

Abstract
In Drosophila melanogaster, there is an excess of genes duplicated by retroposition from the X chromosome to the autosomes. Most of those retrogenes that originated on the X chromosome have testis expression pattern. These observations could be explained by natural selection favoring genes that avoided spermatogenesis X inactivation or by sexual antagonistic effects favoring the fixation of male beneficial mutations on the autosomes. If natural selection played the essential role in distributing male-related genes, then the out-of-the-X chromosomal gene movement should not be limited to retrogenes. Here, we studied DNA-based interchromosome gene movement patterns by analyzing relocated genes that were previously identified in 12 Drosophila genome sequences. We found a significant excess of gene movement out of the X chromosome. In addition, we were able to extend previous retrogene movement analysis to species and branches other than those involving D. melanogaster, confirming the pervasiveness of gene movement out of the X chromosome. Also, for X chromosome-to-autosome (X→A) movement, we observed high testis expression of relocated genes as opposed to the low testis expression of parental genes, corroborating the involvement of the male germ line on the gene movement process. These analyses of both DNA-based and RNA-based gene relocations reveal that the out-of-the-X movement of testis-expressed genes is a general pattern in the Drosophila genus.
Genes that are differentially expressed in females and males (in somatic tissues or gonads) have been extensively identified in various species, including worm (Reinke et al. 2000), fruitfly (Meiklejohn et al. 2003; Parisi et al. 2003, 2004; Ranz et al. 2003), mouse (Khil et al. 2004), and human (Wang et al. 2005). Intriguingly, genes that are highly expressed in males (in somatic tissues or testis of flies and late spermatogenic cells of mammals) are under-represented on the X chromosome (Meiklejohn et al. 2003; Parisi et al. 2003, 2004; Ranz et al. 2003; Khil et al. 2004). One possible evolutionary mechanism that explains the demasculinization of the X chromosome is the movement of male-biased genes out of X chromosome into the autosomes. The existence of such gene movement has been supported by the study of retrogene movement patterns in fly and mammals (Betrán et al. 2002; Emerson et al. 2004). Retroposition can create new genes through the reverse transcription of mRNAs, followed by insertion in the genome. These new genes (retrogenes) can be inserted into new genomic positions and could be a source of new functions (Long et al. 2003). Retrogene studies have demonstrated that there is a significant excess of retrogenes generated by X-linked parental genes escaping to the autosomes (Betrán et al. 2002; Emerson et al. 2004). Moreover, those retrogenes that escape the X have a higher potential to be expressed in testis and develop a male-biased expression likely driven by positive selection (Betrán et al. 2002; Emerson et al. 2004). Recently, other studies were able to expand the number of retrogenes (Bai et al. 2006; Dai et al. 2006) and to analyze the age of the retrotransposition events using the 12 Drosophila genomes (Bai et al. 2006). Meanwhile, Sturgill et al. (2007) have shown that the X chromosomes of the other six Drosophila species have an under-representation of male-biased genes. Remarkably, they also found that the neo-X chromosome in D. pseudoobscura has recently developed a paucity of X-linked male-biased genes.
The excess of retroposed genes that have moved from the X chromosome to the autosomes and the paucity of X-linked male-biased genes could be explained by natural selection favoring genes that avoided spermatogenesis X-inactivation or by sexual antagonism (Lifschytz and Lindsley 1972; Rice 1984; Charlesworth et al. 1987; Wu and Xu 2003). On one hand, inactivation of X-linked genes in the male germ line would favor accumulation of male-related genes on the autosomes because they could still be expressed if they are not located on the X chromosome (Lifschytz and Lindsley 1972; Betrán et al. 2002). Alternatively, the sexual antagonism hypothesis proposes that the X chromosome demasculization is a product of the longer time spent by the X chromosome in females rather than males (Rice 1984; Wu and Xu 2003). Additionally, population genetic studies have shown that the sexual antagonism hypothesis predicts that the probability of fixing a mutation with beneficial effects in males and harmful effects in females on the X chromosome or on the autosomes will, respectively, depend on its recessive or dominant genetic status (Charlesworth et al. 1987). Contrasting Drosophila findings, different aspects of those selective hypotheses, have been extensively studied in mammals (Khil et al. 2004). Genes expressed during mitotic phases are enriched on the X chromosome as predicted for sexually antagonistic recessive mutations, whereas those expressed later in spermatogenesis are enriched on autosomes in agreement with meiotic male germ line X inactivation (Khil et al. 2004).
If natural selection, rather than the mutation process, played the essential role in distributing sex-biased genes, then the pattern of interchromosomal gene movement should not be limited to retrogenes. The out-of-the-X movement pattern should also affect genes created by other processes causing gene movement, including DNA level mechanisms like illegitimate recombination and TE-intermediated transposition (Li 1997; Long et al. 2003; Betrán et al. 2004; Arguello et al. 2007). Since DNA-based mechanisms do not require an intermediate RNA step, they may be more likely to happen during evolution (Zhou et al. 2008). Thus, the movement pattern of genes generated by DNA-based mechanisms may represent a more general picture of gene movement in the genome and can further be used to test the role of natural selection in distributing genes in the genome.
Although Drosophila male-biased gene chromosomal distribution (Parisi et al. 2003, 2004; Ranz et al. 2003; Sturgill et al. 2007) and D. melanogaster retrogene chromosomal movement have been extensively studied (Betrán et al. 2002; Bai et al. 2006; Dai et al. 2006), the movement pattern of genes caused by DNA-based mechanisms has not been analyzed. In contrast to RNA-based movement, like retrogenes, where the offspring gene is a single-exon gene and the parental gene contains multiple exons, DNA-based movement does not have obvious features to distinguish derived from ancestral states, which are necessary to identify the direction of the movement. However, the availability of genomic sequences for 12 Drosophila species (Drosophila 12 Genomes Consortium 2007), and the respective syntenic information, allowed us to date DNA-based gene movements and to identify the ancestral gene. With this information we were able to identify the direction of movement and study the movement pattern of genes generated by mechanisms other than just retroposition.
Here, we studied DNA-based interchromosome gene movement patterns by analyzing relocated genes that were previously identified in the 12 Drosophila genome sequences (Bhutkar et al. 2007). We found a significant excess of gene movement out of the X chromosome. Furthermore, we were able to extend previously RNA-based movement analysis to species and branches other than those involving D. melanogaster, confirming the pervasiveness of gene movement out of the X chromosome. We also observed a trend of high testis expression of X chromosome-to-autosome (X→A) relocated genes opposing the low testis expression of their parental genes, which corroborates the suggestion that the underlying force behind this movement involves the male germ line. We thus have shown that biased movement from the X chromosome to the autosomes is common to gene relocations that have occurred through both DNA- and RNA-based mechanism.
Results and Discussion
We used genes that were identified as being positionally relocated across Muller elements (Bhutkar et al. 2007) as the primary data set to look for gene movement between chromosomes. Direction was assigned whenever derived and ancestral states were clearly identified using phylogenetic information (Methods) (Fig. 1). Since relocated genes sometimes represent the only copy of the gene in the derived species or branch, we could not distinguish between gene duplication (that can be followed by gene loss) and gene translocation. Hence, we identified both situations as gene movement or relocations. Gene interchromosome relocations were classified as RNA and DNA based, depending on the mechanism underlying the movement. In addition, the movement pattern was also assessed separately for external branches, using a similar approach Petrov et al. (1996) to detect possible recent evolutionary patterns. Finally, we also performed functionality tests based on a Likelihood Ratio Test framework and found that the majority (87%) of the relocated genes tested show significant functional constraint (Methods; Supplemental Table 1).
Figure 1.

Gene movement assignment. Example of gene duplicated from Muller element A to E in D. willistoni. Direction is assigned by comparing chromosome locations between D. willistoni and its sister group and an outgroup. Movements can be a product of gene duplication or gene translocation. In some cases, the parental gene can be lost in species where the relocation occurred.
RNA-based movement out of the X chromosome
RNA-based movement represents ∼25% of the data set of interchromosome relocations. We excluded all gene movements involving the branches leading toward D. melanogaster to determine whether the gene movement out of the X was also found for the other Drosophila species and branches. In agreement with what was previously found for D. melanogaster retrogenes (Betrán et al. 2002; Bai et al. 2006; Dai et al. 2006), we observed an excess of relocations out of the X chromosome, even for external branches (χ2 test: P = 1.32 × 10−8, P = 6.27 × 10−6, respectively) (Table 1; Fig. 2; Supplemental Table 1). Hence, the observation of an excess of out-of-the-X gene movement has been extended to other Drosophila lineages, in agreement with the selective advantage hypothesis.
Table 1.
Analysis of RNA-based relocations

Figure 2.

Distribution of gene movements in the Drosophila phylogenetic tree. Relocations based on RNA and DNA are located above and below the branch lines, respectively. Movements between chromosomes are presented as follows: (△) X→A; (○) A→X (⋄) A→A. The average expected proportions of these relocations are 21:23:56, respectively. For species bearing neo-X chromosome the average expected proportions are 35:34:31.
DNA-based movement out of the X chromosome
If there is a selective advantage for gene relocation out of the X chromosome, we would expect to observe this movement pattern for other types of gene relocation beside retroposition. Hence, we decided to investigate the pattern of chromosome DNA-based gene relocations. In agreement with previous studies (Zhou et al. 2008), we found that DNA-based relocations outnumber RNA-based events, corresponding to 75% of our data set. We found a significant excess of DNA-based relocations out of the X chromosome (Table 2; χ2 test: P = 1.19 × 10−6), even considering only external branches (Table 2; χ2 test: P = 1.62 × 10−4). The pattern also can be individually observed for most species where there were enough numbers of relocations (Fig. 2). There are a few species and branch exceptions to this trend, namely D. simulans, D. willistoni, and branches H and D (Fig. 2), on which selective forces might be relaxed. Alternatively, additional selective forces, such as those driving sexual antagonistic recessive mutations, might have governed relocations more often to the X chromosome (Charlesworth et al. 1987). D. simulans is a clear out-of-the-X pattern outlier, bearing ∼70% autosome-to-autosome relocations (most Muller D to Muller C relocations). There is no specific evidence to explain why those particular species or branches are subject to different selective forces. So, further careful investigation on those particular lineages could shed more light on our observation.
Table 2.
Analysis of DNA-based relocations

(X) X chromosome; (A) autosomes; Excess = [(O – E)/E] × 100, where E is expected and O is observed.
One hypothesis can be proposed that the different recombination rate might impact the insertion of new gene duplicates. Therefore, the chromosomes that are associated with different recombination rate could have different amounts of new gene duplicates. However, the fourth chromosome and autosomes that differ significantly in recombination rates (Berry et al. 1991; Wang et al. 2002) have a similar level of new duplicate polymorphisms (Emerson et al. 2008) rejecting this hypothesis. Transposable elements (TE), on the other hand, seem to play a significant role in the generation of new duplicate copies, suggesting an underlying replication-involved molecular mechanism (Fiston-Lavier et al. 2007; Yang 2007). No significant difference in TE frequency was observed between X chromosome and autosome (Bergman et al. 2006). These analyses do not support the mechanistic interpretation for the observed pattern of the out-of-X gene movement.
Movement out of the neo-X chromosomes
After D. willistoni speciation and after the Drosophila obscura group split, independent fusions between Muller elements D and A occurred (Tamura et al. 2004; Richards et al. 2005). Since then, the fused Muller D has started to segregate as a neo-X chromosome (Tamura et al. 2004; Richards et al. 2005). Interestingly, we found also an excess of RNA- and DNA-based movements out of the neo-X chromosome (Table 3). Except, again, for D. willistoni, all species and branches involved in these chromosome fusions present such a trend. It seems that as soon as genes located in the Muller D element became sex linked, selective forces drove their relocation to the autosomes.
Table 3.
Neo-X chromosome relocations

aχ2 test pooling together classes A→D and A→A.
(D) Muller element D; (A) autosomes; Branch G, Obscura group.
Male expression
Most of D. melanogaster retrogenes that originated on the X chromosome have testis expression patterns (Betrán et al. 2002; Bai et al. 2006; Dai et al. 2006). These observations could be explained by natural selection favoring genes that avoided the spermatogenesis X-inactivation or by sexual antagonistic effects favoring the fixation of male-biased dominant mutations on the autosomes (Lifschytz and Lindsley 1972; Rice 1984; Charlesworth et al. 1987; Wu and Xu 2003; Hense et al. 2007). Bhutkar et al. (2007) observed a general excess of testis expression for gene relocations that occurred within the D. melanogaster subgroup. Unfortunately, there is no testis/ovary microarray or large-scale testis EST data available for other Drosophila species beside D. melanogaster (Methods).
The use of D. melanogaster data precludes the direct and pairwise expression comparison between relocated and ancestral copies, since only one relocation is a duplication, i.e., has both copies present in D. melanogaster (otu [CG12743], CG3251; Supplemental Table 1). However, we compared X→A to A→ (i.e., autosomes to autosomes or to the X chromosome) expression separately for each data set (parental and relocated copies) (Tables 4, 5). For DNA-based relocated copies, we observed a higher male-biased expression for X→A movement in comparison to A→ (Table 4), although not significant due to small sample size. However, we found the statistically significant opposite pattern for DNA-based parental copies. X→A parental copies have significant lower male-biased expression compared with A→ cases (Fisher's exact test, P = 0.03; Table 4). The same trend is observed for our RNA-based relocations (Fisher's exact test, P ≤ 0.001; Table 5). These results are in agreement with lower male-biased expression on the X chromosome, favoring the accumulation of male-related genes on autosomes and suggest that evolutionary forces acting on the testis are involved in the movement out-of-the-X.
Table 4.
DNA-based relocations expression analysis

Male-biased expression was obtained by comparison of testis to whole fly expression (Supplemental Table 1). Note that for parental genes analysis, Muller D→A relocations in D. willistoni, D. persimilis, D. pseudoobscura, and Branch D were considered as A→A relocations, since Muller D in D. melanogaster is an autosome.
aExcluding relocation occurring on the D. simulans external branch.
(Muller A) Muller element A; (A) autosomes; (A→) movement from autosomes to autosomes or to the X chromosome.
Table 5.
RNA-based relocation expression analysis

Male-biased expression is obtained by comparison of testis to whole fly expression. Note that for parental genes analysis, Muller D→A relocations in D. willistoni, D. persimiliss, D. pseudoobscura, and Branch D were considered as A→A relocations, since Muller D in D. melanogaster is an autosome.
(Muller A) Muller element A; (A) autosomes; (A→) movement from autosomes to autosomes or to the X chromosome.
In conclusion, our analyses revealed that gene relocation out of the X chromosome into the autosomes is a general phenomenon occurring for both DNA-based and RNA-based gene relocations. Such an observation rules out any mutational biased intrinsic related to the retroposition mechanisms as the cause for chromosomal distribution bias for duplicated genes. In addition, our work extends the observation for retrogene movement out of the X to Drosophila branches other than those that include D. melanogaster, supporting the idea that this pattern is a general, recent, and on-going process of Drosophila gene evolution.
Methods
Direction of interchromosome gene relocations
Bhutkar et al. (2007) identified Drosophila genes that were positionally relocated across Muller elements using the recently generated genome sequences of 12 species. We selected the single-gene relocations that occurred between Muller elements that correspond to different D. melanogaster chromosomes (X, 2, 3, and 4). We excluded all multiple gene relocations to avoid violating statistical test assumptions of independency of events. In order to assign directionality to the relocations, we selected those cases where the derived state is clear: In a group of ortholog genes, one species or one branch shows a different chromosome location compared with its sister group and one more outgroup (Fig. 1). For example, relocations between subgenus Sophophora and subgenus Drosophila were discarded from our analysis since direction cannot be predicted. Derived state refers to the gene relocated to a different chromosome, whereas ancestral state refers to the genes in the sister group and in one more outgroup.
In D. pseudoobscura, D. persimilis, D. willistoni, and Branch G (Fig. 2), Muller elements A and D were counted as the X chromosome. Relocations on external branches were analyzed separately (Tables 1, 2; Supplemental Table S1).
Classification of relocation movement: RNA or DNA based
The molecular processes that relocate genes can be classified into two categories: (1) processes occurring at the DNA level (e.g., duplication by illegimate recombination, duplication mediated by transposable elements), and (2) processes occurring at the RNA level (retroposition). Cases where the ancestral state is a multiple-exon gene and the derived state is a single-exon gene were classified as RNA-based relocations, whereas cases with multiple-exon-derived state genes were classified as DNA-based relocations, irrespective of the number of exons in the ancestral state. Intron loss and gain might mislead the assignment of genes in relocation categories. However, low rates of intron loss and gain in the Drosophila genus (∼7% and 1%, respectively; Coulombe-Huntington and Majewski 2007), could not play a major role in the out-of-the-X pattern observed both in DNA- or RNA-based relocations. Additionally, single-exon to multiple-exon relocations are useful to estimate other types of intron incorporation such as the creation of chimeric genes by recruiting other intronic regions. Five cases observed demonstrate how rare an intron incorporation is and could not account for significant changes in the out-of-the-X movement observed in DNA-based relocations.
Cases where both ancestral and derived state are single-exon genes were analyzed separately, since it is not possible to determine their relocation mechanism basis: DNA or RNA level. Nonetheless, we were able to extend the out-of-the-X observation to those single-exon relocations (Supplemental Table S2).
Expected number of interchromosomal gene movements
We used similar calculations to those described in Betrán and colleagues (2002). In brief, based on a random model, we assumed that (1) the number of interchromosomal gene relocations generated by a given chromosome (source chromosome) is proportional to its number of genes, and (2) the capacity of a given chromosome to accept inserted genes is proportional to its size (target chromosome). Such assumptions could be described as the following formula. Let F and T be in {X, A} with A itself being in {2, 3, 4}. Then, the expected proportion of a given chromosomal movement can be written as
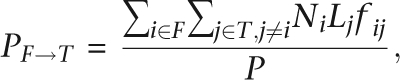 |
where P = PX→A + PA→X + PA→A.
Gene chromosome numbers (N) and euchromatic chromosome sizes (L) across different species were retrieved from Bhutkar et al. (2008) and Schaeffer et al. (2008) (Supplemental Tables S3, S4). Chromosome sizes for D. melanogaster were obtained from UCSC genome browser, version dm2 (Kuhn et al. 2007). In D. pseudoobscura, D. persimilis, D. willistoni, and Branch G (Fig. 2), Muller elements A and D were pooled together to calculate expectations for movements in and out of the X chromosome. Expectation values were calculated for each species based on its chromosome sizes and its gene numbers. D. melanogaster values were used to calculate expectation values for branches B, C, D, E, and F, whereas D. pseudoobscura and D. mojavensis were used for branches G and H, respectively. Final expectation values were calculated weighing all individual expectation values according to the number of observed relocations found in each species or branch.
In the case of RNA-based relocations, we considered the frequency of retroposition to a given chromosome in the population (fij) to calculate the expected values of relocations. The frequency depends on the relative population sizes of the chromosomes, which is 0.75 when the X chromosome is the target. Assuming complete dosage compensation, the relative population size of X chromosome when considered as the source chromosome is 1.
For DNA-based relocations, meiotic recombination hot spots could potentially increase the frequency of chromosomal rearrangements, such as translocations (affecting both source and target chromosomes). Since there is no crossing-over in Drosophila males, recombination rates are the same for the X chromosomes and autosomes as it is a product of population size (X = 3/4; A = 1) and the time spent in females (X = 2/3; A = 1/2) (the recombining sex) (Betrán et al. 2002). Therefore, the term (fij) plays no role in the equation and was considered equal to 1.
Recombination rates of the target chromosome could affect both DNA- and RNA-based relocation in an opposite way. Regions of low recombination are more prone to inserted elements because they are less likely to be deleted by unequal recombination (Langley et al. 1988; Sniegowski and Charlesworth 1994). As it is a product of population size and time spent in females, recombination rates of X and autosomes remains the same.
Although some species across the Drosophila genus do not show male crossing-over as D. mediopunctata from a Drosophila subgroup (Carvalho et al. 1997), we calculated expectation values for RNA- and DNA-based movements considering the range of zero to regular male meiotic recombination. There were no qualitative changes from previously observed patterns.
Expression analysis
Male expression data (EST or microarray) for non-melanogaster genomes are not complete. EST male-related data are available for five Drosophila species, but mostly in low numbers. Microarray data for male/female comparisons recovered male-biased expression for just 30% of previously reported retrogenes (Bai et al. 2006; Sturgill et al. 2007). In addition, there is no microarray expression data available for almost 50% of our DNA-based relocations, probably due to reduced probe numbers covering lineage or species-specific young genes of non-melanogaster genomes. Therefore, we concluded that D. melanogaster microarray testis expression data would be more adequate to test our DNA-based and RNA-based movement.
In order to verify the male expression of genes relocated in the Drosophila genus, we analyzed only cases where the derivate state is present in D. melanogaster (Supplemental Table S1). For male expression of parental genes, we analyzed only the cases where D. melanogaster corresponded to the ancestral state. We obtained testis expression from a FlyAtlas tissue search (http://www.flyatlas.org; Chintapalli et al. 2007). Male-biased expression between X→A and A→ (autosome to autosome or to the X chromosome) relocations were assessed using Fisher's exact tests.
Functionality test
Although there is no prior reason to assume these relocated genes are pseudogenes, we performed the functionality test based on sequence alignment to ensure these protein-coding gene models are indeed of high quality.
We downloaded the updated ortholog mappings and sequence annotations from FlyBase (June 2008). Based on the accession number for D. melanogaster, we can retrieve all ortholog genes together with their corresponding coding sequences (CDS) or protein sequences for the other 11 species. We then constructed codon-based alignments and performed the likelihood ratio test (LRT) using the Codeml program in the PAML package (Yang 2007).
Specifically, we classified all cases into two groups, translocation and duplication. As for the former, we tested the null model with an expectation of Ka/Ks equal to 1. In contrast, following previous works, for paralogs the null model is a Ka/Ks equal to 0.5 (Betrán et al. 2002; Emerson et al. 2004). Selecting which sequences to compare is not straightforward.
For branch translocations, we compared the most unrelated relocated genes within the branch. For example, in the case of CG2657, its relocation occurred at Branch B. Hence, we compared the orthologs from D. melanogaster and D. persimilis, which correspond to the longest evolutionary distance. As for species translocations, we compared the species-specific relocated gene with its closest ortholog. For example, for pck (CG14779), whose relocation is D. ananassae specific, we compared D. ananassae and D. erecta sequences.
For all duplications, considering numerous changes of accession numbers in the 11 Drosophila species, we inferred the relocated paralogs semiautomatically. Specifically, we searched the D. melanogaster ortholog based on within-species BLASTP and defined the paralog as the best hit. A subsequent manual check was performed to ensure that they were derived from different chromosomes. For branch duplications, we performed LRT tests for the species with the lowest protein identity or the highest Ka values between the parental and offspring genes. Our goal was to perform a conservative test to investigate whether these pairs of genes are under constraint even if they have diverged at the protein level. Finally, for species-specific duplications, we performed tests for between interchromosome paralogs.
Given protein sequences and CDS, bl2seq in the BLAST package was used to construct protein alignments, which were converted into codon-based alignment using PAL2NAL (Suyama et al. 2006). The results of LRT like Ka, Ks, and P-values are shown in Supplemental Table 1. For comparison, Ka and Ks calculated with an NG model in BioPerl (Stajich et al. 2002) is also included. We were unable to perform functionality tests based on a LRT framework for ∼9% of relocations due to the following scenarios: (1) possible wrong gene model or wrong homology assignment and (2) identical sequence comparisons.
Acknowledgments
We thank all members of the M. Long laboratory for comments, review, and support on this work, especially Jun Wang, Margarida Cardoso-Moreira, Benjamin H. Krinsky, and J. Roman Arguello. We also thank Esther Betran and Hedibert Lopes for discussions regarding the calculation of gene movement expectation numbers. This work was supported by a USA National Science Foundation CAREER award (MCB0238168) and by USA National Institutes of Health R01 grants (R01GM065429-01A1 and 1R01GM078070-01A1) to M.L. M.D.V. was supported by a Pew Latin America Fellowship.
Footnotes
[Supplemental material is available online at www.genome.org.]
Article is online at http://www.genome.org/cgi/doi/10.1101/gr.088609.108.
References
- Arguello J.R., Fan C., Wang W., Long M. Origination of chimeric genes through DNA-level recombination. Genome Dyn. 2007;3:131–146. doi: 10.1159/000107608. [DOI] [PubMed] [Google Scholar]
- Bai Y., Casola C., Feschotte C., Betrán E. Comparative genomics reveals a constant rate of origination and convergent acquisition of functional retrogenes in Drosophila. Genome Biol. 2006;8:R11. doi: 10.1186/gb-2007-8-1-r11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergman C.M., Quesneville H., Anxolabéhère D., Ashburner M. Recurrent insertion and duplication generate networks of transposable element sequences in the Drosophila melanogaster genome. Genome Biol. 2006;7:R112. doi: 10.1186/gb-2006-7-11-r112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry A.J., Ajioka J.W., Kreitman M. Lack of polymorphism on the Drosophila fourth chromosome resulting from selection. Genetics. 1991;129:1111–1117. doi: 10.1093/genetics/129.4.1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betrán E., Thornton K., Long M. Retroposed new genes out of the X in Drosophila. Genome Res. 2002;12:1854–1859. doi: 10.1101/gr.604902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betrán E., Emerson J.J., Kaessmann H., Long M. Sex chromosomes and male functions: Where do new genes go? Cell Cycle. 2004;3:873–875. [PubMed] [Google Scholar]
- Bhutkar A., Russo S.M., Smith T.F., Gelbart W.M. Genome-scale analysis of positionally relocated genes. Genome Res. 2007;17:1880–1887. doi: 10.1101/gr.7062307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhutkar A., Schaeffer S.W., Russo S.M., Xu M., Smith T.F., Gelbart W.M. Chromosomal rearrangement inferred from comparisons of 12 Drosophila genomes. Genetics. 2008;179:1657–1680. doi: 10.1534/genetics.107.086108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho A.B., Vaz S.C., Klaczko L.B. Polymorphism for Y-linked suppressors of sex-ratio in two natural populations of Drosophila mediopunctata. Genetics. 1997;146:891–902. doi: 10.1093/genetics/146.3.891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth B., Coyne A.J., Barton N.H. The relative rates of evolution of sex chromosomes and autosomes. Am. Nat. 1987;130:113–146. [Google Scholar]
- Chintapalli V.R., Wang J., Dow J.A.T. Using FlyAtlas to identify better Drosophila models of human disease. Nat. Genet. 2007;39:715–720. doi: 10.1038/ng2049. [DOI] [PubMed] [Google Scholar]
- Coulombe-Huntington J., Majewski J. Intron loss and gain in Drosophila. Mol. Biol. Evol. 2007;24:2842–2850. doi: 10.1093/molbev/msm235. [DOI] [PubMed] [Google Scholar]
- Dai H., Yoshimatsu T.F., Long M. Retrogene movement within- and between-chromosomes in the evolution of Drosophila genomes. Gene. 2006;385:96–102. doi: 10.1016/j.gene.2006.04.033. [DOI] [PubMed] [Google Scholar]
- Drosophila 12 Genomes Consortium. Evolution of genes and genomes on the Drosophila phylogeny. Nature. 2007;450:203–218. doi: 10.1038/nature06341. [DOI] [PubMed] [Google Scholar]
- Emerson J.J., Kaessmann H., Betrán E., Long M. Extensive gene traffic on the mammalian X chromosome. Science. 2004;303:537–540. doi: 10.1126/science.1090042. [DOI] [PubMed] [Google Scholar]
- Emerson J.J., Cardoso-Moreira M., Borevitz J.O., Long M. Natural selection shapes genome-wide patterns of copy-number polymorphism in Drosophila melanogaster. Science. 2008;320:1629–1631. doi: 10.1126/science.1158078. [DOI] [PubMed] [Google Scholar]
- Fiston-Lavier A.S., Anxolabehere D., Quesneville H. A model of segmental duplication formation in Drosophila melanogaster. Genome Res. 2007;17:1458–1470. doi: 10.1101/gr.6208307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hense W., Baines J.F., Parsch J. X chromosome inactivation during Drosophila spermatogenesis. PLoS Biol. 2007;5:e273. doi: 10.1371/journal.pbio.0050273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khil P.P., Smirnova N.A., Romanienko P.J., Camerini-Otero R.D. The mouse X chromosome is enriched for sex-biased genes not subject to selection by meiotic sex chromosome inactivation. Nat. Genet. 2004;36:642–646. doi: 10.1038/ng1368. [DOI] [PubMed] [Google Scholar]
- Kuhn R.M., Karolchik D., Zweig A.S., Trumbower H., Thomas D.J., Thakkapallayil A., Sugnet C.W., Stanke M., Smith K.E., Siepel A., et al. The UCSC genome browser database: Update 2007. Nucleic Acids Res. 2007;35:D668–D673. doi: 10.1093/nar/gkl928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langley C.H., Montgomery E., Hudson R., Kaplan N., Charlesworth B. On the role of unequal exchange in the containment of transposable element copy number. Genet. Res. 1988;52:223–235. doi: 10.1017/s0016672300027695. [DOI] [PubMed] [Google Scholar]
- Li W.-H. Molecular evolution. Sinauer Associates; Sunderland, MA: 1997. [Google Scholar]
- Lifschytz E., Lindsley D.L. The role of X-chromosome inactivation during spermatogenesis. Proc. Natl. Acad. Sci. 1972;69:182–186. doi: 10.1073/pnas.69.1.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long M., Betrán E., Thornton K., Wang W. The origin of new genes: Glimpses from the young and old. Nat. Rev. Genet. 2003;4:865–875. doi: 10.1038/nrg1204. [DOI] [PubMed] [Google Scholar]
- Meiklejohn C.D., Parsch J., Ranz J.M., Hartl D.L. Rapid evolution of male-biased gene expression in Drosophila. Proc. Natl. Acad. Sci. 2003;100:9894–9899. doi: 10.1073/pnas.1630690100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parisi M., Nuttall R., Naiman D., Bouffard G., Malley J., Andrews J., Eastman S., Oliver B. Paucity of genes on the Drosophila X chromosome showing male-biased expression. Science. 2003;299:697–700. doi: 10.1126/science.1079190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parisi M., Nuttall R., Edwards P., Minor J., Naiman D., Lü J., Doctolero M., Vainer M., Chan C., Malley J., et al. A survey of ovary-, testis-, and soma-biased gene expression in Drosophila melanogaster adults. Genome Biol. 2004;5:R40. doi: 10.1186/gb-2004-5-6-r40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrov D.A., Lozovskaya E.R., Hartl D.L. High intrinsic rate of DNA loss in Drosophila. Nature. 1996;384:346–349. doi: 10.1038/384346a0. [DOI] [PubMed] [Google Scholar]
- Ranz J.M., Castillo-Davis C.I., Meiklejohn C.D., Hartl D.L. Sex-dependent gene expression and evolution of the Drosophila transcriptome. Science. 2003;300:1742–1745. doi: 10.1126/science.1085881. [DOI] [PubMed] [Google Scholar]
- Reinke V., Smith H.E., Nance J., Wang J., Van Doren C., Begley R., Jones S.J.M., Davis E.B., Scherer S., Ward S., et al. A global profile of germline gene expression in C. elegans. Mol. Cell. 2000;6:605–616. doi: 10.1016/s1097-2765(00)00059-9. [DOI] [PubMed] [Google Scholar]
- Rice W.R. Sex chromosomes and the evolution of sexual dimorphism. Evol. Int. J. Org. Evol. 1984;38:735–742. doi: 10.1111/j.1558-5646.1984.tb00346.x. [DOI] [PubMed] [Google Scholar]
- Richards S., Liu Y., Bettencourt B.R., Hradecky P., Letovsky S., Nielsen R., Thornton K., Hubisz M.J., Chen R., Meisel R.P., et al. Comparative genome sequencing of Drosophila pseudoobscura: Chromosomal, gene, and cis-element evolution. Genome Res. 2005;15:1–18. doi: 10.1101/gr.3059305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaeffer S.W., Bhutkar A., McAllister B.F., Matsuda M., Matzkin L.M., O'Grady P.M., Rohde C., Valente V.L.S., Aguade M., Anderson W.W. Polytene chromosomal maps of 11 Drosophila species: The order of genomic scaffolds inferred from genetic and physical maps. Genetics. 2008;179:1601–1655. doi: 10.1534/genetics.107.086074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sniegowski P.D., Charlesworth B. Transposable element numbers in cosmopolitan inversions from a natural population of Drosophila melanogaster. Genetics. 1994;137:815–827. doi: 10.1093/genetics/137.3.815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stajich J.E., Block D., Boulez K., Brenner S.E., Chervitz S.A., Dagdigian C., Fuellen G., Gilbert J.G., Korf I., Lapp H., et al. The Bioperl toolkit: Perl modules for the life sciences. Genome Res. 2002;12:1611–1618. doi: 10.1101/gr.361602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturgill D., Zhang Y., Parisi M., Oliver B. Demasculinization of X chromosomes in the Drosophila genus. Nature. 2007;8:238–241. doi: 10.1038/nature06330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suyama M., Torrents D., Bork P. PAL2NAL: Robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 2006;34:W609–W612. doi: 10.1093/nar/gkl315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K., Subramanian S., Kumar S. Temporal patterns of fruitfly (Drosophila) evolution revealed by mutation clocks. Mol. Biol. Evol. 2004;21:36–44. doi: 10.1093/molbev/msg236. [DOI] [PubMed] [Google Scholar]
- Wang W., Thornton K., Berry A., Long M. Nucleotide variation along the Drosophila melanogaster fourth chromosome. Science. 2002;295:134–137. doi: 10.1126/science.1064521. [DOI] [PubMed] [Google Scholar]
- Wang P.J., Page D.C., McCarrey J.R. Differential expression of sex-linked and autosomal germ-cell-specific genes during spermatogenesis in the mouse. Hum. Mol. Genet. 2005;14:2911–2918. doi: 10.1093/hmg/ddi322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C.I., Xu E.Y. Sexual antagonism and X inactivation—The SAXI hypothesis. Trends Genet. 2003;19:243–247. doi: 10.1016/s0168-9525(03)00058-1. [DOI] [PubMed] [Google Scholar]
- Yang Z. PAML4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007;24:1586–1591. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- Yang S., Arguello J.R., Li X., Ding Y., Zhou Q., Chen Y., Zhang Y., Zhao R., Brunet F., Peng L., et al. Repetitive element- mediated recombination as a mechanism for new gene origination in Drosophila. PLoS Genet. 2008;4:e3. doi: 10.1371/journal.pgen.0040003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q., Zhang G., Zhang Y., Xu S., Zhao R., Zhan Z., Li X., Ding Y., Yang S., Wang W. On the origin of new genes in Drosophila. Genome Res. 2008;8:1446–1455. doi: 10.1101/gr.076588.108. [DOI] [PMC free article] [PubMed] [Google Scholar]