

Abstract
Iron-sulfur cluster-dependent interconversion of iron regulatory protein 1 (IRP1) between its RNA binding and cytosolic aconitase (c-acon) forms controls vertebrate iron homeostasis. Cluster removal from c-acon is thought to include oxidative demetallation as a required step, but little else is understood about the process of conversion to IRP1. In comparison with c-aconWT, Ser138 phosphomimetic mutants of c-acon contain an unstable [4Fe-4S] cluster and were used as tools to further define the pathway(s) of iron-sulfur cluster disassembly. Under anaerobic conditions cluster insertion into purified IRP1S138E and cluster loss on treatment with NO regulated aconitase and RNA binding activity over a similar range as observed for IRP1WT. However, activation of RNA binding of c-aconS138E was an order of magnitude more sensitive to NO than for c-aconWT. Consistent with this, an altered set point between RNA-binding and aconitase forms was observed for IRP1S138E when expressed in HEK cells. Active c-aconS138E could only accumulate under hypoxic conditions, suggesting enhanced cluster disassembly in normoxia. Cluster disassembly mechanisms were further probed by determining the impact of iron chelation on acon activity. Unexpectedly EDTA rapidly inhibited c-aconS138E activity without affecting c-aconWT. Additional chelator experiments suggested that cluster loss can be initiated in c-aconS138E through a spontaneous nonoxidative demetallation process. Taken together, our results support a model wherein Ser138 phosphorylation sensitizes IRP1/c-acon to decreased iron availability by allowing the [4Fe-4S]2+ cluster to cycle with [3Fe-4S]0 in the absence of cluster perturbants, indicating that regulation can be initiated merely by changes in iron availability.
Iron regulatory protein 1 (IRP1)3 is a cytosolic iron-regulated RNA-binding protein that post-transcriptionally regulates the synthesis of proteins required for the maintenance of iron homeostasis in animal cells. IRP1 dictates mRNA fate by binding to iron-responsive elements (IREs) in at least six mRNA encoding key proteins that directly control the uptake and metabolic fate of iron. Excess iron promotes inactivation of IRP1 RNA binding through two mechanisms. The first involves insertion of a [4Fe-4S] cluster converting IRP1 to the cytosolic isoform of aconitase (c-acon), the so-called iron-sulfur switch. A second mechanism relies on iron-mediated degradation of the IRP1 apoprotein (1–3). Several studies have demonstrated that dysregulation of IRP expression can be deleterious and even lethal, thereby further focusing efforts on the elucidation of the mechanisms through which these central regulators of iron metabolism are controlled (4, 5). This is particularly relevant to c-acon/IRP1 because the c-acon form can be up to 100-fold more abundant than the IRP1 RNA-binding form (6, 7), predicting harmful consequences should unregulated loss of the iron-sulfur cluster occur (4, 5). The mechanism(s) of conversion of c-acon to IRP1 is the subject of this investigation.
Generation of IRE RNA binding activity from c-acon requires complete removal of the iron-sulfur cluster along with reduction of critical Cys (8). The generally held view is that iron-sulfur cluster loss from c-acon is not kinetically feasible in response to iron deficiency without the action of agents that directly promote this process (9). Studies to date have focused on the role of cluster perturbants such as reactive oxygen and nitrogen species in promoting cluster removal (2, 10–12). With oxidants, a [3Fe-4S]+ cluster is initially formed by oxidative removal of Fea (13–15). When NO is the perturbant, the [3Fe-4S]+ cluster is not an obligatory intermediate, and complete disruption has been observed (16, 17).
Although both solvent accessibility of the iron-sulfur cluster in aconitases and the lack of direct ligation of the fourth iron of the cluster (Fea) to a protein moiety facilitates cluster removal by perturbants, many questions remain concerning this process (12, 18). For example, it is not clear whether perturbant action is required for generation of RNA binding activity. It is notable that members of the aconitase family differ substantially with respect to the stability of their iron-sulfur cluster, ranging from the spontaneous instability observed in Escherichia coli aconitase B (AcnB) to mammalian c-acon, which appears to have one of the most stable iron-sulfur clusters in this protein family (19, 20). Although the relative stability of c-acon may limit maladaptive responses to cluster perturbants, it raises questions concerning the efficacy of the iron-sulfur switch in controlling IRP1 function. Consequently, a mechanism to enhance the sensitivity of the iron-sulfur cluster in a pool of c-acon to perturbants could provide a key physiological means for generating IRE binding activity while limiting its potential pathological impact.
Previous studies suggest that Ser138 phosphorylation of IRP1 provides a mechanism to control the stability of the iron-sulfur cluster in c-acon and to alter the set point for iron regulation of IRP1 (4, 10, 21). First, Ser138 phosphomimetic mutants of IRP1 (IRP1S138D and IRP1S138E) can assemble a [4Fe-4S] cluster and exhibit aconitase activity as the purified protein or when supporting aconitase-dependent growth in yeast in vivo. In both cases the iron-sulfur cluster displayed markedly increased sensitivity to oxygen or reactive oxygen species and altered accumulation of a likely intermediate in the conversion of c-acon to IRP1 (10, 21). Second, the capacity of both IRP1S138D and IRP1S138E to support aconitase-dependent growth in yeast was more sensitive to iron depletion than that of IRP1WT, implying that the phosphomimetic mutants exhibit increased rates of cluster removal in vivo (21). Third, in mammalian cells Ser138 phosphomimetic mutants fail to accumulate as c-acon (4, 22), suggesting that enhanced cluster disassembly alters the balance between c-acon and IRP1. Taken together, these studies indicate that the Ser138 phosphomimetic mutants would be useful tools for further elucidating the mechanisms responsible for the conversion of c-acon to IRP1.
In this study we have investigated the impact of cluster perturbants and iron chelators on the capacity for cluster-dependent regulation of Ser138 phosphomimetic mutants of IRP1. In vitro cluster reconstitution studies revealed that IRP1S138E exhibits reciprocal regulation of RNA binding and aconitase activities but with markedly greater sensitivity to NO. Interestingly, c-aconS138E was strongly inhibited by iron chelators that were without effect on c-aconWT. Following anaerobic incubation with EDTA, conversion of c-aconS138E from the [4Fe-4S] to a [3Fe-4S] form was detected, suggesting enhanced 4Fe-to-3Fe cycling and spontaneous nonoxidative demetallation of the iron-sulfur cluster in the Ser138 phosphomimetic mutant. Our results support a model wherein Ser138 phosphorylation allows for regulation of IRP1 via the iron-sulfur switch mechanism in a manner that enhances responsiveness to changes of intracellular iron status or oxidative/nitrosative stress while preventing potentially lethal overaccumulation of IRP1 through iron-mediated degradation of the protein.
EXPERIMENTAL PROCEDURES
Materials—Unless noted otherwise, all of the reagents were obtained from Sigma. Ferrous ethylene diammonium sulfate was from GFS Chemicals (Powell, OH). NifS protein was a generous gift from Dennis Dean (Virginia Tech). Diethylamine NONOate was from Caymen Chemical (Ann Arbor, MI). Trisodium citrate and 2-mercaptoethanol were from Fluka (Milwaukee, WI). Tris was from Fisher. BCA protein assay and chemiluminescent substrate (Super Signal) for immunoblots were from Pierce.
IRP1 Purification and Iron-Sulfur Cluster Reconstitution—IRP1 was expressed in and purified from yeast as described (10). The [4Fe-4S] cluster was reconstituted by anaerobic addition of a 10-fold molar excess of Fe2+ (ferrous ethylene diammonium sulfate) and l-cysteine, plus dithiothreitol (5–10 mm), and NifS protein (final concentration, 0.1 mg/ml) at 25 °C in an anaerobic chamber (Coy, Grass Lake, MI; 95% N2, 5% H2; <5 ppm O2) (23). After 16–20 h the reconstitution salts were removed by anaerobic gel filtration over Sephadex G-50 (medium, 50–150-μm particle size) in 90 mm Hepes, pH 7.5, 10% glycerol. Total IRP1/c-acon protein after desalting was quantified by Coomassie-stained SDS-PAGE using a bovine serum albumin standard curve.
Aconitase Activity—Aconitase activity was determined by monitoring the conversion of isocitrate to cis-aconitate (ε = 3.6 mm–1 cm–1) at 240 nm (25, 26). A unit of activity is defined as converting 1 μmol of substrate/min at 25 °C. The assay was performed in duplicate in Tris buffer (pH 8.0, 0.1 m) containing 20 mm dl-isocitrate against a blank lacking protein. Units of activity were converted to moles of c-acon using a conversion factor of 3.3 units/nmol cluster (24). Control reactions lacking iron yielded no aconitase activity.
Nitric Oxide and EDTA Treatments of Reconstituted c-acon—In an anaerobic chamber varying concentrations of the NO source diethylamine NONOate (DEANO, stabilized in pH 8.7 TAPS buffer or 0.01 n NaOH at 4 °C) were diluted into a solution of c-acon (5–6 units/ml aconitase, in 90 mm Hepes, 10% glycerol, pH 7.5). Upon dilution into pH 7.5 Hepes buffer at 25 °C, DEANO decays to produce NO (measured t½ = 10–12 min). As a control the aconitase activity of the protein solution in the absence of DEANO was monitored concurrently. For EDTA experiments c-aconWT or c-aconS138E was diluted to an enzymatic activity of 3–6 units/ml, and EDTA or buffer control was added. After anaerobic incubation with EDTA, aconitase activity was measured.
Electron Paramagnetic Resonance Spectroscopy—X-band EPR spectra were obtained at liquid helium temperatures on a Bruker E500 ELEXYS spectrometer with 100-kHz field modulation, equipped with an Oxford Instrument ESR-9 helium flow cryostat and a DM-0101 cavity.
RNA Binding Activity—For purified IRP1 and iron-sulfur cluster-reconstituted c-acon, the electrophoretic mobility shift assay (EMSA) method was performed as described except that EDTA was omitted from the gel and the running buffer because of its reactivity with IRP1S138E (27). Binding reactions between IRP1/c-acon and [32P]RNA (l-ferritin 73-nucleotide mRNA fragment containing IRE) were set up, incubated anaerobically, and loaded onto the gel outside the anaerobic chamber. Filter binding assays were performed as described (28), using 25-mm nitrocellulose filters (Protran BA 85, Whatman/Schleicher & Schuell) except that all steps were carried out in the anaerobic chamber. Briefly, the protein samples were diluted into binding buffer containing a 5–10-fold molar excess of radiolabeled IRE and incubated for 10 min on ice before passing through a nitrocellulose filter. The filter was washed once with fresh binding buffer. The filter and the flow-through/wash were collected in separate scintillation vials and counted. For extracts of HEK 293 cells an RNA binding supershift assay was performed (4). The lysates were anaerobically incubated in binding buffer with 1 μg of anti-Myc antibody for 2 h at 4 °C. [32P]RNA (final concentration, 2 nm) was added and incubated for 10 min. After the addition of heparin (0.5 mg/ml final), the reactions were loaded onto a 8% polyacrylamide gel prepared in Tris borate buffer, pH 8.0, lacking EDTA and run at 4 °C to separate free, proteinbound (endogenous IRP1 and IRP2) and supershifted (IRP1 antibody-bound) RNA.
Mammalian Cell Culture—HEK 293 cells expressing Myc-tagged IRP1WT or Myc-tagged IRP1S138E were plated at 0.5–1.0 × 106/ml, induced with tetracycline (+tet) or not (–tet) for 36 h under hypoxia (1% O2/5% CO2/94% N2) or under normoxia (normal incubator conditions), and harvested anaerobically in phosphate-buffered saline with 1 mm citrate (4). The cells were lysed inside the anaerobic chamber in four volumes of lysis buffer (LB, 20 mm Hepes, pH 7.5, 100 mm NaCl, 1% Triton X-100, 1 mm citrate, and protease inhibitor mixture as described (4) but omitting EDTA). After centrifugation at 14,000 rpm for 10 min using O-ring-sealed tubes, the clarified lysate was assayed for aconitase activity within 2 h of lysis. Protein content was determined by the BCA assay. To calculate aconitase activity caused by recombinant protein, the endogenous (–tet) aconitase activity was subtracted from the (+tet) aconitase activity. Tet treatment had no impact on aconitase activity in nontransfected cells.4 Immunoblot analysis confirmed the induction of Myc-IRP1 in lysates of tet-induced cells, and no Myc-IRP1 was detected in lysates from uninduced cells.
RESULTS
A Ser138 Phosphomimetic Mutant of IRP1 Is Regulated by the Iron-Sulfur Switch in Vitro—Previous studies had suggested that Ser138 phosphorylation might completely uncouple IRP1 from the iron-sulfur switch mechanism, allowing for its regulation only through iron-mediated protein degradation (22). However, we reasoned that although the iron-sulfur cluster in Ser138 phosphomimetic mutants of c-acon is extremely sensitive to disruption, they would still be regulated by the iron-sulfur switch mechanism depending on the level of cluster perturbants present. Thus, these mutants could serve as useful models allowing further elucidation of the process(es) of cluster disassembly. An important premise for this hypothesis is that Ser138 phosphomutants of IRP1 should be able to undergo the traditional iron-sulfur switch mechanism in vitro and exhibit a reciprocal relationship between RNA binding and aconitase activities (8, 29). To test this, we used IRP1S138E because it displays the strongest instability phenotype. In the following sections cluster-containing IRP1 is referred to as c-acon.
After anaerobic iron-sulfur cluster reconstitution, the IRP1WT and IRP1S138E displayed substantial aconitase activity (Fig. 1A). No aconitase activity was observed for either protein in the absence of reconstitution (data not shown). The impact of cluster reconstitution on RNA binding activity was determined by EMSA (Fig. 1, B and C). RNA binding activity of IRP1WT and IRP1S138E was decreased by 81 and 53%, respectively, after cluster reconstitution. Previous studies have shown that treatment of c-acon with 2% 2-mercaptoethanol allows c-acon to bind RNA, permitting determination of the total amount of RNA binding activity in a mixture of IRP1 and c-acon (30). As expected total 2-mercaptoethanol-induced RNA binding activity of IRP1WT and IRP1S138E was unaffected by reconstitution (Fig. 1C). Hence, under anaerobic conditions the classical iron-sulfur switch can operate in vitro in IRP1S138E over a similar range as observed for IRP1WT.
FIGURE 1.

Regulation of RNA binding activity in IRP1WT and IRP1S138E by the iron-sulfur switch. Anaerobic reconstitution of IRP1 with Fe2+, l-cysteine, dithiothreitol, and NifS leads to 40–60% yield of aconitase activity and a decrease in RNA binding activity. The data were averaged from independent experiments (n = 4 for IRP1WT; n = 8 for IRP1S138E). The bars represent the means. The error bars show the S.E. A, aconitase activity of cluster-reconstituted protein. B, representative EMSA in the absence (–) or presence (+) of 2% 2-mercaptoethanol (2-ME) (details under “Experimental Procedures”). C, RNA binding activity of cluster-reconstituted IRP1WT and IRP1S138E expressed as a percentage of control (reduced apo) protein in the absence and presence of 2% 2-mercaptoethanol. An asterisk indicates value significantly different from control (Student's t test, two-tailed, p ≤ 0.05). WT, wild type; C, control; R, reconstituted.
Altered Set Point for Regulation of RNA Binding and Aconitase Activity in Ser138 Phosphomimetic Mutants—We then asked whether the set point for regulation of the iron-sulfur switch by NO, an established cluster perturbant (16, 17, 31), was altered in Ser138 phosphomimetic mutants. We first compared the NO sensitivity of the iron-sulfur cluster in c-aconWT and the control, c-aconS138A, with that in the phosphomimetic mutants c-aconS138E and c-aconS138D. The NO generator DEANO was used.
In the presence of a 200-fold molar excess of DEANO, c-aconWT and c-aconS138A were stable, with more than 90% of aconitase activity being retained over the 80-min experiment (Fig. 2A). From these results the half-life of active aconitase enzyme for c-aconWT and c-aconS138A was estimated to be greater than 10 h. In contrast, the Ser138 phosphomimetic mutants c-aconS138D and c-aconS138E displayed greatly enhanced sensitivity to NO. The half-lives of enzymatically active c-aconS138D and c-aconS138E were 7 and 2 min, respectively, 2 orders of magnitude shorter than for IRP1WT (Fig. 2A).
FIGURE 2.

Differential dose-dependent effect of nitric oxide on aconitase and RNA binding activity of IRP1WT and IRP1S138E. A, effect of a 200-fold molar excess of DEANO, relative to c-acon, on acon activity of reconstituted c-aconWT, c-aconS138A, c-aconS138D, and c-aconS138E ([c-acon], ∼1.7 μm). Best fit lines and standard deviations were calculated using the data shown (51). Half-lives (±S.D.) were >10 h for c-aconWT and c-aconS138A, 7 ± 3 min for c-aconS138D, and 2 ± 1 min for c-aconS138E. B, aconitase activity was measured after 30 min of exposure to DEANO or carrier buffer (control) and is expressed as fraction of activity remaining relative to control. Starting [c-acon] was 1.5–2.0 μm. IC50 is defined as the molar ratio of DEANO to enzyme that effects a 50% loss of aconitase activity after 30 min. IC50 values, calculated using best fit method as in A, are 224 ± 83 for c-aconWT, 267 ± 121 for c-aconS138A, 36 ± 7 for c-aconS138D, and 7 ± 1 for c-aconS138E. C and D, reconstituted c-aconWT or c-aconS138E was treated with DEANO in varying concentrations and analyzed for acon activity and RNA binding by filter binding under anaerobic conditions. Aconitase (filled symbols) and RNA binding (open symbols) are shown. The data represent the average values from two or three independent experiments. For c-aconWT (C) a range of [DEANO] from 0 to 1 mm is shown. For c-aconS138E (D) a range of [DEANO] from 0 to 0.5 mm is shown. WT, wild type.
Based on the striking differences in half-lives of the iron-sulfur cluster in c-aconS138D and c-aconS138E compared with c-aconWT after exposure to NO, a more thorough characterization of the instability was desired. Hence, the dose dependence of the DEANO effect was determined. A 50% loss of aconitase activity in c-aconWT required a ratio of DEANO to c-aconWT of 220:1. In contrast, only a 36:1 ratio was required to achieve the same percentage loss of activity in c-aconS138D and only a 7:1 ratio for c-aconS138E (Fig. 2B). We conclude that on a dosage basis, the phosphomimetic mutants are 6–30-fold more sensitive to the cluster perturbant NO than is WT.
Given the greater sensitivity to NO-dependent cluster disassembly in the Ser138 phosphomimetic mutants of c-acon, we asked whether this translated into enhanced generation of RNA binding activity at low NO concentration. A strictly anaerobic filter binding assay was used to determine RNA binding activity. RNA binding activity of IRP1WT was maximally increased by 4-fold at 1.0 mm DEANO (Fig. 2C). The RNA binding activity of IRP1S138E was maximally increased 2-fold in the presence of 0.1 mm DEANO, a 10-fold lower concentration than observed for c-aconWT (Fig. 2D). The observation that aconitase activity declined more than the RNA binding activity increased for both IRP1WT and IRP1S138E likely indicates the presence of additional forms of IRP1/c-acon not active in RNA binding or aconitase activity (16, 17, 31).
In HEK Cells IRP1S138E Displays an Altered Response to Hypoxia and Nitric Oxide—Because the experiments with purified protein demonstrated that IRP1S138E can be regulated by the iron-sulfur switch and that the set point for this regulation is altered, we further probed the effect of enhanced iron-sulfur cluster disassembly on the function and regulation of IRP1. To accomplish this we determined the impact of hypoxia versus normoxia on IRP1, c-acon, and related species for IRP1WT and IRP1S138E expressed in HEK cells. It has been shown that hypoxia generally causes a shift of IRP1 to the aconitase form with a decrease in RNA binding activity in several cell lines, although the impact of hypoxia on IRP1 may depend on cell type (12, 32–34). This decrease in RNA binding activity is in contrast to IRP2, which is stabilized in hypoxia, increasing its RNA binding activity (35). Using activity assays and immunoblots, we determined the impact of oxygen tension on the abundance of the RNA-binding, aconitase, and inactive forms of IRP1 in HEK cells expressing IRP1WT and IRP1S138E from a tetracycline-inducible promoter. Finally, we determined whether the c-aconS138E generated in cells recapitulated the NO sensitivity phenotype observed with in vitro reconstituted protein.
HEK 293 cells were exposed to normoxia or to hypoxia (1% O2) for 36 h with or without tet. The aconitase activity attributable to the IRP1WT or IRP1S138E transgenes was determined by subtracting the activity obtained in the absence of tet (endogenous cellular activity) from the total activity obtained in the presence of tet (endogenous plus tet-induced activity). RNA binding was determined by antibody supershift EMSA (see supplemental Figs. S1 and S2). IRP1WT displayed a pattern of increased aconitase and reduced RNA binding activity in response to hypoxia similar to what others have observed (32, 34), although the differences were not statistically significant (Fig. 3A). In contrast, no aconitase activity was detected for IRP1S138E in normoxic conditions. However, significant albeit low levels of aconitase activity were observed for c-aconS138E in hypoxia, indicating that it is a bona fide substrate for the iron-sulfur cluster biogenesis machinery in cells. Interestingly, RNA binding activity attributable to IRP1S138E displayed an unanticipated lack of response to hypoxia (Fig. 3A). Significantly, the binding activity of IRP1S138E from cells grown in hypoxia was 18-fold higher than for IRP1WT.
FIGURE 3.

Effect of hypoxia on aconitase and RNA binding activity of IRP1WT and IRP1S138E expressed in HEK 293 cells. A, calculated distribution of aconitase and RNA-binding forms for WT (n = 3) and S138E (n = 4) in cells grown under normoxic (N) or hypoxic (Hy) conditions. The activities and protein levels are for the tet-induced IRP1; endogenous (–tet) aconitase activities or protein levels have been subtracted. RNA binding was determined using antibody supershift EMSA with the anti-Myc antibody. The mass of the c-acon pool was determined using the established specific activity of purified c-acon, and the mass of the RNA binding pool was determined by using the specific radioactivity of the [32P]RNA used for EMSA and assumed a 1:1 IRP1/IRE ratio under saturating conditions (see “Experimental Procedures”). Panel i, aconitase activity attributable to IRP1WT or IRP1S138E was determined. For normoxic S138E lysate acon activity, the resulting value was below zero, denoted by #. Panel ii, effect of hypoxia on RNA binding activity of IRP1WT and IRP1S138E was also determined. Although the mean RNA binding appeared to decrease for IRP1WT in hypoxia, the difference was not statistically significant. The mean RNA binding activity of IRP1S138E was not reduced by hypoxia. Panel iii, total active forms of IRP1/c-acon was determined by summing the aconitase and RNA binding activity for the wild type and S138E phosphomutant under each condition using the data in Panels i and ii. Panel iv, IRP1 protein was quantified in lysates by anti-IRP1 immunoblot using purified recombinant IRP1 for the standard curve. An asterisk indicates hypoxia value significantly different from corresponding normoxia control (Student's t test, p ≤ 0.05). B, pie chart representation of the data in A. The percentages of forms were calculated by dividing the active form (pmol/mg lysate protein) by the total IRP1 protein (pmol/mg lysate protein) from the quantitative immunoblot. When activity was below the endogenous background, which occurred only for acon activity of S138E lysates in normoxia, a value of zero percent was assigned. C, freshly prepared lysates from HEK cells were anaerobically treated with DEANO or an equivalent volume of carrier buffer (control). After a 15-min incubation each reaction was quenched in assay mix (0.1 m Tris, pH 8.0, 20 mm dl-isocitrate), and aconitase activity was measured. Endogenous activity (–tet) was subtracted from total activity (+tet) to determine the activity caused by c-acon encoded by the transgene. Starting aconitase activity for c-aconWT was 40.3 ± 12.4 millinunits/mg protein, whereas for c-aconS138E it was 8.7 ± 1.8 milliunits/mg protein (means ± S.E., n = 3). An asterisk denotes c-aconS138E activity significantly lower than corresponding c-aconWT activity (Student's t test, two-tailed, unpaired, n = 3, p < 0.05). WT, wild type.
To further understand the impact of hypoxia on IRP1WT and IRP1S138E, we quantified the size of the different pools of IRP1 protein under normoxic and hypoxic conditions. The reciprocal pattern observed in aconitase and RNA binding activity for IRP1WT noted above, with no significant change in IRP1WT protein level, is consistent with what others have observed (Fig. 3A) (32, 34). When comparing the total amount of active forms relative to the amount of total immunoreactive IRP1 protein, we observed that 54% of IRP1WT was present as an inactive form(s) under normoxia, which decreased to 30%, in hypoxia (Fig. 3B). Inactive forms of IRP1 that lack both enzymatic and RNA binding activities have been observed (16, 31, 36). These include the oxidized apo and the [3Fe-4S]+ forms of the protein (8, 21, 30, 37). For the S138E protein a different picture emerged. First, total active forms for the phosphomimetic mutant increased almost 2-fold in hypoxia (Fig. 3A). Second, in normoxia a greater fraction of IRP1S138E was in the RNA-binding form (46%) than was the case for IRP1WT (13%), whereas the same fractional amount of both proteins (54%) was not active for either aconitase or RNA binding (Fig. 3B). However, the inactive pool of IRP1S138E appeared to be more strongly affected by hypoxia because it decreased to 19% of total IRP1 protein, a 65% decrease (Fig. 3B).
Because we showed enhanced sensitivity to NO for c-aconS138E in an in vitro system (Fig. 2), we wished to confirm the phenotype with c-acon generated in cells. Lysates from cells grown hypoxically were exposed to DEANO. A substantial loss of activity was observed for c-aconS138E even at 0.01 mm DEANO, with a corresponding IC50 of 0.02 mm DEANO. Use of 1 mm DEANO abolished detectable c-aconS138E activity (Fig. 3C). In contrast, c-aconWT retained most of its activity, and only showed significant losses at a level of 1 mm DEANO, with a corresponding IC50 of 2.6 mm DEANO. On the basis of these IC50 values, the cluster in c-aconS138E was about 100-fold more sensitive than that in c-aconWT. This confirms a NO sensitivity phenotype for an Ser138 phosphomimetic mutant of c-acon generated under cellular conditions.
Taken together, these results underline the tendency of IRP1S138E to accumulate in the RNA-binding form as a consequence of increased disassembly of the iron-sulfur cluster in c-aconS138E, whereas IRP1WT accumulates as c-acon. We suggest that Ser138 phosphorylation sensitizes IRP1 to regulation by oxidative or nitrosative stress.
The [4Fe-4S] Cluster of c-aconS138E Displays Enhanced Sensitivity to Iron Chelation—The enhanced instability of the iron-sulfur cluster in c-aconS138E is reminiscent of the properties of other aconitases with unique physiological roles (19, 20). For example, in contrast to mammalian m-acon, which is stable in the presence of EDTA (25), bacterial AcnB loses activity rapidly in the presence of EDTA (19). The instability of the cluster in AcnB allows greater sensitivity to environmental change.
Because c-aconS138E is more responsive to changes in iron status relative to c-aconWT (21), we determined whether it also exhibited enhanced sensitivity to iron chelators. We determined the effect of EDTA on aconitase activity in cluster-reconstituted c-aconWT and c-aconS138E under anaerobic conditions. There was no effect of 5 mm EDTA on c-aconWT (Fig. 4A). In contrast, c-aconS138E was much more sensitive to EDTA, showing an estimated half-life of less than 10 min under the same conditions. In the presence of citrate, which stabilizes the cluster by binding to Fea (25, 37), the EDTA-dependent loss of activity was inhibited (Fig. 4A). When c-aconS138E was treated with 1 mm EDTA, the half-time for loss of activity was less than 10 min (Fig. 4B). Under these conditions, RNA binding activity was stimulated nearly 2-fold after treatment of c-aconS138E for 30 min.5 The inactivation of c-acon was substantially blocked by 1 mm Fe2+ (Fig. 4B). For c-aconS138E a molar ratio of EDTA/c-aconS138E as low as 40 inactivated the enzyme (Fig. 4C). The sensitivity of c-aconS138E to EDTA was notable in light of the different responses of stable and unstable aconitases to EDTA (19, 25).
FIGURE 4.

Differential effect of EDTA on the [4Fe-4S] Cluster in c-aconS138Eversus c-aconWT. A, anaerobic loss of aconitase activity. The data were normalized to a control reaction with either c-aconWT or c-aconS138E that did not contain EDTA. Filled diamonds, c-aconWT + 5 mm EDTA; filled circles, c-aconS138E + 5 mm EDTA; open circles, c-aconS138E + 5 mm EDTA + 2 mm citrate (cit). B, anaerobic exposure of c-aconS138E to 1 mm EDTA (circles), 1 mm Fe2+ (squares), or 1 mm EDTA plus 1 mm Fe2+ (triangles). C, effect of EDTA concentration on activity of c-aconS138E after 30 min. D, anaerobic dialysis of c-aconS138E or c-aconWT against 50 mm Tris, pH 8.0, with 0.5% (w/v) chelex. Acon activity in dialyzed samples and control were measured, normalized to protein concentration determined after dialysis, and expressed as percentages of control. The bars represent the means. The error bars show S.E. The percentage of activity remaining in c-aconS138E was significantly different from in c-aconWT, denoted by an asterisk (Student's t test, two-tailed, unpaired, p ≤ 0.05). E, EPR spectra. Top spectrum, c-aconWT (10 μm) following titration with [Fe(CN)6]3–. Middle spectrum, c-aconS138E (18 μm) following treatment with EDTA (2 mm) for 2.5 h. Bottom spectrum, c-aconS138E (18 μm) without treatment with EDTA. All of the samples were treated anaerobically in the presence of tricarballylate (1 mm, substrate analog). The samples were desalted, transferred to 4 mm O.D. quartz tubes under N2 in the presence of 1 mm tricarballylate, frozen in liquid nitrogen, and kept frozen on dry ice during transportation. The spectrometer conditions were: microwave frequency, 9.6336 GHz; modulation amplitude, 5 G; sweep time, 41.94 s; time constant, 40.96 s; microwave power, 0.1 milliwatt; temperature, 7.5 K. WT, wild type.
Whether EDTA was attacking the cluster directly by entering the active site cleft or scavenging Fe2+ ions released spontaneously could not be distinguished by the experiments described thus far. Although m-acon does not bind EDTA (25), whether c-aconS138E can bind the chelator is not known. Therefore, we wished to find out whether it was possible for iron to be lost from cluster without the direct action of a chelator. An anaerobic dialysis experiment was performed in which a metal chelating resin (Chelex) was present in the dialysis chamber but separated from the protein by a membrane. Under these conditions, c-aconS138E lost 26% of its enzymatic activity in 30 min (Fig. 4D), suggesting spontaneous release of iron from the cluster in c-aconS138E. When c-aconWT was treated in the same way, there was no significant loss of aconitase activity (Fig. 4D). Hence, for c-aconS138E, aconitase activity can be lost without direct contact of a chelator.
[3Fe-4S]-c-aconS138E Is among the Products When c-aconS138E Is Treated with EDTA—We reasoned that if EDTA were trapping iron (Fea) released from the c-aconS138E, a [3Fe-4S] cluster should be formed in c-aconS138E under mild conditions. Previous experiments failed to reveal [3Fe-4S]+ by EPR spectroscopy when c-aconS138E was treated with the strong oxidant ferricyanide6 or when yeast cells expressing IRP1S138E were exposed to H2O2 (21). We treated c-aconS138E anaerobically with EDTA until at least 50% of its aconitase activity had been lost and then attempted to observe a signal for a [3Fe-4S] cluster by EPR. A signal at g = 2.01 indicative of an oxidized [3Fe-4S]+ cluster was observed (Fig. 4E, middle spectrum). By comparison with a positive [3Fe-4S]+ control prepared by ferricyanide titration of c-aconWT (Fig. 4E, top spectrum), we estimate that the [3Fe-4S]+ signal for the c-aconS138E represents ∼50% of the total c-aconS138E. A sample of c-aconS138E with the same starting concentration was incubated in parallel without EDTA, did not lose activity, and did not generate a detectable [3Fe-4S]+ signal (Fig. 4E, bottom spectrum). Therefore, we conclude that the [3Fe-4S]+ signal is correlated with EDTA-dependent loss of aconitase activity. Because the proportion of enzyme inactivated was nearly the same as the proportion giving rise to the [3Fe-4S]+ signal, subsequent oxidation of [3Fe-4S]0 in the frozen solution appears to have been surprisingly quantitative. A plausible oxidation mechanism would be the interaction of diffused O2 with iron complexes to produce reactive oxygen species capable of the required univalent oxidation (38). Experiments are underway to determine the oxidation pathway. Our observations suggest that iron chelation shifts the equilibrium in favor of [3Fe-4S]-c-aconS138E, which is readily oxidized to [3Fe-4S]+-c-aconS138E. Detection of the [3Fe-4S]+ cluster after treatment with a chelator supports the concept that there is enhanced 4Fe to 3Fe cycling in Ser138 phosphomimetic mutants of c-acon.
DISCUSSION
The mechanism through which IRP1 RNA binding activity is generated from c-acon remains one of the critical unresolved issues concerning how the activity of this central regulator of iron metabolism is modulated. In this paper we have used phosphomimetic mutants of IRP1 that assemble a highly unstable iron-sulfur cluster to address the key issue of how iron-sulfur cluster disassembly in c-acon occurs and can be controlled. We approached this in two ways. First, using the IRP1S138E phosphomimetic mutant we showed that loss of the iron-sulfur cluster in c-acon may be initiated not only through perturbant-mediated (e.g. NO) pathways but also through a perturbant-independent process. This latter process represents a previously unrecognized mechanism through which changes in cellular iron status alone could be sufficient to initiate the conversion of c-acon to IRP1. Additionally, we observed that the set point for reciprocal regulation of aconitase and RNA binding activity by NO is altered in Ser138 phosphomimetic mutants in that they are significantly sensitized to low and presumably more physiological, concentrations of perturbant (39–41). Second, our findings suggest that Ser138 phosphorylation significantly alters the functional distribution of IRP1/c-acon in cells favoring accumulation of the RNA-binding form. Taken together, our studies expand the regulatory scenarios available for the control of IRP1 RNA binding activity and further substantiate its key role in animal cell iron metabolism.
Unique Roles of Aconitases Are Tied to the Stability of Their [4Fe-4S] Clusters—The inherent difference in iron-sulfur cluster stability in aconitases has a key role in the adaptive responses of cells to changes in growth state, oxidative stress, and iron status (20, 42). This idea is clearly illustrated through the selective expression of different aconitases in E. coli. AcnB, which has an unstable iron-sulfur cluster, is profoundly sensitive to iron depletion and oxidative stress. In iron deficiency, AcnB is inactivated by spontaneous nonoxidative demetallation of the iron-sulfur cluster. This allows accumulation and cellular export of citrate, which may facilitate scavenging and uptake of iron by the ferric citrate iron importer (19, 42). In contrast, AcnA, which has the more stable iron-sulfur cluster, is induced in oxidative stress, presumably allowing greater citric acid cycle flux than would AcnB under this situation. Thus, selective use of aconitase isoforms allows control of metabolism that is responsive to the environment (19, 42).
In eukaryotes the reduced stability of the iron-sulfur cluster in m-acon relative to c-acon is thought to permit enhanced responsiveness of the mitochondrial isoform to oxygen radicals as part of a feedback loop between the respiratory chain and the citric acid cycle (20). The stable nature of the iron-sulfur cluster in c-acon has raised questions about its role as an iron sensor: how is the stable cluster of c-acon disassembled to generate RNA binding activity in response to low iron conditions (3, 12, 18)? Initial removal of the labile iron atom, Fea, which is not directly ligated by a protein moiety, followed by full loss of the cluster is one model. In this model, the initial conversion of the [4Fe-4S]2+ form to the [3Fe-4S]+ form requires oxidative demetallation (13, 38, 43). Consequently cluster perturbants have been viewed as required participants in the activation of IRP1 RNA binding activity from c-acon even during iron deficiency. However, the relative stability of (nonphosphorylated) c-acon, especially in the presence of substrate, implies that high levels of perturbants are likely required for this process (8, 12, 24). On the basis of the work described herein and in previous reports (10, 21), Ser138 phosphorylation appears to provide a mechanism to tune the sensitivity of the iron-sulfur switch in c-acon so that a fraction of the protein (the phosphorylated fraction) can respond with more rapidity and sensitivity to changes in the level of iron and reactive oxygen or nitrogen species. Support for this concept comes from the finding that only a small portion (<5%) of rat liver c-acon responded to iron deficiency through apparent conversion to IRP1 (6). The vast majority of enzyme was unaffected, suggesting the existence of pools of c-acon that differ with regard to the stability of their iron-sulfur cluster (6). Taken together, these observations suggest that mechanisms such as Ser138 phosphorylation are critical for regulation of IRP1 by the iron-sulfur switch.
Evidence for a Novel Regulated Pathway of Cluster Disassembly in Ser138 Phosphorylated c-acon—We have shown that instability of the cluster in c-aconS138E drives it to exist preferentially in the RNA-binding form in cells, although it can maintain full aconitase activity in vitro under strict anaerobic conditions. The mechanism of this instability is likely to be complex. We propose a mechanism that involves nonoxidative demetallation (Fig. 5C), a pathway apparently not available to c-aconWT.
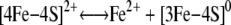 |
SCHEME 1 |
The fate of the [3Fe-4S]0 form would likely be immediate oxidation
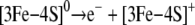 |
SCHEME 2 |
followed by complete loss of the cluster.
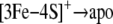 |
SCHEME 3 |
The iron that is released is not likely to be reincorporated if there are any reactive oxygen or nitrogen species or chelators present.
FIGURE 5.

A model for pathways of cluster disassembly in phosphorylated c-acon. In Ser138-phosphorylated c-acon the [4Fe-4S]2+ cluster can undergo oxidative (A) or NO-induced (B) disassembly as its nonphosphorylated counterpart does or lose Fea prior to oxidation (C). OX stands for oxidant. Although the loss of Fea is shown as a reversible process, the instability of the [3Fe-4S]0 cluster and the possible interaction of Fe2+ with other solutes are likely to render the process irreversible in the presence of reactive species, coordinating buffers, or chelators.
The equilibrium constant for Scheme 1 must be negligibly low in c-aconWT but is increased in c-aconS138E as indicated by the effect of dialysis. The addition of EDTA has no effect on c-aconWT but shifts the equilibrium to the right by removing iron from solution in the case of c-aconS138E, resulting in an observable loss of aconitase activity. The EDTA effect is inhibited by citrate or by iron as expected. Evidence for the [3Fe-4S]+ form in Scheme 2 comes from our observation of a c-aconS138E [3Fe-4S]+ cluster by EPR. Evidence for conversion to the apo form (Scheme 3) comes from the finding that the RNA-binding form preferentially accumulates in cells expressing IRP1S138E.
Nonoxidative loss of Fea has not been observed for native m-acon (25, 37), and most mechanisms of 4Fe-3Fe cycling in aconitases invoke an oxidant (37, 38, 44). However, the precedent for spontaneous nonoxidative cluster cycling lies with AcnB (19, 45). Cycling of c-aconS138E between [4Fe-4S] and [3Fe-4S] cluster forms then opens the door to ready disassembly of the unstable [3Fe-4S] cluster (21) and provides a direct link to cellular iron levels.
Multiple Pathways Contribute to the Tendency of Phosphorylated c-acon
to Lose Its [4Fe-4S] Cluster—It has been suggested that the
response of c-acon/IRP1 to iron is tied to oxidants
(43). The oxidation pathway
includes a [3Fe-4S]+ intermediate
(13,
38) or oxidized apo protein
(8). In cells further oxidation
of the [3Fe-4S]+ intermediate and/or additional factors must play a
role in generating apo IRP1 that is active for RNA binding
(9). The increased sensitivity
of Ser138 phosphomimetic mutants of c-acon/IRP1 to the oxidation
pathway (Fig. 5A) is
indicated by the hypoxia experiments described herein, as well as previous
work in vitro and in cells
(4,
10,
21). A pathway involving NO
has also been shown to activate IRP1 RNA binding
(11,
16,
31,
46,
47). In contrast to oxidants
(e.g. 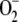 ,
ONOO–), NO is able to act independently of the
[3Fe-4S]+ intermediate and can directly increase RNA binding
activity (16,
17,
48). We have observed that
sensitivity to NO is heightened by an order of magnitude or more in the
Ser138 phosphomimetic mutants
(Fig. 5B), providing a
second mechanism of enhanced cluster loss. The cycling of 4Fe and 3Fe clusters
without a perturbant has been observed for AcnB but not for c-acon
(19). Our proposal that
phosphorylated c-acon can lose Fea without prior oxidation adds an
important and unanticipated third pathway, which is direct sensitivity to iron
depletion (Fig. 5C).
Furthermore, the greater ease with which c-aconS138E loses
Fea raises the intriguing concept that Ser138
phosphorylation may also enhance conversion of c-acon to IRP1 by uncoupling it
from the stabilizing effect of citrate in vivo because the [3Fe-4S]
forms would not be expected to bind substrate as efficiently.
,
ONOO–), NO is able to act independently of the
[3Fe-4S]+ intermediate and can directly increase RNA binding
activity (16,
17,
48). We have observed that
sensitivity to NO is heightened by an order of magnitude or more in the
Ser138 phosphomimetic mutants
(Fig. 5B), providing a
second mechanism of enhanced cluster loss. The cycling of 4Fe and 3Fe clusters
without a perturbant has been observed for AcnB but not for c-acon
(19). Our proposal that
phosphorylated c-acon can lose Fea without prior oxidation adds an
important and unanticipated third pathway, which is direct sensitivity to iron
depletion (Fig. 5C).
Furthermore, the greater ease with which c-aconS138E loses
Fea raises the intriguing concept that Ser138
phosphorylation may also enhance conversion of c-acon to IRP1 by uncoupling it
from the stabilizing effect of citrate in vivo because the [3Fe-4S]
forms would not be expected to bind substrate as efficiently.
Thus, we propose that Ser138-phosphorylated IRP1 more easily converts to the RNA-binding form under conditions in which nonphosphorylated IRP1 would tend to exist in the enzymatically active [4Fe-4S]2+ or inactive but stable [3Fe-4S]+ c-acon form. A feedback loop to turn off RNA binding activity for the phosphorylated protein in iron replete conditions, rather than cluster reassembly, could be iron-induced degradation of the IRP1 apoprotein as occurs for IRP2 (4, 22, 49, 50).
Cellular Role of Ser138 Phosphorylation—Our study further focuses interest on the physiological scenarios where IRP1 is phosphorylated at Ser138. Ser138 is phosphorylated by protein kinase C, and the RNA-binding form, rather than c-acon, is the likely substrate for kinase action (27, 52). IRP1 is Ser138-phosphorylated in normally growing tissue culture cells, and phosphorylation is stimulated by the pharmacological activator of protein kinase C, phorbol 12-myristate-13-acetate (4, 27). Protein kinase C isoforms are well established to be involved in the regulation of cell proliferation. Given that increased iron uptake and reduced iron storage are supportive pathways in the program of cell proliferation, Ser138 phosphorylation of IRP1 could provide a mechanism to promote formation of essential iron-containing proteins needed for proliferation. Our results presented here with c-aconS138E argue that enhanced destabilization of the iron-sulfur cluster, through nonoxidative demetallation and elevated action of perturbants, provides an inducible mechanism to favor accumulation of RNA binding activity. In this manner, IRP1 could alter the metabolic fate of iron to meet the unique needs of proliferating cells.
Conclusions and Future Directions—We have studied the unstable phosphomimetic mutants of IRP1/c-acon as a model for the phosphorylation at Ser138. Our studies have confirmed the cluster instability phenotype in several ways and have provided insight into the mechanisms of this instability. Thus, Ser138 phosphorylation of a pool of IRP1/c-acon renders the [4Fe-4S] cluster highly sensitive to both perturbants and iron concentration. Cycling between [4Fe-4S] and [3Fe-4S] forms, a means of sensing cellular iron requirements, is enhanced by an additional pathway that can be initiated under nonoxidative conditions. Future studies will be directed at a better understanding of specific pathways of disassembly for [3Fe-4S] clusters and toward characterizing the inactive species of IRP1/c-acon observed upon loss of the cluster from c-aconWT versus c-aconS138E.
Supplementary Material
Acknowledgments
We acknowledge Dennis Dean for NifS protein; William Walden, Helmut Beinert, Patricia Kiley, and James Imlay for helpful discussions; David Eide for comments on the manuscript; Christopher Nizzi, Xin Wei Sarah Luo and Nathan Johnson for experimental assistance; and Judith Kozminski for assistance with manuscript preparation.
This paper is dedicated to the memory of Helmut Beinert.
This work was supported in part by National Institutes of Health Grants DK 66600 (to R. S. E.) and National Biomedical ESR Center Grant EB001980. This work was also supported by United States Department of Agriculture Cooperative States Research Education and Extension Service Grant 2006-35200-16604 and University of Wisconsin-Madison Hatch Project 4885 (to R. S. E.).
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1 and S2.
Footnotes
The abbreviations used are: IRP, iron regulatory protein; IRE, iron-responsive element; c-acon, cytosolic aconitase; HEK, human embryonic kidney; Acn, bacterial aconitase; m-acon, mitochondrial aconitase; DEANO, diethylamine NONOate; EMSA, electrophoretic mobility shift assay; TAPS, 3-{[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]amino}-1-propanesulfonic acid; tet, tetracycline.
S. L. Clarke and K. M. Deck, unpublished results.
K. M. Deck, unpublished results.
M. C. Kennedy, personal communication.
References
- 1.Muckenthaler, M. U., Galy, B., and Hentze, M. W. (2008) Annu. Rev. Nutr. 28 197–213 [DOI] [PubMed] [Google Scholar]
- 2.Wallander, M. L., Leibold, E. A., and Eisenstein, R. S. (2006) Biochim. Biophys. Acta 1763 668–689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rouault, T. A. (2006) Nat. Chem. Biol. 2 406–414 [DOI] [PubMed] [Google Scholar]
- 4.Clarke, S. L., Vasanthakumar, A., Anderson, S. A., Pondarre, C., Koh, C. M., Deck, K. M., Pitula, J. S., Epstein, C. J., Fleming, M. D., and Eisenstein, R. S. (2006) EMBO J. 25 544–553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shaw, G. C., Cope, J. J., Li, L., Corson, K., Hersey, C., Ackermann, G. E., Gwynn, B., Lambert, A. J., Wingert, R. A., Traver, D., Trede, N. S., Barut, B. A., Zhou, Y., Minet, E., Donovan, A., Brownlie, A., Balzan, R., Weiss, M. J., Peters, L. L., Kaplan, J., Zon, L. I., and Paw, B. H. (2006) Nature 440 96–100 [DOI] [PubMed] [Google Scholar]
- 6.Chen, O. S., Schalinske, K. L., and Eisenstein, R. S. (1997) J. Nutr. 127 238–248 [DOI] [PubMed] [Google Scholar]
- 7.Meyron-Holtz, E. G., Ghosh, M. C., Iwai, K., LaVaute, T., Brazzolotto, X., Berger, U. V., Land, W., Ollivierre-Wilson, H., Grinberg, A., Love, P., and Rouault, T. A. (2004) EMBO J. 23 386–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Haile, D. J., Rouault, T. A., Harford, J. B., Kennedy, M. C., Blondin, G. A., Beinert, H., and Klausner, R. D. (1992) Proc. Natl. Acad. Sci. U. S. A. 89 11735–11739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Beinert, H., Kennedy, M. C., and Stout, C. D. (1996) Chem. Rev. 96 2335–2374 [DOI] [PubMed] [Google Scholar]
- 10.Brown, N. M., Anderson, S. A., Steffen, D. W., Carpenter, T. B., Kennedy, M. C., Walden, W. E., and Eisenstein, R. S. (1998) Proc. Natl. Acad. Sci. U. S. A. 95 15235–15240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bouton, C., and Drapier, J. C. (2003) Sci. STKE 2003, pe17. [DOI] [PubMed]
- 12.Meyron-Holtz, E. G., Ghosh, M. C., and Rouault, T. A. (2004) Science 306 2087–2090 [DOI] [PubMed] [Google Scholar]
- 13.Gardner, P. R. (1997) Biosci. Rep. 17 33–42 [DOI] [PubMed] [Google Scholar]
- 14.Imlay, J. A. (2006) Mol. Microbiol. 59 1073–1082 [DOI] [PubMed] [Google Scholar]
- 15.Rouault, T. A., and Klausner, R. D. (1996) EXS 77 183–197 [DOI] [PubMed] [Google Scholar]
- 16.Soum, E., and Drapier, J. C. (2003) J. Biol. Inorg. Chem. 8 226–232 [DOI] [PubMed] [Google Scholar]
- 17.Kennedy, M. C., Antholine, W. E., and Beinert, H. (1997) J. Biol. Chem. 272 20340–20347 [DOI] [PubMed] [Google Scholar]
- 18.Eisenstein, R. S., Kennedy, M. C., and Beinert, H. (1998) in Metal Ions in Gene Regulation (Silver, S., and Walden, W. eds) pp. 157–216, Chapman & Hall, New York
- 19.Varghese, S., Tang, Y., and Imlay, J. A. (2003) J. Bacteriol. 185 221–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Walden, W. E. (2002) Proc. Natl. Acad. Sci. U. S. A. 99 4138–4140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brown, N. M., Kennedy, M. C., Antholine, W. E., Eisenstein, R. S., and Walden, W. E. (2002) J. Biol. Chem. 277 7246–7254 [DOI] [PubMed] [Google Scholar]
- 22.Fillebeen, C., Chahine, D., Caltagirone, A., Segal, P., and Pantopoulos, K. (2003) Mol. Cell. Biol. 23 6973–6981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Basilion, J. P., Kennedy, M. C., Beinert, H., Massinople, C. M., Klausner, R. D., and Rouault, T. A. (1994) Arch. Biochem. Biophys. 311 517–522 [DOI] [PubMed] [Google Scholar]
- 24.Kennedy, M. C., Mende-Mueller, L., Blondin, G. A., and Beinert, H. (1992) Proc. Natl. Acad. Sci. U. S. A. 89 11730–11734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kennedy, M. C., Emptage, M. H., Dreyer, J. L., and Beinert, H. (1983) J. Biol. Chem. 258 11098–11105 [PubMed] [Google Scholar]
- 26.Henson, C. P., and Cleland, W. W. (1967) J. Biol. Chem. 242 3833–3838 [PubMed] [Google Scholar]
- 27.Schalinske, K. L., and Eisenstein, R. S. (1996) J. Biol. Chem. 271 7168–7176 [DOI] [PubMed] [Google Scholar]
- 28.Barton, H. A., Eisenstein, R. S., Bomford, A., and Munro, H. N. (1990) J. Biol. Chem. 265 7000–7008 [PubMed] [Google Scholar]
- 29.Haile, D. J., Rouault, T. A., Tang, C. K., Chin, J., Harford, J. B., and Klausner, R. D. (1992) Proc. Natl. Acad. Sci. U. S. A. 89 7536–7540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hentze, M. W., Rouault, T. A., Harford, J. B., and Klausner, R. D. (1989) Science 244 357–359 [DOI] [PubMed] [Google Scholar]
- 31.Cairo, G., Ronchi, R., Recalcati, S., Campanella, A., and Minotti, G. (2002) Biochemistry 41 7435–7442 [DOI] [PubMed] [Google Scholar]
- 32.Hanson, E. S., and Leibold, E. A. (1998) J. Biol. Chem. 273 7588–7593 [DOI] [PubMed] [Google Scholar]
- 33.Tacchini, L., Bianchi, L., Bernelli-Zazzera, A., and Cairo, G. (1999) J. Biol. Chem. 274 24142–24146 [DOI] [PubMed] [Google Scholar]
- 34.Schneider, B. D., and Leibold, E. A. (2003) Blood 102 3404–3411 [DOI] [PubMed] [Google Scholar]
- 35.Hanson, E. S., Foot, L. M., and Leibold, E. A. (1999) J. Biol. Chem. 274 5047–5052 [DOI] [PubMed] [Google Scholar]
- 36.Bouton, C. (1999) Cell. Mol. Life Sci. 55 1043–1053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Emptage, M. H., Dreyer, J. L., Kennedy, M. C., and Beinert, H. (1983) J. Biol. Chem. 258 11106–11111 [PubMed] [Google Scholar]
- 38.Imlay, J. A. (2003) Annu. Rev. Microbiol. 57 395–418 [DOI] [PubMed] [Google Scholar]
- 39.Beckman, J. S., and Koppenol, W. H. (1996) Am. J. Physiol. 271 C1424–C1437 [DOI] [PubMed] [Google Scholar]
- 40.Cooper, C. E. (1999) Biochim. Biophys. Acta 1411 290–309 [DOI] [PubMed] [Google Scholar]
- 41.Tarpey, M. M., and Fridovich, I. (2001) Circ. Res. 89 224–236 [DOI] [PubMed] [Google Scholar]
- 42.Jordan, P. A., Tang, Y., Bradbury, A. J., Thomson, A. J., and Guest, J. R. (1999) Biochem. J. 344 739–746 [PMC free article] [PubMed] [Google Scholar]
- 43.Rouault, T. A., and Klausner, R. D. (1996) Trends Biochem. Sci 21 174–177 [PubMed] [Google Scholar]
- 44.Gardner, P. R., Raineri, I., Epstein, L. B., and White, C. W. (1995) J. Biol. Chem. 270 13399–13405 [DOI] [PubMed] [Google Scholar]
- 45.Kiley, P. J., and Beinert, H. (2003) Curr. Opin. Microbiol. 6 181–185 [DOI] [PubMed] [Google Scholar]
- 46.Weiss, G., Goossen, B., Doppler, W., Fuchs, D., Pantopoulos, K., Werner-Felmayer, G., Wachter, H., and Hentze, M. W. (1993) EMBO J. 12 3651–3657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Drapier, J. C., Hirling, H., Wietzerbin, J., Kaldy, P., and Kuhn, L. C. (1993) EMBO J. 12 3643–3649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Soum, E., Brazzolotto, X., Goussias, C., Bouton, C., Moulis, J. M., Mattioli, T. A., and Drapier, J. C. (2003) Biochemistry 42 7648–7654 [DOI] [PubMed] [Google Scholar]
- 49.Guo, B., Phillips, J. D., Yu, Y., and Leibold, E. A. (1995) J. Biol. Chem. 270 21645–21651 [DOI] [PubMed] [Google Scholar]
- 50.Samaniego, F., Chin, J., Iwai, K., Rouault, T. A., and Klausner, R. D. (1994) J. Biol. Chem. 269 30904–30910 [PubMed] [Google Scholar]
- 51.Press, W. H., Flannery, B. P., Teukolsky, S. A., and Vetterling, W. T. (1986) Numerical Recipes: The Art of Scientific Computing, 1st Ed., Cambridge University Press, New York
- 52.Schalinske, K. L., Anderson, S. A., Tuazon, P. T., Chen, O. S., and Eisenstein, R. S. (1997) Biochemistry 36 3950–3958 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.