

Abstract
A new role is reported for CP12, a highly unfolded and flexible protein, mainly known for its redox function with A4 glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Both reduced and oxidized CP12 can prevent the in vitro thermal inactivation and aggregation of GAPDH from Chlamydomonas reinhardtii. This mechanism is thus not redox-dependent. The protection is specific to CP12, because other proteins, such as bovine serum albumin, thioredoxin, and a general chaperone, Hsp33, do not fully prevent denaturation of GAPDH. Furthermore, CP12 acts as a specific chaperone, since it does not protect other proteins, such as catalase, alcohol dehydrogenase, or lysozyme. The interaction between CP12 and GAPDH is necessary to prevent the aggregation and inactivation, since the mutant C66S that does not form any complex with GAPDH cannot accomplish this protection. Unlike the C66S mutant, the C23S mutant that lacks the N-terminal bridge is partially able to protect and to slow down the inactivation and aggregation. Tryptic digestion coupled to mass spectrometry confirmed that the S-loop of GAPDH is the interaction site with CP12. Thus, CP12 not only has a redox function but also behaves as a specific “chaperone-like protein” for GAPDH, although a stable and not transitory interaction is observed. This new function of CP12 may explain why it is also present in complexes involving A2B2 GAPDHs that possess a regulatory C-terminal extension (GapB subunit) and therefore do not require CP12 to be redox-regulated.
CP12 is a small 8.2-kDa protein present in the chloroplasts of most photosynthetic organisms, including cyanobacteria (1, 2), higher plants (3), the diatom Asterionella formosa (4, 5), and green (1) and red algae (6). It allows the formation of a supramolecular complex between phosphoribulokinase (EC 2.7.1.19) and glyceraldehyde-3-phosphate dehydrogenase (GAPDH),3 two key enzymes of the Calvin cycle pathway, and was recently shown to interact with fructose bisphosphate aldolase, another enzyme of the Calvin cycle pathway (7). The phosphoribulokinase·GAPDH·CP12 complex has been extensively studied in Chlamydomonas reinhardtii (8, 9) and in Arabidopsis thaliana (10, 11). In the green alga C. reinhardtii, the interaction between CP12 and GAPDH is strong (8). GAPDH may exist as a homotetramer composed of four GapA subunits (A4) in higher plants, cyanobacteria, and green and red algae (6, 12), but in higher plants, it can also exist as a heterotetramer (A2B2), composed of two subunits, GapA and GapB (13, 14). GapB, up to now, has exclusively been found in Streptophyta, but recently two prasinophycean green algae, Ostreococcus tauri and Ostreococcus lucimarinus, were also shown to possess a GapB gene, whereas CP12 is missing (15). The GapB subunit is similar to the GapA subunit but has a C-terminal extension containing two redox-regulated cysteine residues (16). Thus, although the A4 GAPDHs lack these regulatory cysteine residues (13, 14, 17–20), they are also redox-regulated through its interaction with CP12, since the C terminus of this small protein resembles the C-terminal extension of the GapB subunit. The regulatory cysteine residues for GapA are thus supplied by CP12, as is well documented in the literature (1, 8, 11, 16).
CP12 belongs to the family of intrinsically unstructured proteins (IUPs) (21–26). The amino acid composition of these proteins causes them to have no or few secondary structures. Their total or partial lack of structure and their high flexibility allow them to be molecular adaptors (27, 28). They are often able to bind to several partners and are involved in most cellular functions (29, 30). Recently, some IUPs have been described in photosynthetic organisms (31, 32).
There are many functional categories of IUPs (22, 33). They can be, for instance, involved in permanent binding and have (i) a scavenger role, neutralizing or storing small ligands; (ii) an assembler role by forming complexes; and (iii) an effector role by modulating the activity of a partner molecule (33). These functions are not exclusive; thus, CP12 can form a stable complex with GAPDH, regulating its redox properties (8, 34, 35), and can also bind a metal ion (36, 37). IUPs can also bind transiently to partners, and some of them have been found to possess a chaperone activity (31, 38). This chaperone function was first shown for α-synuclein (39) and for α-casein (40), which are fully disordered. The amino acid composition of IUPs is less hydrophobic than those of soluble proteins; hence, they lack hydrophobic cores and do not become insoluble when heated. Since CP12 belongs to this family, we tested if it was resistant to heat treatment and finally, since it is tightly bound to GAPDH, if it could prevent aggregation of its partner, GAPDH, an enzyme well known for its tendency to aggregate (41–44) and consequently a substrate commonly used in chaperone studies (45, 46).
Unlike chaperones, which form transient, dynamic complexes with their protein substrates through hydrophobic interactions (47, 48), CP12 forms a stable complex with GAPDH. The interaction involves the C-terminal part of the protein and the presence of negatively charged residues on CP12 (35). However, only a site-directed mutagenesis has been performed to characterize the interaction site on GAPDH. Although the mutation could have an indirect effect, the residue Arg-197 was shown to be a good candidate for the interaction site (49).
In this report, we accordingly used proteolysis experiments coupled with mass spectrometry to detect which regions of GAPDH are protected by its association with CP12. To conclude, the aim of this report was to characterize a chaperone function of CP12 that had never been described before and to map the interaction site on GAPDH using an approach that does not involve site-directed mutagenesis.
EXPERIMENTAL PROCEDURES
Production and Purification of Recombinant CP12s and GAPDH—Recombinant wild-type or mutated (C23S, C66S, and W35A) CP12s, with the His tag, were expressed in Escherichia coli BL21(DE3)pLysS and purified by Ni2+-nitrilotriacetic acid-agarose (Qiagen, Courtaboeuf, France), as previously described (8). For the W35A CP12 mutant, site-directed mutagenesis was performed using the QuikChange™ kit (Stratagene, La Jolla, CA). The primer was as follows: W35A, 5′-CTGCGCCGTGGCCGCGGACACCGTTGAGG-3′. The mutation was confirmed by sequencing. After purification, the CP12 proteins were dialyzed against 50 mm Tris, 100 mm NaCl at pH 8 and stored at –20 °C. GAPDH was expressed in E. coli BL21(DE3)pLysS and purified by a DEAE-Trisacryl column using a protocol slightly altered from Ref. 9. The enzyme was dialyzed against 30 mm Tris, 100 mm NaCl, 2 mm EDTA, 0.1 mm NAD at pH 7.9 and stored with 10% glycerol at –80 °C. We checked, using Ellman's reagent (50), that in all experiments, GAPDH has four thiol groups per monomer, whereas oxidized CP12 has two disulfide bridges. Protein concentrations were determined with the Bio-Rad reagent protein assay using BSA as a standard (51).
Activity Measurements—To determine NADPH-dependent activity of GAPDH, 1,3-bisphosphoglyceric acid was synthesized, and its concentration was determined as in Ref. 9. Kinetic measurements were performed in 50 mm glycyl glycine, 50 mm KCl, 15 mm MgCl2, 0.5 mm EDTA at pH 7.7 using 0.2 mm NADPH. The activity was followed by measuring the change in absorbance at 340 nm using a Pye Unicam UV2 spectrophotometer (Cambridge, UK).
Thermal Aggregation and Inactivation of GAPDH—The heat-induced inactivation and aggregation of 0.8 μm GAPDH was followed in 30 mm Tris, 100 mm NaCl, 2 mm EDTA, pH 7.9, alone or in the presence of either 0.2–1.6 μm oxidized or 1.6 μm reduced WT CP12, 1.6 μm C23S, 1.6 μm C66S, 1.6 μm W35A, 1.6 μm oxidized thioredoxin from Spirulina sp. (Sigma) or 1.6 μm BSA, for thermal inactivation only. Reduced CP12 was obtained after incubation with 20 mm DTT for 30 min. A general heat shock protein chaperone, oxidized Hsp33 (1.6 μm) (52–54), was also tested on the aggregation of 0.8 μm GAPDH and of 1 μm catalase (as a control). The samples were incubated at 43 °C in a water bath. At various intervals up to a maximum time of 1 h, turbidity was measured. In parallel, aliquots were withdrawn to follow the GAPDH activity. The aggregation was followed by measuring the increase of the absorbance at 650 nm and using a Pye Unicam 8625 UV-visible spectrophotometer (Cambridge, UK). Aggregation of GAPDH was also tested in the presence of a 10 mm concentration of either oxidized or reduced glutathione.
Analysis of Thermal Aggregation and Inactivation of GAPDH—For the aggregation experiments, data were fitted to theoretical curves with Sigma Plot version 10.0 (Systat Software GmbH, Erkrath, Germany) using a single exponential equation,
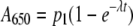 |
(Eq. 1) |
where A650 represents the absorbance at 650 nm, t is the reaction time, and p1 and λ are the amplitude and the time constant of the aggregation process, respectively.
For inactivation experiments, data were fitted to a single exponential equation,
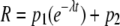 |
(Eq. 2) |
where R represents the residual activity (as a percentage of the maximal activity) of NADPH-GAPDH activity, t is the reaction time, and p1 (equal to 100 – p2) is the amplitude of the inactivation process. λ is the time constant of the inactivation process, and p2 is the residual activity at the end of the inactivation process.
Chaperone Activity Assays—The anti-aggregation activity of CP12 was checked with several proteins known to aggregate. The anti-aggregation activity of CP12 (50 μm) was tested on 7 μm alcohol dehydrogenase (EC 1.1.1.1) from Saccharomyces cerevisiae (Sigma) at 50 °C. The aggregation of 3.3 μm catalase (EC 1.11.1.6) from bovine liver (GE Healthcare) was tested at 43 °C in the presence of 10 μm CP12, whereas the anti-aggregation activity of CP12 (10 μm) on lysozyme (35.5 μm) from chicken egg white (EC 3.2.1.17; Sigma) was tested in the presence of 20 mm DTT at 25 °C. Alcohol dehydrogenase aggregation was tested in a 50 mm phosphate buffer at pH 7.5, whereas aggregation of all other enzymes was tested in 30 mm Tris, 100 mm NaCl, 2 mm EDTA at pH 7.9 for 1 h. Aggregation was followed by measuring the change in absorbance at 650 nm with a Pye Unicam UV2 spectrophotometer.
Fluorescence Measurements—CP12 proteins (1.6 μm) and/or GAPDH (0.8 μm) were diluted in 30 mm Tris, 100 mm NaCl, 2 mm EDTA, pH 7.9. GAPDH fluorescence has also been studied in the presence of 6 m guanidinium chloride at 23 °C. Fluorescence spectra were recorded on a Fluorolog-3 Jobin Yvon-Spex spectrometer (HORIBA Jobin Yvon SAS, Longjumeau, France) connected with a circulating water bath at either 43 or 23 °C from 290 to 450 nm with an excitation wavelength of 280 nm. Measurements were performed with a scan speed of 1 nm/s. All spectra were recorded with an emission slit of 2 nm.
Circular Dichroism Studies—Spectra were recorded on a Jasco J-815 CD spectrometer (JASCO, Bouguenais, France). All spectra were recorded from 260 to 190 nm with a 20 nm/min scan rate at 43 °C in a 10 mm Na2HPO4-NaH2PO4 buffer at pH 7.9. Measurements were performed using 0.2-cm quartz vials in a final volume of 400 μl with a 0.8 μm concentration of GAPDH and/or 1.6 μm CP12 proteins.
Surface Plasmon Resonance and in Vitro GAPDH·CP12 Reconstitution—Reduced CP12 (50 μg/ml) and oxidized CP12 were coupled to a biosensor chip (CM5; BiaCore) following the manufacturer's instructions. In the case of oxidized CP12, 210 resonance units (RU) were immobilized, whereas 117 RU were immobilized in the case of reduced CP12. We then studied the interactions of GAPDH with immobilized reduced or oxidized CP12 using HBS-EP running buffer (BiaCore), 0.1 mm NAD, pH 7.5, at 20 μl/min using a BiaCore 2000 apparatus (Uppsala, Sweden). The analyte (GAPDH) interacts with the ligand (CP12) to give the association phase, which continues until a plateau is reached, indicating that association and dissociation rates are in equilibrium. The analyte begins to dissociate as soon as injection is stopped and replaced by buffer. The association and dissociation phases were used to derive the dissociation and association rate constants (kd and ka, respectively). Global fits, using the BiaEvaluation software (version 2.1; BiaCore), of the exponential curves (sensorgrams), gave both kd and ka values and the response at equilibrium Req. Eight and seven concentrations of tetrameric GAPDH ranging from 0.5 to 470 nm and from 0.1 to 200 nm for reduced and oxidized CP12, respectively, were used. Req was then plotted as a function of GAPDH concentration and fitted to a hyperbola (Equation 3),
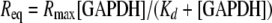 |
(Eq. 3) |
The formation of the GAPDH·CP12 complex using either oxidized or reduced CP12 was checked using native PAGE performed on a 4–15% minigel using a GE Healthcare Phastsystem apparatus. The reconstitution was carried out as described in Ref. 35, using 0.03 nmol of CP12 and 0.03 nmol of tetrameric GAPDH.
Immunoblotting Experiments—GAPDH (0.8 μm) or 0.8 μm GAPDH plus 1.6 μm CP12 were incubated for 20 min at 43 °C in 30 mm Tris, 100 mm NaCl, 2 mm EDTA, pH 7.9. The samples (1 ml) were centrifuged for 25 min at 13,000 rpm. 15 μl of the 20 mm DTT reduced or unreduced supernatant were loaded on a 12% SDS-polyacrylamide gel. The pellets were washed, adding 500 μl of buffer, and then another centrifugation at 13,000 rpm for 25 min was performed. The resulting pellets were resuspended in 45 μl of 0.2 m Tris-HCl, 20% glycerol, 0.05% bromphenol blue buffer, pH 6.8, and mixed or not with 20 mm DTT, and 10 μl were loaded on the gel. Proteins were then transferred onto a nitrocellulose membrane (0.45 μm; Schleicher & Schüll) by passive diffusion for 16 h. The membrane was blocked for 1 h in phosphate-buffered saline (PBS; 137 mm NaCl, 2.7 mm KCl, 4.1 mm Na2HPO4, 1.8 mm KH2PO4, 0.1% Tween 20 (v/v), pH 7.4) amended with 5% fat-free milk powder. The membrane was rinsed twice with PBS and incubated with a rabbit antiserum directed against recombinant C. reinhardtii CP12 (1:20,000 dilution in PBS) or recombinant C. reinhardtii GAPDH (1:20,000 dilution in PBS) for 1 h at room temperature and rinsed twice briefly and then once for 15 min and three times for 1 min in PBS. Antigen-antibody complexes were detected using a 1:10,000 dilution (in PBS, 1-h incubation) of a donkey anti-rabbit secondary antibody conjugated with a horseradish peroxidase and rinsed as before. Cross-reacting proteins were visualized using an enhanced chemiluminescence kit (GE Healthcare) following the manufacturer's guidelines.
Tryptic Digestion Experiments—GAPDH (10 μg) alone or with CP12 (10 μg) were digested using 0.1 μg of trypsin (EC 3.4.21.4) from bovine pancreas (Sigma) for 30 s to 20 min at 37 °C in a 25 mm NH4HCO3, 5 mm CaCl2 buffer at pH 8. In experiments concerning the robustness of the CP12 protection, 0.1, 1, or 10 μg of trypsin was also used for 30 s. For defined time points (30 s and 1, 3, 5, 15, and 20 min) after the addition of trypsin, the reactions were immediately stopped by adding SDS-PAGE loading buffer containing 20 mm DTT and heated for 3 min at 100 °C. Peptides were then separated by 12% SDS-PAGE performed according to Laemmli (55), using a Bio-Rad Mini Protean system and stained with Coomassie Brilliant Blue R250. Molecular mass markers were from the Precision Plus protein standard dual color kit (Bio-Rad).
Mass Spectrometry and Peptide Identification—Spots were excised, and peptides were treated by a liquid-handling work station (Evo, Tecan, Switzerland) for a final extraction of the peptides and for reduction, alkylation, and tryptic digestion steps. Following these steps, matrix-assisted laser desorption ionization time-of-flight (MALDI-TOF) analysis was performed on the Ultraflex II mass spectrometer (Bruker, Germany) in a positive reflectron mode. A saturated solution of α-cyano-4-hydroxycinnamic acid in 70% acetonitrile and 0.1% trifluoroacetic acid was used as matrix. A peak list was generated by the PMF_PS method on the FlexAnalysis software and then manually checked. Spectra were internally calibrated with the theoretical values of tryptic peptides from GAPDH. Proteins were identified by using the Mascot Search Data Base (MSDB_20060831) on the Mascot (version 2.2.03) website with the following settings: fixed modification of cysteine (carbamidomethylcysteine), optional oxidation of methionine, one mis-cleavage, and a mass tolerance of 45 ppm. Proteins were considered as identified if the Mascot score was greater than 65 (p < 0.05) and the number of identified peptides was greater than 5.
RESULTS
Protection Effect of CP12 on Heat-induced GAPDH Aggregation and Inactivation—At 43 °C, CP12 did not aggregate over a time course of 1 h (data not shown). When GAPDH was incubated alone, the half-time (t½ = (ln2)/λ) for the aggregation process was 5.7 min (Fig. 1A, curve 1), and the enzyme was completely inactivated in less than 5 min (Fig. 1B, curve 1). As shown in curves 2–4 of Fig. 1, the presence of CP12 protects GAPDH from aggregation and inactivation, and the protection depends on the ratio of GAPDH to CP12. When a molar ratio of 2.0 (tetrameric GAPDH/CP12) was used, 63% of GAPDH (Fig. 1A, curve 2) aggregated, and the half-time was 7.1 min. The aggregation can be fully abolished in the presence of CP12, especially at a molar ratio of 1.0 (Fig. 1A, curve 3) or 0.5 (Fig. 1A, curve 4). In agreement with these findings, the NADPH-dependent activity of GAPDH alone decreased as a function of time and reached zero (Fig. 1B, curve 1). In the presence of CP12, this loss of activity was only slightly affected for 1.0 and 0.5 (tetrameric GAPDH/CP12) molar ratios (Fig. 1B, curves 3 and 4), although for the 2.0 (tetrameric GAPDH/CP12) molar ratio, 46% of the NADPH dependent activity of GAPDH remained (Fig. 1B, curve 2). Data were fitted to Equation 2, and the half-time of the inactivation process of GAPDH alone was less than 1 min. In the presence of CP12, this inactivation was slowed down when molar ratios (GAPDH/CP12) of 0.5 (Fig. 1B, curve 4) and 1 (Fig. 1B, curve 3) were used, and 80% of the activity remained even after a 1-h incubation. The redox state of CP12 does not affect its protecting effect, since reduced CP12, like oxidized CP12, was also able to fully prevent aggregation and inactivation of GAPDH. Aggregation of GAPDH was also followed in the presence of either oxidized or reduced glutathione, and only the oxidized form had an effect and fully protected GAPDH from aggregation (data not shown).
FIGURE 1.

Effect of CP12 on heat-induced aggregation and thermal inactivation of GAPDH. GAPDH aggregation (A) or inactivation (B) was induced at 43 °C in 30 mm Tris, 100 mm NaCl, 2 mm EDTA, pH 7.9. GAPDH (0.8 μm) was used alone (•, curve 1) or with different concentrations of CP12 (0.4 μm (○, curve 2), 0.8 μm (▿, curve 3), 1.6 μm (▵, curve 4)). A, aggregation was followed by measuring absorbance at 650 nm (A650 nm). The continuous lines correspond to the best fits to Equation 1, except for curves 3 and 4, where no aggregation is observed. The parameters are as follows: curve 1, p1 = 0.151 ± 0.006 and t½ = 5.7 ± 0.8 min; curve 2, p1 = 0.095 ± 0.004 and t½ = 7.1 ± 1.0 min. B, the NADPH-GAPDH activity, R, is given as a percentage of the value at zero time. The continuous lines correspond to the best fits to Equation 2 except at 0.8 or 1.6 μm CP12, where almost no inactivation was observed. The parameters are as follows: curve 1, p1 = 100 ± 2%, p2 = 0, and t½ = 0.59 ± 0.04 min; curve 2, p1 = 53 ± 5%, p2 = 46 ± 5%, and t½ = 13.4 ± 3.7 min. All data points are means ± S.D. of five experiments (for 0.0 or 1.6 μm CP12) or of three experiments (0.4 or 0.8 μm CP12).
Specificity of CP12 to Protect GAPDH Aggregation and Inactivation—In order to test the specificity of the protecting effect of CP12 on GAPDH, we tested other proteins. Fig. 2 shows the effect of BSA and thioredoxin. The inactivation process was also slowed down using oxidized thioredoxin (t½ ∼10 min; Fig. 2, curve 3), using a 0.5 molar ratio (tetrameric GAPDH/thioredoxin) and was almost unaffected in the presence of BSA (t½ ∼1.7 min; Fig. 2, curve 2).
FIGURE 2.

Specificity of CP12 upon thermal inactivation of NADPH-GAPDH. 0.8 μm GAPDH was incubated at 43 °C alone (curve 1) or with a 1.6 μm concentration of either BSA (curve 2) or thioredoxin (curve 3) in 30 mm Tris, 100 mm NaCl, 2 mm EDTA, pH 7.9. The enzyme activity (R) is given as the percentage of the value at zero time. All data points are means ± S.D. of five experiments for GAPDH alone and of two experiments for GAPDH plus BSA or thioredoxin. The continuous lines correspond to the best fits to Equation 2, and the parameters are as follows: curve 1, p1 = 100 ± 2%, p2 = 0, and t½ = 0.59 ± 0.04 min; curve 2, p1 = 98 ± 5%, p2 = 0, and t½ = 1.7 ± 0.2 min; curve 3, p1 = 99 ± 2%, p2 = 0, t½ = 10.1 ± 0.6 min.
Therefore, proteins such as BSA do not offer any protection, and thioredoxin only slows this process down slightly. A general chaperone, Hsp33, was also tested on the aggregation of GAPDH and of catalase (Fig. 3). Although Hsp33 fully protected catalase against aggregation (Fig. 3A), it only delayed the aggregation of GAPDH from about 5 to 20 min for 50% aggregation and from 20 to 30 min for total aggregation (Fig. 3B). These data suggest that GAPDH requires the presence of CP12 in order to be efficiently protected against inactivation and aggregation induced by heat treatment.
FIGURE 3.

Hsp33 less effective than CP12. Hsp33 prevents thermal precipitation of catalase (A) but not of GAPDH (B). Catalase (1 μm) or tetrameric GAPDH (0.8 μm) was incubated at 43 °C in the absence of any protein (filled circles) or in the presence of Hsp33 in a molar ratio of 2 (empty circles).
Effect of CP12 on Heat-induced Aggregation of Other Proteins—There was no significant effect of CP12 on catalase, lysozyme, or alcohol dehydrogenase (data not shown). Thus, CP12 does not behave as a general chaperone and seems to protect specifically and efficiently tetrameric GAPDH from aggregation, and this protection requires at least a 1.0 molar ratio.
Binding Mechanism of CP12 and Link to Protection of GAPDH—We used a cysteine mutant, with a substitution in the C-terminal disulfide bridge of CP12 (with a cysteine residue at position 66 replaced by a serine, C66S) that is unable to form the GAPDH·CP12 complex (8, 35). As a control, a cysteine mutant with a substitution in the N-terminal bridge (cysteine residue at position 23 replaced by a serine, C23S) that is able to form the GAPDH·CP12 complex (8, 35) was also used (Fig. 4). In the presence of the C66S mutant at a 0.5 molar ratio (tetrameric GAPDH·C66S), no protection against heat-induced inactivation was detected (Fig. 4A, curve 2), and GAPDH behaved as it did alone, whereas in the presence of the C23S mutant, the inactivation process was slightly slowed down, and a residual GAPDH activity of 35% was measured (Fig. 4A, curve 3). In agreement with these findings, the heat-induced aggregation of GAPDH in the presence of the C66S mutant was about the same (t½ = 1.4 min; Fig. 4B, curve 2) as the one observed with GAPDH alone (t½ about 0.6 min; Fig. 4B, curve 1), whereas, in the presence of the C23S, it decreased and slowed down with a half-time of about 23 min (Fig. 4B, curve 3). In the case of C23S, the aggregation of 70% of GAPDH is in agreement with the remaining activity. The protection of GAPDH against aggregation seems to involve the GAPDH·CP12 complex formation. Although the presence of DTT prevented the formation of the ternary complex between GAPDH·CP12 and phosphoribulokinase (8), we checked that the presence of reducing agent still allowed the formation of the GAPDH·CP12 subcomplex. We performed surface plasmon resonance with immobilized reduced CP12 using GAPDH as analyte, and the dissociation constant (Kd) was found to be 0.98 ± 0.01 nm (Fig. 5A, curve 1). The dissociation constant value previously obtained (8) between oxidized CP12 and GAPDH was 0.440 ± 0.002 nm (see Fig. 5A, curve 2). The two Kd values between GAPDH and CP12 either reduced or oxidized, therefore, are within the same range (nanomolar), indicating that the pseudoaffinity of GAPDH and both forms of CP12 is high. The formation of the subcomplex GAPDH·reduced CP12 was checked and found using an in vitro reconstitution experiment (Fig. 5B).
FIGURE 4.

Effect of CP12 mutants on the thermal inactivation and heat-induced aggregation of GAPDH. GAPDH inactivation (A) or aggregation (B) was induced at 43 °C in 30 mm Tris, 100 mm NaCl, 2 mm EDTA, pH 7.9. GAPDH (0.8 μm) was used alone (curve 1, filled circle) or with a 1.6 μm concentration of either C66S mutant (curve 2, filled triangle) or C23S mutant (curve 3). A, the enzyme activity, R, is given as a percentage of the value at zero time. The continuous lines correspond to the best fits to Equation 2, and the parameters are as follows: curve 1, p1 = 100 ± 2%, p2 = 0, and t½ = 0.59 ± 0.04 min; curve 2, p1 = 99 ± 3%, p2 = 0, and t½ = 1.4 ± 0.1 min; curve 3, p1 = 60 ± 6%, p2 = 36 ± 3%, and t½ = 4.8 ± 1.2 min. B, aggregation was followed by measuring absorbance at 650 nm (A650 nm). The continuous lines correspond to the best fits to Equation 1. The parameters are as follows: curve 1, p1 = 0.151 ± 0.006, t½ = 5.7 ± 0.8 min; curve 2, p1 = 0.17 ± 0.01, t½ = 7.4 ± 0.9 min; curve 3, p1 = 0.104 ± 0.005, t½ = 13.6 ± 1.6 min. All data points are means ± S.D. of five experiments for GAPDH alone and of three experiments for GAPDH plus mutant CP12 proteins.
FIGURE 5.

Interaction between GAPDH and reduced CP12. A, surface plasmon resonance binding experiments. The response at equilibrium Req (in RU) was reported as a function of the analyte concentration ([GAPDH]). The experimental points were fitted to a hyperbola, Equation 3. Reduced CP12 (117 RU) was immobilized, and GAPDH was used as analyte (curve 1). Oxidized CP12 (210 RU) was immobilized, and GAPDH was used as analyte (curve 2). B, Western blot analysis of the in vitro reconstitution of GAPDH·CP12 complex. GAPDH and CP12 were mixed in a molar ratio of 1:1. After 1 h at 30 °C, proteins were separated on a native 4–15% gradient gel, transferred to a nitrocellulose membrane, and revealed with antibodies raised against CP12 (1:10,000). Lane 1, CP12 alone (0.03 nmol); lane 2, reconstitution mixture of oxidized CP12 (0.03 nmol) and GAPDH (0.03 nmol); lane 3, reconstitution mixture of reduced CP12 (0.03 nmol) and GAPDH (0.03 nmol). 33 ng of CP12 was loaded in each lane. No band was revealed with recombinant GAPDH alone.
Solubilizing Effect of Wild-type CP12—To check the solubilizing effect of wild-type CP12 on GAPDH, gel electrophoresis followed by immunoblots was carried out (Fig. 6). The presence of either CP12 or GAPDH was detected using antibodies raised against each protein. With the heat-treated samples, in the absence of CP12, most of the GAPDH was found in the pellet (Fig. 6A, lanes 3 and 4), and some bands corresponding to high molecular mass were observed, indicating formation of aggregates. Some of these aggregates, however, disappeared when the sample was treated with a reducing agent (DTT; Fig. 6A, lane 4), showing that some of the aggregates, but not all of them, result from disulfide bridge formation. When CP12 was present in the heat-treated sample, the results were clearly different (Fig. 6, B and C). GAPDH was mainly found in the supernatant corresponding to the soluble fraction (Fig. 6B, lanes 1 and 2), but it was also observed in lesser amounts in the pellet fraction (Fig. 6B, lanes 3 and 4). CP12 was exclusively found in the supernatant fraction, as shown by antibody detection (Fig. 6C). The presence of two bands for CP12 corresponds to the oxidized and reduced form (Fig. 6C, lane 1), since one band clearly disappears upon reduction (Fig. 6C, lane 2).
FIGURE 6.

Western blot analysis of GAPDH partition between soluble and precipitated fractions. GAPDH (0.8 μm) (A) or 0.8 μm of GAPDH plus 1.6 μm CP12 (B and C) were incubated at 43 °C. Soluble fractions not treated (lanes 1) or treated with DTT (lanes 2) and precipitated ones not treated (lanes 3) or treated with DTT (lanes 4), obtained as described under “Experimental Procedures,” were analyzed by immunoblots with antibodies against GAPDH (A and B) or CP12 (C).
Conformation Change of GAPDH upon Heat Treatment Probed by Spectrofluorimetry and CD—We recorded fluorescence and CD spectra for GAPDH alone and in the presence of CP12 at physiological temperature (23 °C) (Fig. 7, A and B) and at high temperature (43 °C) (Fig. 7, C and D) in order to follow the global structure and conformation change of GAPDH. At 23 °C, the wavelength of the maximum of the fluorescence emission for GAPDH was 338 nm (Fig. 7A, curve 1). It was possible to measure the fluorescence of 0.8 μm GAPDH in the presence of 1.6 μm CP12, because the fluorescence of CP12 does not interfere as a consequence of a single tryptophan residue on CP12. A control was carried out, however, with a mutant CP12 in which the tryptophan residue at position 35 was replaced with an alanine residue (W35A; Fig. 7A, curve 2). No fluorescence of the W35A mutant was detected (data not shown). A decrease of the intensity of the fluorescence of GAPDH in the presence of wild-type CP12 or W35A was observed (Fig. 7A, curves 2 and 3). The wavelength of the maximum of fluorescence emission for GAPDH in the presence of either wild-type CP12 or W35A CP12 was always 338 nm. Fluorescence of the tryptophan residues on GAPDH was also measured in the presence of 6 m guanidinium chloride (Fig. 7A, curve 4). A shift was observed leading to a maximum of fluorescence emission at 354 nm, indicating a complete exposure of tryptophan residues to the solvent. The CD spectra were also recorded in the same conditions but in phosphate buffer because of the incompatibility of the Tris buffer with this technique. In these conditions, 0.8 μm GAPDH still aggregated when it was alone but at a different time scale (t½ ∼30 min; data not shown), and again no precipitation was observed in the presence of 1.6 μm CP12 (data not shown). At 23 °C, spectra were recorded for 1.6 μm CP12 alone (Fig. 7B, curve 1), 0.8 μm GAPDH alone (Fig. 7B, curve 2), and GAPDH plus CP12 (Fig. 7B, curve 3). When GAPDH and CP12 were mixed, the spectrum obtained corresponded to the addition of the spectra of both proteins alone, showing that the presence of CP12 did not induce changes in the secondary structure of GAPDH during their interaction.
FIGURE 7.

Fluorescence (A and C) and circular dichroism (B and D) analysis. A, fluorescence spectra, recorded at 23 °C, with an emission slit of 2 nm, correspond to 0.8 μm GAPDH alone (curve 1) or in the presence of 1.6 μm W35A mutant (curve 2) or 1.6 μm WT CP12 (curve 3). Curve 4 corresponds to the spectrum of 0.8 μm GAPDH denatured by 6 m guanidinium chloride. B, CD spectra, at 23 °C, were obtained with 1.6 μm CP12 (curve 1), 0.8 μm GAPDH (curve 2), and 1.6 μm CP12 plus 0.8 μm GAPDH (curve 3). C, fluorescence spectra of 0.8 μm GAPDH alone were recorded at 43 °C, curve 1 corresponding to the initial spectrum and all of the other curves (curve 2) corresponding to records from 10 min to 1 h of incubation. D, CD spectra of 0.8 μm GAPDH alone were recorded at 43 °C immediately and after 10, 20, and 40 min, as indicated by the arrow. The spectra obtained from 40 min to 2 h are the same. These experiments were carried out in a phosphate buffer at pH 7.9. FI, fluorescence intensity expressed in arbitrary units (au).
The conformation change leading to inactivated and aggregated GAPDH upon heat treatment at 43 °C was then probed by spectrofluorimetry and CD experiments (Fig. 7, C and D). A slight increase in fluorescence of the tryptophan residues, during the heat treatment of GAPDH alone, was observed, and there was also a slight shift of the wavelength of the maximum of the fluorescence emission from 338 (native enzyme; Fig. 7C, curve 1) to 342 nm (denatured enzyme; Fig. 7C, curve 2). The red shift of the fluorescence spectrum could correspond to a simple denaturation process, in agreement with the decline of the activity of the enzyme. This red shift is linked to a denaturation process, as mentioned above, when fluorescence of the tryptophan residues on GAPDH was measured in the presence of 6 m guanidinium chloride. The CD spectra were also recorded for 2 h at 43 °C. The absolute values of the two minima at 208 and 222 nm, characteristic of secondary structure, decreased for the first 40 min and then remained constant (Fig. 7D). When the fluorescence and CD spectra of GAPDH were recorded in the presence of CP12, they did not vary during heat treatment, and the spectra were identical to those obtained at 23 °C (Fig. 7, curve 3 in A and B). No shift of the wavelength of the maximum of the fluorescence emission was observed, and it remained at 338 nm (data not shown). This agrees with the activity measurement.
Identification of the Interaction Site between CP12 and GAPDH—To identify the site of interaction between CP12 and GAPDH, GAPDH alone or in the presence of CP12 (GAPDH·CP12) was submitted to a limited digestion by trypsin (Fig. 8A). First, the samples not treated by trypsin were loaded onto a 12% SDS-gel. For the CP12 sample, two bands were observed. The upper one corresponded to reduced CP12, and the lower one corresponded to oxidized CP12, and these bands were also detected in the GAPDH·CP12 sample that contained 10 μg of CP12 and 10 μg of GAPDH (Fig. 8A). After treatment of GAPDH alone with 0.1 μg of trypsin, digestion was observed within 30 s, and two supplementary bands, labeled 2 and 3, were observed with apparent molecular masses of 22 and 13 kDa. In contrast, no digestion was observed with 0.1 μg of trypsin for the GAPDH·CP12 sample, even after a 15-min incubation. However, the bands corresponding to the CP12 detected for shorter incubation times in the GAPDH·CP12 sample disappeared after 3 min of incubation, indicating that these molecules of CP12 were digested (Fig. 8B). Indeed, we used a molar ratio of 1:14 (tetramer GAPDH/CP12), and the molecules of CP12 that were not in interaction with GAPDH were rapidly digested, as has been found previously for IUPs (31). Even after treatment with 1 μg of trypsin, the GAPDH·CP12 sample was not digested after 30 s. After treatment with 10 μg of trypsin, GAPDH plus CP12 was also digested but less than GAPDH alone (data not shown). CP12 thus conferred a high resistance to trypsin. The bands labeled 2 and 3 in the GAPDH sample were cut from the gel, and the fragments were identified by MALDI-TOF mass spectrometry (Tables 1 and 2). The fragment of 22 kDa corresponds to the peptide starting at position 1 of GAPDH ending at the Arg-200 (see Table 1), whereas the one at 13 kDa corresponds to the peptide starting at alanine residue at position 204 (see Table 2 and Fig. 8C).
FIGURE 8.

Tryptic digestion of GAPDH alone or with CP12. A and B, SDS electrophoresis gels. 10 μg of GAPDH were digested alone or in the presence of 10 μg of CP12, with 0.1 μg of trypsin at 37 °C. The reaction was stopped by adding SDS-PAGE loading buffer, and the samples were heated for 3 min at 100 °C. The samples were then charged on a 12% SDS-polyacrylamide gel. The proteins present in each sample and the time of incubation (30 s (A) and from 1 to 20 min (B)) with trypsin are indicated at the top of each lane, N corresponding to the nondigested samples. Three bands at 37 (1), 22 (2), and 13 kDa (3) were observed when GAPDH alone was digested. They were extracted and analyzed by mass spectrometry. On the left part of the gels, the positions and molecular masses of protein standard kit (M) are indicated. On the right part of the gel, the positions of reduced (red) and oxidized (ox) CP12 are shown. C, scheme representing the interaction site between GAPDH and CP12. The arrows delimit the zone on GAPDH that is protected from tryptic digestion in the presence of CP12.
TABLE 1.
Identification of the 22 kDa band generated by limited tryptic digestion of GAPDH
The band was identified by mass spectrometry with an error of 20 ppm, a score of 103, and 31% of coverage of the full-length of the protein. The asterisks correspond to oxidized methionine. The periods correspond to the residue after which trypsin cleaves the sequence.
| Fragments | Observed Mr | Theoretical Mr | Δ | Miscleavage | Sequence |
|---|---|---|---|---|---|
| ppm | |||||
| 4-13 | 1101.6333 | 1101.6407 | -7 | 1 | K.IRVAINGFGR.I |
| 6-13 | 832.4259 | 832.4555 | -36 | 0 | R.VAINGFGR.I |
| 50-62 | 1386.6770 | 1386.6667 | 7 | 0 | K.YDSTLGTFAADVK.I |
| 50-74 | 2652.2639 | 2652.2919 | -11 | 1 | K.YDSTLGTFAADVKIVDDSHISVDGK.Q |
| 63-74 | 1283.6387 | 1283.6358 | 2 | 0 | K.IVDDSHISVDGK.Q |
| 78-90 | 1537.8308 | 1537.8616 | -20 | 1 | K.IVSSRDPLQLPWK.E |
| 83-90 | 995.5795 | 995.5440 | 36 | 0 | R.DPLQLPWK.E |
| 108-120 | 1222.6667 | 1222.6782 | -9 | 1 | K.VGAGKHIQAGASK.V |
| 129-145 | 1894.9389 | 1894.9313 | 4 | 1 | K.DKDIPTFVVGVNEGDYK.H |
| 172-190 | 2115.0255 | 2115.0055 | 9 | 1 | K.FGIVKGTM*TTTHSYTGDQR.L |
| 177-190 | 1554.7012 | 1554.6733 | 18 | 0 | K.GTMTTTHSYTGDQR.L |
| 177-190 | 1570.7070 | 1570.6682 | 25 | 0 | K.GTM*TTTHSYTGDQR.L |
| 191-197 | 810.4040 | 810.4348 | -38 | 0 | R.LLDASHR.D |
| 191-200 | 1194.6477 | 1194.6469 | 1 | 1 | R.LLDASHRDLR.R |
TABLE 2.
Identification of the 13 kDa band generated by limited tryptic digestion of GAPDH
The band was identified by mass spectrometry with an error of 14 ppm, a score of 115, and 32% coverage of the full length of the protein. The asterisks correspond to oxidized methionine.
| Fragments | Observed Mr | Theoretical Mr | Δ | Miscleavage | Sequence |
|---|---|---|---|---|---|
| ppm | |||||
| 204-218 | 1397.7907 | 1397.7878 | 2 | 0 | R.AAALNIVPTTTGAAK.A |
| 219-228 | 1025.6501 | 1025.6485 | 2 | 0 | K.AVSLVLPSLK.G |
| 229-237 | 940.5654 | 940.5818 | -17 | 1 | K.GKLNGIALR.V |
| 231-237 | 755.4557 | 755.4653 | -13 | 0 | K.LNGIALR.V |
| 231-254 | 2545.4693 | 2545.4843 | -6 | 1 | K.LNGIALRVPTPTVSVVDLVVQVEK.K |
| 238-254 | 1808.0009 | 1808.0296 | -16 | 0 | R.VPTPTVSVVDLVVQVEK.K |
| 238-255 | 1936.1715 | 1936.1245 | 24 | 1 | R.VPTPTVSVVDLVVQVEKK.T |
| 255-266 | 1381.6858 | 1381.6990 | -10 | 1 | K.KTFAEEVNAAFR.E |
| 256-266 | 1253.5926 | 1253.6040 | -9 | 0 | K.TFAEEVNAAFR.E |
| 267-274 | 832.4085 | 832.3749 | 40 | 0 | R.EAANGPM*K.G |
| 267-290 | 2552.2904 | 2552.2945 | -2 | 1 | R.EAANGPM*KGVLHVEDAPLVSIDFK.C |
| 275-290 | 1737.9454 | 1737.9301 | 9 | 0 | K.GVLHVEDAPLVSIDFK.C |
| 313-326 | 1771.7954 | 1771.7954 | 0 | 0 | K.VVAWYDNEWGYSQR.V |
| 327-336 | 1043.5812 | 1043.5863 | -5 | 0 | R.VVDLAEVTAK.K |
| 327-337 | 1171.6715 | 1171.6812 | -8 | 1 | R.VVDLAEVTAKK.W |
DISCUSSION
CP12 interacts with GAPDH and belongs to the family of IUPs. Many IUPs have been shown to possess a chaperone function, and thus, this prompted us to examine if CP12 exhibits any chaperone-like activity. We show here for the first time that CP12, both in its reduced and oxidized state, prevents GAPDH aggregation induced by heat treatment, thus fulfilling a chaperone-like task that involves a binding mechanism. This function is clearly independent of the redox properties of CP12 that regulate the activity of A4 GAPDH that are well documented.
It was first thought that the open and exposed character of IUPs makes them particularly vulnerable to aggregation, but their special amino acid composition itself counters this threat. In fact, IUPs are usually highly charged and low in hydrophobic residues, which all act against aggregation and subsequent amyloid formation (56). We showed here that CP12 does not aggregate under heat treatment. In the case of GAPDH, where aggregation was observed, the time required to inactivate GAPDH (t½ = 1 min) is shorter than the time required for the GAPDH to aggregate (t½ = 6 min). The first step is fast and may correspond to the unfolding step, where the fluorescence of GAPDH increases slightly and the wavelength of the maximum of the fluorescence emission shifts from 338 to 342 nm, corresponding to an exposure of the tryptophan residues. Moreover, during this step, as evidenced by CD experiments, there is a decrease of the secondary structure of GAPDH. These changes might induce the exposure of hydrophobic clusters that lead to aggregation of the protein. It is shown here that, upon heat treatment, GAPDH distributions change from the soluble to the insoluble fraction and that very likely GAPDH becomes denatured and precipitates. CP12 seems to have special sequence features to prevent aggregation of GAPDH. BSA, a nonchaperone control protein, does not offer any protection. Thioredoxin is well known for its thermoresistance (57) and in some cases has been described as a chaperone (58). The inactivation and aggregation of GAPDH in the presence of thioredoxin are still observed, although this protein slows down the process. Moreover, the heat shock protein Hsp33, which functions as a potent molecular chaperone capable of recognizing and binding aggregation-sensitive intermediates (54), can prevent aggregation of catalase fully but is only able to slow down aggregation of GAPDH. Thus, CP12 offers efficient protection of GAPDH that is specific, although glutathione, a “chemical” chaperone, in its oxidized form also protects GAPDH against aggregation. In its reduced form, glutathione is a guardian of the cell cycle and is involved in plant growth and also in defense against oxidative stress (59) but, in this state, has no effect on GAPDH aggregation. Glutathione in its oxidized form acts directly on some aggregating proteins, namely βL-crystallin and alcohol dehydrogenase, although this process is probably complex and not yet fully understood (60).
The fact that CP12 could offer protection against aggregation could explain why it is also found associated with the A2B2 form of GAPDH in supramolecular complexes from higher plants (3, 11, 61), although this form is by itself redox-regulated. This association might be even more important, since recombinant B4 GAPDH and the chimeric mutant of GapA subunits fused with the C-terminal extension of GapB subunit were found to be prone to aggregation (62). In land plants, AB-GAPDH gained an autonomous regulatory potential from the fusion of the GapA subunit to the C-terminal part of CP12 to give the GapB subunit (12, 63) but still coexists with a “free” CP12-dependent regulatory mechanism (61). Interestingly, an aggregated A8B8 form that results from the AB-GAPDH also exists and lacks CP12 (61). One can hypothesize from the results reported here that CP12 might also confer anti-aggregation properties to AB-GAPDH.
The protecting effect observed with CP12 might also actually relieve chaperones from the duty of guarding GAPDH under stress conditions, such as high temperature. However, and according to the classical definition of chaperones that are shown to bind only aggregation-prone proteins but not native ones (64), CP12 does not have a general chaperone activity. We show here that the protecting effect is rather specific to GAPDH, since for instance no effect of CP12 was observed on lysozyme, catalase, or alcohol dehydrogenase, proteins that are well known as chaperone substrates (31, 65, 66). Unlike most chaperones, CP12 forms a stable interaction with the native state of GAPDH. Protein chaperones are able to form stable complexes only with denatured proteins and not with native states (67–69). CP12 interacts both with denatured GAPDH at 43 °C (data not shown) and with the native state of GAPDH, and the binding is very strong (8). Thus, CP12 will behave as a chaperone-like protein and will not only interact transitorily with GAPDH.
The C-terminal part of CP12 is involved in the interaction with GAPDH, and it has been shown that, for the C66S mutant, no GAPDH·CP12 complex was observed by immunoblot after a native gel, in contrast to wild-type CP12 or C23S mutant (35). Interestingly, in this study, no solubilizing effect was observed with the C66S mutant, confirming that specific interactions are required to protect GAPDH against aggregation. The C23S mutant that has been shown to interact with GAPDH under the native state is also able to slow down the aggregation of GAPDH and its inactivation. It is clear that to fulfill its anti-aggregation function, CP12 has to form a stable complex with GAPDH. The C23S mutant association might be weaker at 43 °C, because aggregation is observed. Interestingly, reduced CP12 can also interact with GAPDH to form a stable GAPDH·CP12 complex and can protect GAPDH against aggregation. This means that the C-terminal disulfide bridge is not directly involved in the interaction with GAPDH, as was thought before (1, 35). The mutation of the cysteine residue at position 66 into a serine residue (C66S) may prevent CP12 from properly folding upon binding to its partner, so-called induced fit, as described for other IUPs (21), and thus no formation of the complex will be possible. The C-terminal disulfide bridge is thus not necessarily required to form the complex, since reduced CP12 is still able to reconstitute a subcomplex with GAPDH. The formation of the higher supramolecular complex GAPDH·CP12·phosphoribulokinase is, however, dependent on the redox state of CP12, since in the presence of reduced CP12, no formation of this ternary complex was observed (8).
Beside the involvement of the C-terminal part of CP12 in the interaction with GAPDH, it was previously shown by trypsin protection experiments that GAPDH may be bound to the central α-helix of CP12, which includes a highly conserved sequence in CP12 with aspartate and glutamate residues at positions 36 and 39 (35). It was hypothesized that electrostatic interactions and an ionic bridge exist between CP12 and GAPDH. This was also supported by the results obtained with a mutant of GAPDH in which Arg-197 had been replaced (49). This arginine residue is found in a region that is called the S loop. This area corresponds to residues 177–203 (numbered as in spinach GAPDH) or 180–206 (numbered as in Chlamydomonas GAPDH), is located between the β1 and β2 strands, and is close to the NADP nicotinamide moiety (70). The results obtained in this report, using state of the art techniques combining limited proteolysis, gel electrophoresis, and mass spectrometry to map the interaction site of CP12 on GAPDH, clearly show that the S-loop of GAPDH is indeed involved in the binding of CP12.
Like well known IUPs (21), CP12 binding on GAPDH induces its folding. A model of CP12 structure upon complex formation has been described and showed the presence of two α-helices with a particular distribution of charges along the first helix (71). Like many molecular chaperones that have well separated hydrophilic regions (to increase solubility) and hydrophobic regions (to bind the substrates) (72), CP12 also has a highly charged surface that could increase solubility of GAPDH. Moreover, changes of the fluorescence of the tryptophan residues of GAPDH indicate that CP12 may block the hydrophobic surfaces exposed from GAPDH upon heat treatment, since in the presence of CP12, no changes in either the red shift of the wavelength of maximal fluorescence or the fluorescence intensity are observed.
To conclude, CP12 has all of the features to fulfill a chaperone function but, unlike other chaperones, does not bind to the denatured states of its target but fully prevents this state and is rather specific to GAPDH. It remains to be determined if the chaperone function of CP12 is linked to the moonlighting features (73) of GAPDH (74).
Acknowledgments
We thank Dr. Stephen Maberly and Dr. Athel Cornish-Bowden for editing the English. We are also indebted to Maya Belghazi from CAPM-IFR Jean Roche for access to the Ultraflex II mass spectrometer (Bruker, Germany) and to Nicole Boggetto for W35A purification. We are indebted to Dr. U. Jakob and to C. Cremers for the generous gift of Hsp33 and advice to carry out the experiments. We are also grateful to Dr. M. Ilbert and Dr. M. L. Cardenas for helpful discussions and help in writing the manuscript.
Footnotes
The abbreviations used are: GAPDH, glyceraldehyde-3-phosphate dehydrogenase (EC 1.2.1.13); IUP, intrinsically unstructured protein; DTT, dithiothreitol; BSA, bovine serum albumin; Hsp, Heat shock protein; MALDI-TOF, matrix-assisted laser desorption ionization time-of-flight; RU, response units; PBS, phosphate-buffered saline.
References
- 1.Wedel, N., and Soll, J. (1998) Proc. Natl. Acad. Sci. U. S. A. 95 9699–9704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tamoi, M., Miyazaki, T., Fukamizo, T., and Shigeoka, S. (2005) Plant J. 42 504–513 [DOI] [PubMed] [Google Scholar]
- 3.Wedel, N., Soll, J., and Paap, B. K. (1997) Proc. Natl. Acad. Sci. U. S. A. 94 10479–10484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boggetto, N., Gontero, B., and Maberly, S. C. (2007) J. Phycol. 43 1227–1234 [Google Scholar]
- 5.Erales, J., Maberly, S., and Gontero, B. (2008) J. Phycol. 44 1455–1464 [DOI] [PubMed] [Google Scholar]
- 6.Oesterhelt, C., Klocke, S., Holtgrefe, S., Linke, V., Weber, A. P., and Scheibe, R. (2007) Plant Cell Physiol. 48 1359–1373 [DOI] [PubMed] [Google Scholar]
- 7.Erales, J., Avilan, L., Lebreton, S., and Gontero, B. (2008) FEBS J. 275 1248–1259 [DOI] [PubMed] [Google Scholar]
- 8.Graciet, E., Gans, P., Wedel, N., Lebreton, S., Camadro, J. M., and Gontero, B. (2003) Biochemistry 42 8163–8170 [DOI] [PubMed] [Google Scholar]
- 9.Graciet, E., Lebreton, S., Camadro, J. M., and Gontero, B. (2003) Eur. J. Biochem. 270 129–136 [DOI] [PubMed] [Google Scholar]
- 10.Marri, L., Trost, P., Pupillo, P., and Sparla, F. (2005) Plant Physiol. 139 1433–1443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Marri, L., Trost, P., Trivelli, X., Gonnelli, L., Pupillo, P., and Sparla, F. (2008) J. Biol. Chem. 283 1831–1838 [DOI] [PubMed] [Google Scholar]
- 12.Trost, P., Fermani, S., Marri, L., Zaffagnini, M., Falini, G., Scagliarini, S., Pupillo, P., and Sparla, F. (2006) Photosynth. Res. 89 263–275 [DOI] [PubMed] [Google Scholar]
- 13.Brinkmann, H., Cerff, R., Salomon, M., and Soll, J. (1989) Plant Mol. Biol. 13 81–94 [DOI] [PubMed] [Google Scholar]
- 14.Cerff, R., and Chambers, S. E. (1979) J. Biol. Chem. 254 6094–6098 [PubMed] [Google Scholar]
- 15.Robbens, S., Petersen, J., Brinkmann, H., Rouze, P., and Van de Peer, Y. (2007) J. Mol. Evol. 64 601–604 [DOI] [PubMed] [Google Scholar]
- 16.Pohlmeyer, K., Paap, B. K., Soll, J., and Wedel, N. (1996) Plant Mol. Biol. 32 969–978 [DOI] [PubMed] [Google Scholar]
- 17.Baalmann, E., Scheibe, R., Cerff, R., and Martin, W. (1996) Plant Mol. Biol. 32 505–513 [DOI] [PubMed] [Google Scholar]
- 18.Li, A. D., and Anderson, L. E. (1997) Plant Physiol. 115 1201–1209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Scagliarini, S., Trost, P., and Pupillo, P. (1998) J. Exp. Bot. 49 1307–1315 [Google Scholar]
- 20.Sparla, F., Pupillo, P., and Trost, P. (2002) J. Biol. Chem. 277 44946–44952 [DOI] [PubMed] [Google Scholar]
- 21.Dyson, H. J., and Wright, P. E. (2005) Nat. Rev. Mol. Cell Biol. 6 197–208 [DOI] [PubMed] [Google Scholar]
- 22.Tompa, P. (2005) FEBS Lett. 579 3346–3354 [DOI] [PubMed] [Google Scholar]
- 23.Uversky, V. N. (2002) Eur. J. Biochem. 269 2–12 [DOI] [PubMed] [Google Scholar]
- 24.Wright, P. E., and Dyson, H. J. (1999) J. Mol. Biol. 293 321–331 [DOI] [PubMed] [Google Scholar]
- 25.Sickmeier, M., Hamilton, J. A., LeGall, T., Vacic, V., Cortese, M. S., Tantos, A., Szabo, B., Tompa, P., Chen, J., Uversky, V. N., Obradovic, Z., and Dunker, A. K. (2007) Nucleic Acids Res. 35 D786–793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dosztanyi, Z., and Tompa, P. (2008) Methods Mol. Biol. 426 103–115 [DOI] [PubMed] [Google Scholar]
- 27.Dunker, A. K., Lawson, J. D., Brown, C. J., Williams, R. M., Romero, P., Oh, J. S., Oldfield, C. J., Campen, A. M., Ratliff, C. M., Hipps, K. W., Ausio, J., Nissen, M. S., Reeves, R., Kang, C., Kissinger, C. R., Bailey, R. W., Griswold, M. D., Chiu, W., Garner, E. C., and Obradovic, Z. (2001) J. Mol. Graph Model. 19 26–59 [DOI] [PubMed] [Google Scholar]
- 28.Tompa, P. (2002) Trends Biochem. Sci. 27 527–533 [DOI] [PubMed] [Google Scholar]
- 29.Iakoucheva, L. M., Brown, C. J., Lawson, J. D., Obradovic, Z., and Dunker, A. K. (2002) J. Mol. Biol. 323 573–584 [DOI] [PubMed] [Google Scholar]
- 30.Dunker, A. K. (2007) Structure 15 1026–1028 [DOI] [PubMed] [Google Scholar]
- 31.Kovacs, D., Kalmar, E., Torok, Z., and Tompa, P. (2008) Plant Physiol. 147 381–390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yoon, M. K., Shin, J., Choi, G., and Choi, B. S. (2006) Proteins 65 856–866 [DOI] [PubMed] [Google Scholar]
- 33.Csizmok, V., Dosztanyi, Z., Simon, I., and Tompa, P. (2007) Curr. Protein Pept. Sci. 8 173–179 [DOI] [PubMed] [Google Scholar]
- 34.Graciet, E., Lebreton, S., and Gontero, B. (2004) J. Exp. Bot. 55 1245–1254 [DOI] [PubMed] [Google Scholar]
- 35.Lebreton, S., Andreescu, S., Graciet, E., and Gontero, B. (2006) FEBS J. 273 3358–3369 [DOI] [PubMed] [Google Scholar]
- 36.Delobel, A., Graciet, E., Andreescu, S., Gontero, B., Halgand, F., and Laprevote, O. (2005) Rapid Commun. Mass Spectrom. 19 3379–3388 [DOI] [PubMed] [Google Scholar]
- 37.Erales, J., Gontero, B., Whitelegge, J., and Halgand, F. (2009) Biochem. J. 419 75–82 [DOI] [PubMed] [Google Scholar]
- 38.Tompa, P., and Csermely, P. (2004) FASEB J. 18 1169–1175 [DOI] [PubMed] [Google Scholar]
- 39.Kim, T. D., Paik, S. R., and Yang, C. H. (2002) Biochemistry 41 13782–13790 [DOI] [PubMed] [Google Scholar]
- 40.Bhattacharyya, J., and Das, K. P. (1999) J. Biol. Chem. 274 15505–15509 [DOI] [PubMed] [Google Scholar]
- 41.Liang, S. J., Lin, Y. Z., Zhou, J. M., Tsou, C. L., Wu, P. Q., and Zhou, Z. K. (1990) Biochim. Biophys. Acta 1038 240–246 [DOI] [PubMed] [Google Scholar]
- 42.Lin, Y. Z., Liang, S. J., Zhou, J. M., Tsou, C. L., Wu, P. Q., and Zhou, Z. K. (1990) Biochim. Biophys. Acta 1038 247–252 [DOI] [PubMed] [Google Scholar]
- 43.Markossian, K. A., Khanova, H. A., Kleimenov, S. Y., Levitsky, D. I., Chebotareva, N. A., Asryants, R. A., Muronetz, V. I., Saso, L., Yudin, I. K., and Kurganov, B. I. (2006) Biochemistry 45 13375–13384 [DOI] [PubMed] [Google Scholar]
- 44.Cumming, R. C., and Schubert, D. (2005) FASEB J. 19 2060–2062 [DOI] [PubMed] [Google Scholar]
- 45.Huang, G. C., Li, Z. Y., Zhou, J. M., and Fischer, G. (2000) Protein Sci. 9 1254–1261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kramer, G., Rutkowska, A., Wegrzyn, R. D., Patzelt, H., Kurz, T. A., Merz, F., Rauch, T., Vorderwulbecke, S., Deuerling, E., and Bukau, B. (2004) J. Bacteriol. 186 3777–3784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wickner, S., Maurizi, M. R., and Gottesman, S. (1999) Science 286 1888–1893 [DOI] [PubMed] [Google Scholar]
- 48.Bukau, B., Deuerling, E., Pfund, C., and Craig, E. A. (2000) Cell 101 119–122 [DOI] [PubMed] [Google Scholar]
- 49.Graciet, E., Mulliert, G., Lebreton, S., and Gontero, B. (2004) Eur. J. Biochem. 271 4737–4744 [DOI] [PubMed] [Google Scholar]
- 50.Habeeb, A. F. S. A. (1972) Methods Enzymol. 25 457–464 [DOI] [PubMed] [Google Scholar]
- 51.Bradford, M. M. (1976) Anal. Biochem. 72 248–254 [DOI] [PubMed] [Google Scholar]
- 52.Ilbert, M., Horst, J., Ahrens, S., Winter, J., Graf, P. C., Lilie, H., and Jakob, U. (2007) Nat. Struct. Mol. Biol. 14 556–563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jakob, U., Eser, M., and Bardwell, J. C. (2000) J. Biol. Chem. 275 38302–38310 [DOI] [PubMed] [Google Scholar]
- 54.Jakob, U., Muse, W., Eser, M., and Bardwell, J. C. (1999) Cell 96 341–352 [DOI] [PubMed] [Google Scholar]
- 55.Laemmli, U. K. (1970) Nature 227 680–685 [DOI] [PubMed] [Google Scholar]
- 56.Hegyi, H., and Tompa, P. (2008) PLoS Comput. Biol. 4 e1000017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Holmgren, A. (1985) Annu. Rev. Biochem. 54 237–271 [DOI] [PubMed] [Google Scholar]
- 58.Caldas, T., Malki, A., Kern, R., Abdallah, J., and Richarme, G. (2006) Biochem. Biophys. Res. Commun. 343 780–786 [DOI] [PubMed] [Google Scholar]
- 59.Potters, G., De Gara, L., Asard, H., and Horemans, N. (2002) Plant Physiol. Biochem. 40 537–548 [Google Scholar]
- 60.Clark, J. I., and Huang, Q. L. (1996) Proc. Natl. Acad. Sci. U. S. A. 93 15185–15189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Scheibe, R., Wedel, N., Vetter, S., Emmerlich, V., and Sauermann, S. M. (2002) Eur. J. Biochem. 269 5617–5624 [DOI] [PubMed] [Google Scholar]
- 62.Sparla, F., Zaffagnini, M., Wedel, N., Scheibe, R., Pupillo, P., and Trost, P. (2005) Plant Physiol. 138 2210–2219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fermani, S., Sparla, F., Falini, G., Martelli, P. L., Casadio, R., Pupillo, P., Ripamonti, A., and Trost, P. (2007) Proc. Natl. Acad. Sci. U. S. A. 104 11109–11114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Manna, T., Sarkar, T., Poddar, A., Roychowdhury, M., Das, K. P., and Bhattacharyya, B. (2001) J. Biol. Chem. 276 39742–39747 [DOI] [PubMed] [Google Scholar]
- 65.Markossian, K. A., Golub, N. V., Khanova, H. A., Levitsky, D. I., Poliansky, N. B., Muranov, K. O., and Kurganov, B. I. (2008) Biochim. Biophys. Acta 1784 1286–1293 [DOI] [PubMed] [Google Scholar]
- 66.Zhang, X., Fu, X., Zhang, H., Liu, C., Jiao, W., and Chang, Z. (2005) Int. J. Biochem. Cell Biol. 37 1232–1240 [DOI] [PubMed] [Google Scholar]
- 67.Chang, Z., Primm, T. P., Jakana, J., Lee, I. H., Serysheva, I., Chiu, W., Gilbert, H. F., and Quiocho, F. A. (1996) 271 7218–7223 [PubMed]
- 68.Ehrnsperger, M., Graber, S., Gaestel, M., and Buchner, J. (1997) EMBO J. 16 221–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lee, G. J., Roseman, A. M., Saibil, H. R., and Vierling, E. (1997) EMBO J. 16 659–671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fermani, S., Ripamonti, A., Sabatino, P., Zanotti, G., Scagliarini, S., Sparla, F., Trost, P., and Pupillo, P. (2001) J. Mol. Biol. 314 527–542 [DOI] [PubMed] [Google Scholar]
- 71.Gardebien, F., Thangudu, R. R., Gontero, B., and Offmann, B. (2006) J. Mol. Graph. Model. 25 186–195 [DOI] [PubMed] [Google Scholar]
- 72.Guha, S., Manna, T. K., Das, K. P., and Bhattacharyya, B. (1998) J. Biol. Chem. 273 30077–30080 [DOI] [PubMed] [Google Scholar]
- 73.Jeffery, C. J. (1999) Trends Biochem. Sci. 24 8–11 [DOI] [PubMed] [Google Scholar]
- 74.Sriram, G. M. J., McCabe, E. R., Liao, J. C., and Dipple, K. M. (2005) Am. J. Hum. Genet. 76 911–924 [DOI] [PMC free article] [PubMed] [Google Scholar]