the coding DNA sequences for each protein in the unannotated set and in the annotated set and then calculated GC content and codon usage statistics for each set of genes. As shown in Figure 5B, overall GC content is considerably higher for the annotated Sfp coding sequences than it is for the unannotated coding sequences. This difference is especially pronounced at third codon position sites, where GC content is 15% higher in the annotated set.
This difference in GC content is associated with a difference in codon usage between the sets. We calculated relative synonymous codon usage (RSCU) values for each codon. RSCU is a measure of how frequently each individual codon is used, relative to all other codons than encode the same amino acid. RSCU is calculated by summing the total number of codons that encode each amino acid, then dividing the number of times a specific codon is used by the average number of times each codon encoding that amino acid is used. Values greater than 1 indicate a preferred codon, while values less than 1 indicate nonpreferred codons. Of the 59 codons that encode a redundantly encoded amino acid, 30 codons showed a difference in RSCU between the two sets of at least 0.20 (Supplemental Fig. S4). Among these 30 codons, those with a third position A or U were always more preferred in the unannotated Sfp set, while those with a third position C or G were always more preferred in the annotated Sfp set. Similarly, 21 of the 22 codons deemed preferred (Akashi 1995) have higher RSCU values in the annotated set (Supplemental Fig. S4). Taken together, these comparisons of the annotated and unannotated sets of Sfps reveal three features—gene length, rate of evolution, and GC content/codon usage biases—that would be expected to make automated gene prediction more difficult for the unannotated Sfps.
Discussion
In the past several years, proteomics has emerged as a powerful tool to improve genome annotations and aid in gene discovery (Ansong et al. 2008). Here, we combine MS and RACE to identify new proteins in a specific biological process, male reproduction in Drosophila. We have discovered 38 previously unannotated Sfps in three Drosophila species, many of which have readily identifiable orthologs in other species. RT-PCR expression analysis of these orthologs showed that new Sfp genes usually have male-biased expression patterns, consistent with a specific role in male reproduction. Like many reproductive proteins, several new Sfps show signs of adaptive evolution across species, consistent with their intimate involvement in male reproductive success. By investigating individual cases of incorrect or missed gene annotations, we have documented several reasons why these sorts of proteins might have been missed. Furthermore, compared with annotated Sfps (a set of proteins that has remarkable properties in its own right), these new proteins show elevated rates of nonsynonymous substitution and reduced GC content. These attributes suggest that in addition to specific instances of incorrect or missing gene predictions, the novel Sfps as a class have attributes that hinder computational gene identification efforts.
One strength of the MS approach for gene annotation is that in one step, it provides both evidence for an expressed protein (in the form of the peptide(s) identified) and a location in the genome that could encode that protein (by matching the identified peptide[s] back to the genome sequence). From this information, it is straightforward to perform RACE and RT-PCR to determine whether the identified peptide was a true positive, to determine the 5′ and 3′ UTR sequences, and to check for proper splicing and polyadenylation. While other groups have used MS and RT-PCR to refine gene models, it was critical in this study to perform an intermediate RACE step. Sfps almost always contain an N-terminal amino acid motif that targets the polypeptide for secretion and that is removed during the secretion process to form the mature protein. Because we detected Sfps in their mature forms in mated females, it would be impossible to identify these signal sequences, and by inference the 5′ end of the gene's coding region, by MS alone. For instance, because most signal sequences are ∼20 residues in length, it would be impossible to determine the full-length sequence of those proteins cited above that have short coding regions in their first exons. These considerations highlight the utility of focusing on a specific biological process when performing MS-based gene discovery. Because we knew that Sfps are secreted, we knew that the RACE procedure would be necessary to determine the 5′ end of each gene.
Previous experiments in D. yakuba and D. erecta have used EST sequencing of transcripts from male accessory glands to identify putative Sfps (Begun et al. 2006). Many of the putative Sfps identified in that work had characteristics that were similar to those that we have described here, particularly length and lineage-restriction. Indeed, several of the study's transcripts appear to correspond to the proteins we have discovered here. Given the attributes of their putative Sfps, Begun et al. (2006) proposed a model for the de novo evolution of Sfps. They reasoned that at any given time, a Drosophila genome should contain many DNA sequences that could encode short ORFs (defined as 30–100 residues). If the sequence upstream of these ORFs were mutated to create a promoter and if the ORF began with a sequence that encoded a secretion signal, then these ORFs would be expressed and their proteins secreted. In most instances, such expression would be deleterious, but occasionally these small, secreted peptides could perform a useful biological function and be retained by selection. To support this model, Begun et al. (2006) performed a computational analysis of the D. melanogaster genome and determined that it encoded at least 8000 ORFs of at least 40 residues that were predicted by SignalP to be secreted.
Our data allow us to assess and refine this model. (Note that while this model concerns the evolution of new genes from noncoding sequence, we have also observed the importance of tandem gene duplication as another source for new Sfps.) All 38 Sfps have predicted signal sequences (with SignalP hidden Markov model P > 0.95), confirming the importance of the signal sequence for Sfp function. As noted above, 11 of our 38 Sfps are encoded by a single-exon gene and thus represent the most straightforward case of an ORF encoding a new, secreted reproductive protein (though two of these proteins are shorter than 40 residues: SFP79B, 35 residues as translated; SFP96F, 33 residues). More complicated refinements to the model are required to explain the 27 Sfps that contain at least one intron. Of these Sfps, 16 have predicted signal sequences that are encoded entirely by their first exons. One explanation of the origin of these Sfps could be that short ORFs encoding signal sequences arose upstream of sequences of ∼60 nucleotides (the typical intron size in Drosophila) that contained many canonical features of an intron, particularly the 5′ and 3′ splice signals. The reading frame following the proto-intron would then encode the mature Sfp. If this mature peptide were not deleterious and instead performed a useful function in reproduction, selection would then act to increase or fine-tune expression levels and specificity, to improve the efficiency of splicing and/or secretion, and to alter the amino acid sequence for optimal function. It is easy to imagine a similar process working for a subsequent intron and exon for those Sfps (five in our data set) that have a third exon. The final class of Sfps contains those that have a signal sequence that is interrupted by the first intron. In principle, these proto-genes would face the same constraints and selective pressures as those described above, with the additional complication that an intron must be placed such that, upon its removal by splicing, a signal sequence emerges.
Another pattern in our data related to the GC content in Sfp coding regions. Mueller et al. (2005) cataloged GC content at third codon positions (GC3) for a set of 52 genes encoding Acps (the most prominent subset of Sfps) and compared this set to sets of testis-expressed genes and random genes with the same length distribution. In this comparison, D. melanogaster Acps had 51.2% GC3, testis-specific genes had 51.6% GC3, and random genes had 66.6% GC3. We found that the transferred, annotated Sfps (with lengths <200 residues) had a GC3 level (54.5%) similar to the Acps studied by Mueller et al. (2005), yet the newly discovered unannotated Sfps had a substantially reduced GC3 level (39.6%). Thus, while annotated Sfp genes themselves show reduced GC content relative to random genes, unannotated Sfps genes are even less GC-rich. One possible explanation for this pattern comes from the observation that unannotated Sfps have a significantly higher rate of nonsynonymous substitutions (dN) than annotated Sfps (Fig. 5A). Previous reports have observed a negative correlation in Drosophila between dN and optimal codon usage, suggesting that when selection for amino acid replacement changes is strong, the pressure to alter a protein's sequence may be far stronger than the pressure to optimize its translational efficiency by altering the nucleotides at silent sites (Betancourt and Presgraves 2002). Thus, many of the unannotated Sfps may be under such strong selection that amino acid-replacing changes occur so rapidly as to interfere with selection for improved translational efficiency.
We have described a proteomic method to identify new Sfps in several Drosophila species and have shown that while these Sfps were previously unannotated in the genomes of these species, orthologs were readily identified. It is now important to determine whether and how these new proteins affect reproductive success. For Sfps that are especially short in their mature forms (such as SFP33A2, SFP33A4, SFP36F, SFP70A5, SFP79B, and SFP96F, which have mature lengths of 14–29 residues), one method for functional analysis may be to artificially synthesize the mature peptide, inject it into virgin females, and assay for behaviors that are part of the stereotyped female post-mating response (e.g., an increased rate of egg-laying, a change in sperm storage patterns, a shortened life span, increased rejection of male suitors). While seemingly crude, this method was used in the first successful characterization of an accessory gland protein, the sex peptide (SP, also called ACP70A) (Chen et al. 1988). In its mature form, SP is only 36 residues in length, yet it is responsible for a suite of behavioral changes in mated females (for review, see Ravi Ram and Wolfner 2007a). Sfp function may also be analyzed genetically through gene knockouts, RNA interference, or deletion studies (e.g., Ravi Ram and Wolfner 2007b; Mueller et al. 2008; Wong et al. 2008a). These studies, coupled with site-specific mutagenesis, might be especially useful in cases such as SFP24F, an Sfp with a well-predicted function and specific sites under selection. Finally, it should continue to be productive to examine natural and/or outbred populations of Drosophila to determine whether heritable genetic variation in sperm competitive ability is attributable to polymorphisms in Sfp genes (Fiumera et al. 2005; 2007; Friberg et al. 2005). Given the dynamic evolutionary patterns shown by these genes, as well as the extensive evidence that Sfps play a critical role in ensuring male reproductive success, it will be exciting to determine the specific roles that these novel Sfps play during reproduction.
Methods
Flies
Flies were reared on standard media at 25°C except during isotopic labeling (see below). For D. melanogaster, laboratory strains Oregon R and w1118 were used. Strain W89 was used for D. simulans and strain Tai6 was used for D. yakuba.
Isotopic labeling, mating experiments, and MS
Three mating experiments to detect transferred Sfps in three Drosophila species (D. melanogaster, D. simulans, and D. yakuba) were performed for this study, which generally followed previously described methods (Findlay et al. 2008). The D. melanogaster experiment used w1118 females mated to Oregon R males, while the D. simulans and D. yakuba strains were as above. Briefly, adult females were allowed to lay eggs into a paste of isotopically “heavy” yeast that had been grown in 15N media. Adults were then removed and embryos were allowed to develop to adulthood with 15N yeast as the sole food source. Virgin females were collected and aged 3 d. These labeled females were then mated with similarly aged, unlabeled males. After 2 h of mating, lower female reproductive tracts were dissected in cold, 50 mM ammonium bicarbonate (pH 7.4). Soluble, extracellular proteins were isolated and digested with trypsin. These peptides were then analyzed using tandem MS. For each experiment, five technical replicates, each containing ∼5 μg of protein, were injected into an HPLC column that was placed online with an LTQ ion-trap mass spectrometer (ThermoElectron). The 75-μm internal diameter column was packed with 40 cm of Jupiter C12 reversed-phase material and eluted using a 4-h gradient. Data-dependent acquisition was used to acquire tandem mass spectra.
Database searches
In addition to the mass spectra acquired in the experiments described above, we also analyzed sets of spectra from two additional mating experiments conducted for a previous study (Findlay et al. 2008). One of these sets came from a mating experiment with D. simulans strain W89, and the other came from D. yakuba strain Tai6. In all cases, sets of spectra were first searched with Sequest (Eng et al. 1994) against a database containing all annotated proteins from the appropriate species. D. melanogaster spectra were searched against annotated proteins from release 5.5 of the genome (obtained from FlyBase), supplemented with the 19 previously unannotated Sfps that we recently discovered (Findlay et al. 2008). D. simulans and D. yakuba spectra were searched against annotated proteins from the version 1.2 releases of their respective genomes. Spectra were also searched against a database of “decoy” proteins in which each protein sequence of the appropriate species was randomly shuffled. Searches were performed with Sequest and then analyzed with percolator (Kall et al. 2007) to improve peptide-spectrum matches and to set a per-spectrum false discovery rate of 1%, and results were assembled and filtered with DTASelect (Tabb et al. 2002).
We used additional searches to discovered unannotated proteins, as previously described (Findlay et al. 2008). Briefly, we translated each species' genome in all six possible reading frames, generating a set of all possible peptides that could be encoded. Hardklör (Hoopmann et al. 2007) was used to predict which MS2 spectra in each data set arose from a peptide showing a 15N isotope distribution (i.e., those peptides derived from a female protein instead of a transferred male Sfp); these spectra were removed from the data set before searching. These sets of filtered spectra and their search results have been deposited in the PRIDE database under accession numbers 9199–9203. Sequest was used to search each filtered set of spectra against the translated database for the relevant species. The results were filtered with DTASelect such that only identifications with spectra scoring above certain XCorr and deltaCN cut-offs were included (XCorr >2.6 for doubly charged peptides or >3.6 for triply charged peptides; deltaCN >0.20 for all peptides). Identified peptides that matched an annotated protein were discarded, leaving only those peptides that were detected with MS but did not match any annotated protein. A complete list of peptides identified and their quality scores is given in Supplemental Table S1.
Experimental discovery of unannotated genes using RACE
To determine which of the peptides found by the six-frame search were accurate identifications and truly encoded by genes, we used a RACE strategy (Findlay et al. 2008). The peptides not matching an annotated protein were mapped back to the genome, and 5′ and 3′ RACE primers were designed around these regions. These primers were then used in 5′ or 3′ RACE reactions using the SMART RACE and Advantage 2 PCR kits (Clontech-Takara) according to the manufacturer's directions. RACE products were cloned using the TOPO TA Cloning Kit for Sequencing (Invitrogen). Randomly selected colonies were grown in liquid culture, and plasmid DNA was extracted using the Perfectprep Plasmid 96 VAC kit (5Prime). Plasmids were sequenced on a 3100 genetic analyzer using BigDye technology (ABI), and sequences that mapped back to the regions of the genome previously identified were inferred to be the 5′ or 3′ ends of transcripts in that region. Transcripts were confirmed by checking for evidence of splicing, the sequences at intron/exon boundaries (e.g., the canonical GT…AG sequence present at the ends of an intron), the presence of 5′ and 3′ UTRs, and the polyadenylation of 3′ transcripts. To confirm the complete gene sequences, RT-PCR was performed on independently prepared cDNA samples from the relevant species to ensure that overlapping 5′ and 3′ RACE products were derived from the same coding sequence. To ensure that amplification was of the desired target, the specificity of RT-PCR primers was verified with in silico PCR, and products were sequenced after amplification. Chromosomal coordinates of the coding sequences are given in Supplemental Table S9; the set of RACE-determined transcribed sequences has been deposited in GenBank under accession numbers FJ460563–FJ460581. The analyses described below were performed on this new set of genes combined with the 19 previously unannotated Sfps described in Table S6 in the work by Findlay et al. (2008), GenBank accession numbers EU755332–EU755350.
Bioinformatic identification of orthologs
Once novel genes were identified in one species, we used bioinformatic methods and the recently sequenced genomes of 12 Drosophila species (Drosophila 12 Genomes Consortium 2007) to determine whether orthologs were present in additional species. We used a combination of BLAT and TBLASTN searches to identify putative orthologs across the five sequenced species of the melanogaster subgroup (D. melanogaster, D. simulans, D. sechellia, D. yakuba, and D. erecta). Orthologs were assessed for conserved splice junctions and overall full-length homology and were aligned at the amino acid level using ClustalW in the MEGA 4.0 program (Tamura et al. 2007). Alignments were then checked by eye, and aligned coding DNA sequences were used for evolutionary analyses.
Expression analysis
Following the procedures of Findlay et al. (2008), we used RT-PCR to test for expression and to assess the sex-specificity of expression for all 38 Sfps and their predicted orthologs in three species, D. melanogaster, D. simulans, and D. yakuba. Briefly, total RNA from whole male or female flies of each species was isolated using the TRIzol reagent (Invitrogen) according to the manufacturer's instructions. Ten micrograms of total RNA was subjected to rigorous DNase treatment with the Turbo DNase kit (Ambion) and then made into cDNA using the SuperScript III kit (Invitrogen). cDNA from each sex was diluted fivefold and used in PCR reactions. When possible, primers were designed to amplify a region in which splicing was expected to occur. Positive control reactions assayed for expression of the RpL32 gene, while negative control reactions used product from cDNA synthesis reactions performed without reverse transcriptase. In addition to the RT-PCR experiments, our MS data revealed that several of the newly discovered D. melanogaster Sfps were also transferred in the seminal fluid of D. simulans and/or D. yakuba (Supplemental Table S5).
Evolutionary analyses
We tested for positive selection on each newly identified gene with codeml of the PAML version 4 suite (Yang 2007). First, whole-gene, pairwise estimates of the dN/dS ratio (ω) were made between D. melanogaster and D. simulans (or D. sechellia when a D. simulans ortholog could not be identified). Because this test for selection is highly conservative, we then expanded our analysis to include all species for which an ortholog was identified and to test for selection acting on specific sites within the protein-coding sequence (Yang et al. 2000). To conduct this test, we compared the likelihoods of two models of evolution (implemented in codeml). Model M8a is a neutral model of evolution in which each codon in an alignment is assigned to one of 11 classes. Ten codon classes have 0 ≤ ω ≤ 1, with each value estimated from the data, while the eleventh class has ω fixed at 1. Model M8 is identical except that the 11th class of codons is allowed to take any value of ω (again estimated from the data), including those >1, which indicates positive selection. The likelihoods of M8a and M8 are then compared with a likelihood ratio test with one degree of freedom (Swanson et al. 2003). The codeml program also implements a Bayes Empirical Bayes (BEB) analysis (Yang et al. 2005) to estimate ω for each codon assigned to the ω > 1 class in model M8 and the probability of that codon belonging to that positively selected class.
Comparisons of annotated and unannotated Sfps in D. melanogaster
To investigate whether any features of the 27 unannotated Sfps in D. melanogaster differed from those annotated Sfps discovered previously (Findlay et al. 2008), we compared the two sets of proteins using several metrics. Protein length was determined for the unannotated set by counting the number of codons in each D. melanogaster Sfp. For annotated proteins, length was determined by downloading protein sequences from FlyBase and extracting the number in the length field from the header line of each protein's FASTA sequence. When multiple isoforms were present, we arbitrarily selected the first isoform listed unless proteomic evidence (Findlay et al. 2008) suggested that another was present in seminal fluid. Because these length comparisons showed that the unannotated Sfps were, on average, significantly shorter than the annotated set, we restricted further analysis of the annotated set to those proteins with lengths less than 200 residues. (This filter produced a set of 51 annotated proteins after the exclusion of LYSC and CG31758 due to problems in their FlyBase sequences.) We compared the pairwise estimates of dN and dS described above for the unannotated set of Sfps to those previously calculated for the annotated set of proteins. We used DnaSP version 4.00 (Rozas et al. 2003) to examine the nucleotide content of the coding sequences in each set of proteins. Coding sequences from each set were concatenated, giving a string of 2585 codons for the unannotated set of proteins and 5964 codons for the annotated set. GC content and RSCU values were then calculated for each concatenation.
Structural modeling and functional predictions
To determine whether any of the new Sfps fell into any of the previously identified functional classes of Sfps (Mueller et al. 2004; Findlay et al. 2008), we first used BLASTP to determine whether any homology with proteins with known functions was present at the level of primary amino acid sequences. We also used PHYRE (Bennett-Lovsey et al. 2008) to assess whether the new Sfps shared structural similarity with other proteins. We modeled the D. melanogaster SFP24F protein by threading the protein sequence onto the structure of the mouse scavenger receptor C-type lectin domain (Mus musculus), found in the Protein Data Bank (PDB; ID no. 2ox9). The structure was visualized in MacPyMOL, and positively selected residues (as determined by the BEB analysis in codeml) were mapped onto the structure.
Acknowledgments
We thank Celeste Berg, Harmit Malik, and the Bloomington Stock Center for fly lines; Philip Green for discussing gene prediction methods; Jan Aagaard and Stevan Springer provided helpful advice about RACE and structural modeling, respectively; Amanda Larracuente answered several queries about the 12 Drosophila genomes data; Barbara Frewen assisted in setting up the six-frame database searches, and Michael Hoopmann provided guidance in using Hardklör. We also thank the Swanson Laboratory and two anonymous reviewers for comments on the manuscript. This work was funded by NSF grant DEB-0743539 (to W.J.S.); NIH grants HD042563, HD054631, and HD057974 (to W.J.S.); NIH grants DK069386 and HG004263 (to M.J.M.); and NIH training grant T32 HG00035 (to G.D.F.). The views expressed in this manuscript are those of the authors and are not necessarily shared by any of the funding agencies.
Footnotes
[Supplemental material is available online at www.genome.org. The sequence data from this study have been submitted to GenBank (http://www.ncbi.nlm.nih.gov/Genbank/) under accession nos. FJ460563–FJ460581. Mass spectrometry data are available in the PRIDE database under accession nos. 9199–9203.]
Article is online at http://www.genome.org/cgi/doi/10.1101/gr.089391.108.
References
- Akashi H. Inferring weak selection from patterns of polymorphism and divergence at silent sites in Drosophila DNA. Genetics. 1995;139:1067–1076. doi: 10.1093/genetics/139.2.1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansong C., Purvine S.O., Adkins J.N., Lipton M.S., Smith R.D. Proteogenomics: Needs and roles to be filled by proteomics in genome annotation. Brief. Funct. Genomics Proteomics. 2008;7:50–62. doi: 10.1093/bfgp/eln010. [DOI] [PubMed] [Google Scholar]
- Baerenfaller K., Grossmann J., Grobei M.A., Hull R., Hirsch-Hoffmann M., Yalovsky S., Zimmermann P., Grossniklaus U., Gruissem W., Baginsky S. Genome-scale proteomics reveals Arabidopsis thaliana gene models and protein dynamics. Science. 2008;320:938–941. doi: 10.1126/science.1157956. [DOI] [PubMed] [Google Scholar]
- Begun D.J., Lindfors H.A. Rapid evolution of genomic Acp complement in the melanogaster subgroup of Drosophila. Mol. Biol. Evol. 2005;22:2010–2021. doi: 10.1093/molbev/msi201. [DOI] [PubMed] [Google Scholar]
- Begun D.J., Whitley P., Todd B.L., Waldrip-Dail H.M., Clark A.G. Molecular population genetics of male accessory gland proteins in Drosophila. Genetics. 2000;156:1879–1888. doi: 10.1093/genetics/156.4.1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begun D.J., Lindfors H.A., Thompson M.E., Holloway A.K. Recently evolved genes identified from Drosophila yakuba and Drosophila erecta accessory gland expressed sequence tags. Genetics. 2006;172:1675–1681. doi: 10.1534/genetics.105.050336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett-Lovsey R.M., Herbert A.D., Sternberg M.J., Kelley L.A. Exploring the extremes of sequence/structure space with ensemble fold recognition in the program Phyre. Proteins. 2008;70:611–625. doi: 10.1002/prot.21688. [DOI] [PubMed] [Google Scholar]
- Betancourt A.J., Presgraves D.C. Linkage limits the power of natural selection in Drosophila. Proc. Natl. Acad. Sci. 2002;99:13616–13620. doi: 10.1073/pnas.212277199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brent M.R., Guigo R. Recent advances in gene structure prediction. Curr. Opin. Struct. Biol. 2004;14:264–272. doi: 10.1016/j.sbi.2004.05.007. [DOI] [PubMed] [Google Scholar]
- Brunner E., Ahrens C.H., Mohanty S., Baetschmann H., Loevenich S., Potthast F., Deutsch E.W., Panse C., de Lichtenberg U., Rinner O., et al. A high-quality catalog of the Drosophila melanogaster proteome. Nat. Biotechnol. 2007;25:576–583. doi: 10.1038/nbt1300. [DOI] [PubMed] [Google Scholar]
- Burge C., Karlin S. Prediction of complete gene structures in human genomic DNA. J. Mol. Biol. 1997;268:78–94. doi: 10.1006/jmbi.1997.0951. [DOI] [PubMed] [Google Scholar]
- Chen P.S., Stumm-Zollinger E., Aigaki T., Balmer J., Bienz M., Bohlen P. A male accessory gland peptide that regulates reproductive behavior of female Drosophila melanogaster. Cell. 1988;54:291–298. doi: 10.1016/0092-8674(88)90192-4. [DOI] [PubMed] [Google Scholar]
- Clark N.L., Swanson W.J. Pervasive adaptive evolution in primate seminal proteins. PLoS Genet. 2005;1:335–342. doi: 10.1371/journal.pgen.0010035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark N.L., Aagaard J.E., Swanson W.J. Evolution of reproductive proteins from animals and plants. Reproduction. 2006;131:11–22. doi: 10.1530/rep.1.00357. [DOI] [PubMed] [Google Scholar]
- Dorus S., Busby S.A., Gerike U., Shabanowitz J., Hunt D.F., Karr T.L. Genomic and functional evolution of the Drosophila melanogaster sperm proteome. Nat. Genet. 2006;38:1440–1445. doi: 10.1038/ng1915. [DOI] [PubMed] [Google Scholar]
- Dorus S., Freeman Z.N., Parker E.R., Heath B.D., Karr T.L. Recent origins of sperm genes in Drosophila. Mol. Biol. Evol. 2008;25:2157–2166. doi: 10.1093/molbev/msn162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drosophila 12 Genomes Consortium. Evolution of genes and genomes on the Drosophila phylogeny. Nature. 2007;450:203–218. doi: 10.1038/nature06341. [DOI] [PubMed] [Google Scholar]
- Eng J.K., McCormack A.L., Yates J.R. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom. 1994;5:976–989. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- Findlay G.D., Yi X., MacCoss M.J., Swanson W.J. Proteomics reveals novel Drosophila seminal fluid proteins transferred at mating. PLoS Biol. 2008;6:1417–1426. doi: 10.1371/journal.pbio.0060178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiumera A.C., Dumont B.L., Clark A.G. Sperm competitive ability in Drosophila melanogaster associated with variation in male reproductive proteins. Genetics. 2005;169:243–257. doi: 10.1534/genetics.104.032870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiumera A.C., Dumont B.L., Clark A.G. Associations between sperm competition and natural variation in male reproductive genes on the third chromosome of Drosophila melanogaster. Genetics. 2007;176:1245–1260. doi: 10.1534/genetics.106.064915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friberg U., Lew T.A., Byrne P.G., Rice W.R. Assessing the potential for an ongoing arms race within and between the sexes: Selection and heritable variation. Evolution Int. J. Org. Evolution. 2005;59:1540–1551. [PubMed] [Google Scholar]
- Gupta N., Benhamida J., Bhargava V., Goodman D., Kain E., Kerman I., Nguyen N., Ollikainen N., Rodriguez J., Wang J., et al. Comparative proteogenomics: Combining mass spectrometry and comparative genomics to analyze multiple genomes. Genome Res. 2008;18:1133–1142. doi: 10.1101/gr.074344.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holloway A.K., Begun D.J. Molecular evolution and population genetics of duplicated accessory gland protein genes in Drosophila. Mol. Biol. Evol. 2004;21:1625–1628. doi: 10.1093/molbev/msh195. [DOI] [PubMed] [Google Scholar]
- Hong X., Scofield D.G., Lynch M. Intron size, abundance, and distribution within untranslated regions of genes. Mol. Biol. Evol. 2006;23:2392–2404. doi: 10.1093/molbev/msl111. [DOI] [PubMed] [Google Scholar]
- Hoopmann M.R., Finney G.L., MacCoss M.J. High-speed data reduction, feature detection, and MS/MS spectrum quality assessment of shotgun proteomic data sets using high-resolution mass spectrometry. Anal. Chem. 2007;79:5620–5632. doi: 10.1021/ac0700833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iobst S.T., Drickamer K. Binding of sugar ligands to Ca2+-dependent animal lectins. II. Generation of high-affinity galactose bindlng by site-directed mutagenesis. J. Biol. Chem. 1994;269:15512–15519. [PubMed] [Google Scholar]
- Kall L., Canterbury J.D., Weston J., Noble W.S., MacCoss M.J. Semi-supervised learning for peptide identification from shotgun proteomics datasets. Nat. Methods. 2007;4:923–925. doi: 10.1038/nmeth1113. [DOI] [PubMed] [Google Scholar]
- May P., Wienkoop S., Kempa S., Usadel B., Christian N., Rupprecht J., Weiss J., Recuenco-Munoz L., Ebenhoh O., Weckwerth W., et al. Metabolomics- and proteomics-assisted genome annotation and analysis of the draft metabolic network of Chlamydomonas reinhardtii. Genetics. 2008;179:157–166. doi: 10.1534/genetics.108.088336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGowan S.J., Terrett J., Brown C.G., Adam P.J., Aldridge L., Allen J.C., Amess B., Andrews K.A., Barnes M., Barnwell D.E., et al. Annotation of the human genome by high-throughput sequence analysis of naturally occurring proteins. Curr. Proteomics. 2004;1:41–48. [Google Scholar]
- Merrihew G.E., Davis C., Ewing B., Williams G., Kall L., Frewen B.E., Noble W.S., Green P., Thomas J.H., MacCoss M.J. Use of shotgun proteomics for the identification, confirmation, and correction of C. elegans gene annotations. Genome Res. 2008;18:1660–1669. doi: 10.1101/gr.077644.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moy G.W., Springer S.A., Adams S.L., Swanson W.J., Vacquier V.D. Extraordinary intraspecific diversity in oyster sperm bindin. Proc. Natl. Acad. Sci. 2008;105:1993–1998. doi: 10.1073/pnas.0711862105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller J.L., Ripoll D.R., Aquadro C.F., Wolfner M.F. Comparative structural modeling and inference of conserved protein classes in Drosophila seminal fluid. Proc. Natl. Acad. Sci. 2004;101:13542–13547. doi: 10.1073/pnas.0405579101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller J.L., Ram K.R., McGraw L.A., Qazi M.C.B., Siggia E.D., Clark A.G., Aquadro C.F., Wolfner M.F. Cross-species comparison of Drosophila male accessory gland protein genes. Genetics. 2005;171:131–143. doi: 10.1534/genetics.105.043844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller J.L., Linklater J.R., Ravi Ram K., Chapman T., Wolfner M.F. Targeted gene deletion and phenotypic analysis of the Drosophila melanogaster seminal fluid protease inhibitor Acp62F. Genetics. 2008;178:1605–1614. doi: 10.1534/genetics.107.083766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravi Ram K., Wolfner M.F. Seminal influences: Drosophila Acps and the molecular interplay between males and females during reproduuction. Integr. Comp. Biol. 2007a;47:427–445. doi: 10.1093/icb/icm046. [DOI] [PubMed] [Google Scholar]
- Ravi Ram K., Wolfner M.F. Sustained post-mating response in D. melanogaster requires multiple seminal fluid proteins. PLoS Genet. 2007b;3:e238. doi: 10.1371/journal.pgen.0030238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice W.R. Sexually antagonistic male adaptation triggered by experimental arrest of female evolution. Nature. 1996;381:232–234. doi: 10.1038/381232a0. [DOI] [PubMed] [Google Scholar]
- Rozas J., Sanchez-DelBarrio J.C., Messeguer X., Rozas R. DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics. 2003;19:2496–2497. doi: 10.1093/bioinformatics/btg359. [DOI] [PubMed] [Google Scholar]
- Springer S.A., Crespi B.J. Adaptive gamete-recognition divergence in a hybridizing Mytilus population. Evolution Int. J. Org. Evolution. 2007;61:772–783. doi: 10.1111/j.1558-5646.2007.00073.x. [DOI] [PubMed] [Google Scholar]
- Stark A., Lin M.F., Kheradpour P., Pedersen J.S., Parts L., Carlson J.W., Crosby M.A., Rasmussen M.D., Roy S., Deoras A.N., et al. Discovery of functional elements in 12 Drosophila genomes using evolutionary signatures. Nature. 2007;450:219–232. doi: 10.1038/nature06340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson W.J., Vacquier V.D. Reproductive protein evolution. Annu. Rev. Ecol. Syst. 2002;33:161–179. [Google Scholar]
- Swanson W.J., Clark A.G., Waldrip-Dail H.M., Wolfner M.F., Aquadro C.F. Evolutionary EST analysis identifies rapidly evolving male reproductive proteins in Drosophila. Proc. Natl. Acad. Sci. 2001;98:7375–7379. doi: 10.1073/pnas.131568198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson W.J., Nielsen R., Yang Q.F. Pervasive adaptive evolution in mammalian fertilization proteins. Mol. Biol. Evol. 2003;20:18–20. doi: 10.1093/oxfordjournals.molbev.a004233. [DOI] [PubMed] [Google Scholar]
- Swanson W.J., Wong A., Wolfner M.F., Aquadro C.F. Evolutionary expressed sequence tag analysis of Drosophila female reproductive tracts identifies genes subjected to positive selection. Genetics. 2004;168:1457–1465. doi: 10.1534/genetics.104.030478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabb D.L., McDonald W.H., Yates J.R. DTASelect and contrast: Tools for assembling and comparing protein identifications from shotgun proteomics. J. Proteome Res. 2002;1:21–26. doi: 10.1021/pr015504q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K., Dudley J., Nei M., Kumar S. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol. Biol. Evol. 2007;24:1596–1599. doi: 10.1093/molbev/msm092. [DOI] [PubMed] [Google Scholar]
- Tanner S., Shen Z.X., Ng J., Florea L., Guigo R., Briggs S.P., Bafna V. Improving gene annotation using peptide mass spectrometry. Genome Res. 2007;17:231–239. doi: 10.1101/gr.5646507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagstaff B.J., Begun D.J. Comparative genomics of accessory gland protein genes in Drosophila melanogaster and D. pseudoobscura. Mol. Biol. Evol. 2005a;22:818–832. doi: 10.1093/molbev/msi067. [DOI] [PubMed] [Google Scholar]
- Wagstaff B.J., Begun D.J. Molecular population genetics of accessory gland protein genes and testis-expressed genes in Drosophila mojavensis and D. arizonae. Genetics. 2005b;171:1083–1101. doi: 10.1534/genetics.105.043372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagstaff B.J., Begun D.J. Adaptive evolution of recently duplicated accessory gland protein genes in desert Drosophila. Genetics. 2007;177:1023–1030. doi: 10.1534/genetics.107.077503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker M.J., Rylett C.M., Keen J.N., Audsley N., Sajid M., Shirras A.D., Isaac R.E. Proteomic identification of Drosophila melanogaster male accessory gland proteins, including a pro-cathepsin and a soluble gamma-glutamyl transpeptidase. Proteome Sci. 2006;4:9. doi: 10.1186/1477-5956-4-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfner M.F., Harada H.A., Bertram M.J., Stelick T.J., Kraus K.W., Kalb J.M., Lung Y.O., Neubaum D.M., Park M., Tram U. New genes for male accessory gland proteins in Drosophila melanogaster. Insect Biochem. Mol. Biol. 1997;27:825–834. doi: 10.1016/s0965-1748(97)00056-8. [DOI] [PubMed] [Google Scholar]
- Wong A., Albright S.N., Giebel J.D., Ravi Ram K., Ji S., Fiumera A.C., Wolfner M.F. A role for Acp29AB, a predicted seminal fluid lectin, in female sperm storage in Drosophila melanogaster. Genetics. 2008a;180:921–931. doi: 10.1534/genetics.108.092106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong A., Turchin M.C., Wolfner M.F., Aquadro C.F. Evidence for positive selection on Drosophila melanogaster seminal fluid protease homologs. Mol. Biol. Evol. 2008b;25:497–506. doi: 10.1093/molbev/msm270. [DOI] [PubMed] [Google Scholar]
- Yang Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007;24:1586–1591. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- Yang Z.H., Nielsen R., Goldman N., Pedersen A.M.K. Codon-substitution models for heterogeneous selection pressure at amino acid sites. Genetics. 2000;155:431–449. doi: 10.1093/genetics/155.1.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z., Wong W.S.W., Nielsen R. Bayes empirical Bayes inference of amino acid sites under positive selection. Mol. Biol. Evol. 2005;22:1107–1118. doi: 10.1093/molbev/msi097. [DOI] [PubMed] [Google Scholar]