

Abstract
The presence of extensive reciprocal conformational freedom between the catalytic and the hemopexin-like domains of full-length matrix metalloproteinase-1 (MMP-1) is demonstrated by NMR and small angle x-ray scattering experiments. This finding is discussed in relation to the essentiality of the hemopexin-like domain for the collagenolytic activity of MMP-1. The conformational freedom experienced by the present system, having the shortest linker between the two domains, when compared with similar findings on MMP-12 and MMP-9 having longer and the longest linker within the family, respectively, suggests this type of conformational freedom to be a general property of all MMPs.
Matrix metalloproteinases (MMP)2 are extracellular hydrolytic enzymes involved in a variety of processes including connective tissue cleavage and remodeling (1–3). All 23 members of the family are able to cleave simple peptides derived from connective tissue components such as collagen, gelatin, elastin, etc. A subset of MMPs is able to hydrolyze more resistant polymeric substrates, such as cross-linked elastin, and partially degraded collagen forms, such as gelatin and type IV collagens (4). Intact triple helical type I–III collagen is only attacked by collagenases MMP-1, MMP-8, and MMP-13 and by MMP-2 and MMP-14 (5–12). Although the detailed mechanism of cleavage of single chain peptides by MMP has been largely elucidated (13–19), little is known about the process of hydrolysis of triple helical collagen. In fact, triple helical collagen cannot be accommodated in the substrate-binding groove of the catalytic site of MMPs (9).
All MMPs (but MMP-7) in their active form are constituted by a catalytic domain (CAT) and a hemopexin-like domain (HPX) (20–22). The CAT domain contains two zinc ions and one to three calcium ions. One zinc ion is at the catalytic site and is responsible for the activity, whereas the other metal ions have structural roles. The isolated CAT domains retain full catalytic activity toward simple peptides and single chain polymeric substrates such as elastin, whereas hydrolysis of triple helical collagen also requires the presence of the HPX domain (9, 23–25). It has been shown that the isolated CAT domain regains a small fraction of the activity of the full-length (FL) protein when high amounts of either inactivated full-length proteins or isolated HPX domains are added to the assay solution (9). Finally, it has been shown that the presence of the HPX domain alone alters the CD spectrum of triple helical collagen in a way that suggests its partial unwinding (26, 27). It is tempting to speculate that full-length collagenases attack collagen by first locally unwinding the triple helical structure with the help of the HPX domain and then cleaving the resulting, exposed, single filaments (9, 28).
Until 2007, three-dimensional structures of full-length MMPs had been reported only for collagenase MMP-1 (29–31) and gelatinase MMP-2 (32). The structures of the two proteins are very similar and show a compact arrangement of the two domains, which are connected by a short linker (14 and 20 amino acids, respectively). It is difficult to envisage that rigid and compact molecules of this type can interact with triple helical collagen in a way that can lead to first unwinding and then cleavage of individual filaments. It has been recently suggested that such concerted action could occur much more easily if the two domains could enjoy at least a partial conformational independence (9). Slight differences in the reciprocal orientation of the CAT and HPX domains of MMP-1 in the presence (29) and absence (30, 31) of the prodomain were indeed taken as a hint that the two domains could experience relative mobility (29).
Two recent solution studies have shown that conformational independence is indeed occurring in gelatinase MMP-9 (33) and elastase MMP-12 (34), whereas the x-ray structure of the latter (34) is only slightly less compact than those of MMP-1 (29–31) and MMP-2 (32). Among MMPs, MMP-9 features an exceptionally long linker (68 amino acid) (33, 35), which in fact constitutes a small domain by itself (the O-glycosylated domain) (33), and therefore, this inspiring observation can hardly be taken as evidence that conformational freedom is a general characteristic of the two-domain MMPs. MMP-12 features a much more normal 16-amino acid linker, thereby making more probable a general functional role for this conformational freedom (34). However, both MMP-9 and MMP-12 retain their full catalytic activity against their substrates even when deprived of the HPX domain (9). Therefore, the question remains of whether conformational freedom is also a required characteristic for those MMPs that are only active as full-length proteins, i.e. collagenases. Interestingly, the three collagenases (MMP-1, MMP-8, and MMP-13) have the shortest linker (14 amino acids) among all MMPs. Demonstrating or negating the presence of conformational freedom in one of these collagenases would therefore constitute a significant step forward to formulate mechanistic hypotheses on their collagenolytic activity.
Our recent studies on MMP-12 in solution (34) have shown that a combination of NMR relaxation studies and small angle x-ray scattering (SAXS) is enough to show the presence and the extent of the relative conformational freedom of the two domains of MMPs. Here we apply the same strategy to full-length MMP-1 and show that sizable conformational freedom is indeed experienced even by this prototypical collagenase, although somewhat less pronounced than that observed for MMP-12.
EXPERIMENTAL PROCEDURES
Preparation of Protein Samples—The cDNA encoding the sequence (Asn-106–Asn-469) of the MMP-1 full-length protein was amplified from TrueClone cDNA (OriGene) by PCR and cloned into the pET21 (Novagen) expression vector using NdeI and XhoI (New England Biolabs) as restriction enzymes. One additional methionine at position 105 was present in the final expression product. The recombinant vector was transformed into Escherichia coli strain BL21(DE3) CodonPlus RIPL (Stratagene), and colonies were selected for ampicillin and chloramphenicol resistance. The bacteria were grown in 2×YT medium. When a cell density corresponding to 0.6 A was reached, the expression of the protein was induced by adding 0.5 mm isopropyl-β-d-thiogalactoside, and the incubation at 37 °C was continued for another 5 h. The full-length MMP-1 precipitated in the inclusion bodies, and these were solubilized, after lysis of the cells, in a solution of 8 m urea, 20 mm dithiothreitol, and 20 mm Tris-HCl, (pH 8.2). The protein was diluted with a buffer containing 6 m urea, 10 mm CaCl2, 0.1 mm ZnCl2, 20 mm cystamine, 20 mm Tris-HCl (pH 8) and refolded by using a multistep dialysis against solutions containing 50 mm Tris-HCl (pH 8), 4 m urea, 10 mm CaCl2, 0.1 mm ZnCl2, 5 mm β-mercaptoethanol, 1 mm 2-hydroxyethyl disulfide, then against a solution containing 50 mm Tris-HCl (pH 7.2), 2 m urea, 10 mm CaCl2, 0.1 mm ZnCl2, 0.3 m NaCl, and then against the same solution without urea. The protein was purified by size-exclusion chromatography on the HiLoad 26/60 Superdex 75 pg (Amersham Biosciences). For expression of 15N- and 13C-enriched FL-MMP-1, the bacteria were grown in minimal medium containing 15N-enriched (NH4)2SO4 and 13C-enriched glucose (Cambridge Isotope Laboratories). The resulting inclusion bodies were solubilized in 8 m urea, 20 mm Tris-HCl (pH 8), 20 mm dithiothreitol. Protein molecular weight and purity were checked on a 17% gel by SDS-PAGE and by mass spectroscopy (matrix-assisted laser desorption). The E219A mutant of FL-MMP-1 was produced using the QuikChange site-directed mutagenesis kit (Qiagen), and the expression and purification of the protein and of its 15N- and 13C-15N-enriched versions were completed using the same procedure described above. Samples of 2H-13C-15N-enriched full-length human MMP-1 protein were obtained by adapting E. coli cells in medium with different percentages of deuterium until reaching 100%, and then growing the cells in OD2 Silantes media carbon, deuterium, nitrogen (CDN). Samples of cadmium(II)-substituted FL-MMP-1 protein were prepared by exhaustive dialysis against a buffer containing 20 mm Tris, pH 7.2, 10 mm CaCl2, 0.3 m NaCl, 0.2 m acetohydroxamic acid, and 0.3 mm CdCl2 (36). Equimolar concentrations of N-isobutyl-N-[4-methoxyphenylsulfonyl] glycyl hydroxamic acid (NNGH) were added to the samples to further increase the protein stability. The cDNA encoding for the HPX domain (Thr-274–Asn-469) was generated by polymerase chain reaction and cloned into pET21a using NdeI and XhoI as restriction enzymes. The expression vector was then transformed into competent E. coli BL21(DE3) CodonPlus RIPL strain, and the colonies were selected for ampicillin and chloramphenicol resistance. Protein refolding both for non-enriched samples and for 13C- and/or 15N-enriched samples was carried out by using the same protocols described previously for the preparation of FL-MMP-1 samples. Samples of the zinc(II) catalytic domain (Asn-106–Gly-261, E219A inactive mutant) were prepared as described previously (37).
NMR Measurements and Protein Assignment—The experiments for the protein assignment and mobility measurements of the isolated domains (CAT and HPX) were performed on protein samples at concentrations ranging between 0.3 and 0.7 mm (pH 7.2). For FL-MMP-1, all NMR experiments were performed on samples at a concentration of 0.3 mm (pH 7.2). NMR experiments were performed at 298 and 306 K and acquired on Bruker AVANCE 900, AVANCE 800, AVANCE 700, AVANCE 600, and DRX 500 spectrometers equipped with triple resonance cryo-probes. All spectra were processed with the Bruker TOPSPIN software packages and analyzed by the program CARA (Computer Aided Resonance Assignment, ETH Zürich) (38)). The backbone resonance assignment of HPX domain was obtained by the analysis of HNCA, HNCACB, and CBCA-(CO)NH spectra performed at 500 and 900 MHz, whereas the assignment of the 2H-, 15N-, and 13C-enriched FL-MMP-1 was obtained by the analysis TROSY-HNCA, TROSY-HNCACB TROSY-HNCOCACB performed on an 800-MHz spectrometer. The obtained assignments are reported in supplemental Table S1 for the full-length protein, in supplemental Table S2 for the catalytic domain, and in supplemental Table S3 for the hemopexin-like domain.
The protein assignment and the mobility measurements on FL-MMP-1 were performed on the NNGH-inhibited, E219A mutant, due to its high stability to the self-hydrolysis. Mobility measurements on the catalytic domain were performed on the NNGH-inhibited form of the protein.
R1,R2, and NOE Measurements—The experiments for the determination of 15N longitudinal and transverse relaxation rates and 1H-15N NOE were recorded at 306 K and 700 MHz on 15N-enriched samples. The 15N longitudinal relaxation rates (R1) were measured using a sequence modified to remove cross correlation effects during the relaxation delay (39). Inversion recovery times ranging between 2.5 and 3000 ms, with a recycle delay of 3.5 s, were used for the experiments. The 15N transverse relaxation rates (R2) were measured using a Carr-Purcell-Meiboom-Gill (CPMG) sequence (39, 40) with delays ranging between 8.5 and 237.4 ms for the CAT domain, between 8.5 and 203.5 ms for the HPX domain, and finally between 8.5 and 135.7 ms for the FL-MMP-1 protein with a refocusing delay of 450 μs. The relaxation data are reported in supplemental Table S4. R1 and R2 data measured on the full-length protein were found nosier and less uniform with respect to those of the single catalytic and hemopexin-like domains. This is related to the overlap of the signals in such a large protein and to the relative low solubility of the full-length construct.
Paramagnetic Relaxation Enhancement Measurements—The paramagnetic relaxation enhancements of the backbone NH protons were obtained by adding a stock water solution (50 mm) of Gd(DTPA·BMA) to a solution of FL-MMP-1 (200 μm protein in a buffer containing 20 mm Tris, pH 7.2, 10 mm CaCl2, 0.3 m NaCl, 0.2 m acetohydroxamic acid, 1 mm ZnCl2, and 3 mm NNGH) up a final concentration of 1.4 mm. NH proton longitudinal relaxation times were measured through an inversion recovery HSQC sequence obtained by introducing a 1H 180°-pulse followed by a variable delay τ in front of a standard 1H-15N-HSQC sequence. T1 values were obtained from a series of spectra obtained with the following τ values (in ms): 20, 50, 80, 100, 200, 400, 500, 800, 1000, 1400, 2000, 2200, 3000, and 3500. The measurement was repeated on the free protein and after each addition of the paramagnetic complex. Peak volumes as a function of the τ value were fitted to a monoexponential recovery with a three-parameter fit.
SAXS Experiments and Data Analysis—Small Angle x-ray scattering data from solutions of the NNGH-inhibited, cadmium(II)-substituted E219A mutant of FL-MMP-1 were collected on the X33 beamline of the European Molecular Biology Laboratory (EMBL) (Deutsches Elektronen Synchrotron (DESY), Hamburg) (41) using a MAR345 image plate detector. The scattering patterns were measured with a 2-min exposure time for several solute concentrations in the range from 0.8 to 8.3 mg/ml. To check for radiation damage, two 2-min exposures were compared, and no changes were detected. Using the sample-detector distance of 2.7 m, a range of momentum transfer of 0.01 < s < 0.5 Å–1 was covered (s = 4π sin(θ)/λ, where 2 θ is the scattering angle and λ = 1.5 Å is the x-ray wavelength). The data were processed using standard procedures and extrapolated to infinite dilution using the program PRIMUS (42). The forward scattering, I(0), and the radius of gyration, Rg, were evaluated using the Guinier approximation (43), assuming that at very small angles (s < 1.3/Rg), the intensity is represented as I(s) = I(0)exp((–s2R 2g)/3). The values of I(0) and Rg, as well as the maximum dimension, Dmax, and the interatomic distance distribution functions, (p(r)), were also computed using the program GNOM (44). The scattering from the high resolution models was computed using the program CRYSOL (45). Given the atomic coordinates, the program predicts the theoretical scattering pattern and fits the experimental intensity by adjusting the excluded volume of the particle and the contrast of the hydration layer to minimize the discrepancy
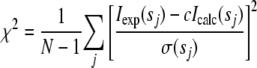 |
where N is the number of experimental points, c is a scaling factor, Iexp(sj) and Icalc(sj) are the experimental and calculated intensities, respectively, and σ(sj) is the experimental error at the momentum transfer sj. To assess the conformational variability of MMP-1, an ensemble optimization method (EOM) was used (46), allowing for the coexistence of multiple conformations in solution. About 10,000 randomized models of FL-MMP-1 differing by the conformation of the interdomain linker were generated, and their scattering patterns were computed using the program RanCh of the EOM package. These models formed a pool of possible structures for which the scattering patterns were computed by CRYSOL. The EOM program employs a genetic algorithm to select from the pool a small number (usually about 20) of representative structures such that the average scattering from the selected ensemble fits the experimental data. Multiple runs of EOM were performed, and the results were averaged to provide quantitative information about the flexibility of the protein in solution (in particular, about the Rg distribution in the selected ensembles).
RESULTS AND DISCUSSION
Full-length human MMP-1 and its CAT-(106–261) and HPX-(274–469) domains were expressed as described under “Experimental Procedures.” In both FL-MMP-1 and its CAT domain, an E219A mutation was introduced. Such mutation ensures a dramatic lowering of the catalytic activity (13, 19), which is needed in the present investigation to prevent self-hydrolysis. In addition, the strong active site-directed hydroxamic inhibitor NNGH (15) was always added. Finally, when needed, cadmium(II) was substituted for zinc(II) in the catalytic site to further reduce the residual activity as it was already shown for FL-MMP-12 and its CAT domain (34). 15N-, 13C-15N-, and 2H-13C-15N-enriched samples were used for NMR investigations. The samples were stable under the NMR experimental conditions for several months. For the present mobility studies, the assignment of the 1H-15N HSQC spectra of the three constructs was needed. The two-dimensional and three-dimensional experiments that were used for this purpose are described under “Experimental Procedures.” The 1H-15N-TROSY-HSQC spectra of the three constructs, acquired at 306 K, are superimposed in Fig. 1. The spectral quality, especially for the FL and HPX domains, is better at 306 K than at 298 K. This prompted us to perform all subsequent work at 306 K. Moreover, the latter temperature is closer to the physiological value of 310 K, so the resulting mobility data will be more meaningful.
FIGURE 1.

1H-15N-TROSY-HSQC spectra of FL-MMP-1 (2H-13C-15N-labeled NNGH-inhibited, E219A mutant) (black), superimposed with CAT-MMP-1 (15N-labeled NNGH-inhibited, E219A mutant) (red) and with HPX-MMP-1 (15N-labeled) (green).
Analysis of the Chemical Shift Differences between FL-MMP-1 and Its Isolated Domains—For clarity, the spectrum of the full-length protein in Fig. 1 is from a triply labeled 2H, 13C, 15N sample, which displays much sharper lines. From visual inspection, it already appears that the FL spectrum is largely the superposition of the CAT and HPX spectra as the majority of the signals in the FL spectrum overlap with either a CAT or an HPX signal. There are a number of additional signals in the FL spectrum that are attributed to the portion of the interdomain linker (262–275), which is missing in both the CAT and the HPX isolated domains. Furthermore, a few more signals undergo a modest shift on passing from the isolated domains to the FL protein. These signals could arise from contacts between each domain and the linker, contacts between the two domains, or both. In the latter two cases, evidence of interdomain contacts would imply that either the FL length structure is rigid or, that even in the presence of conformational freedom, “closed” compact structures contribute to the overall description of the molecule.
The assignments for the three constructs are reported in supplemental Tables S1–S3. From a comparison of the 1H and 15N chemical shifts in the 1H-15N-TROSY-HSQC spectra for the FL and the two isolated domains, a mean shift difference plot (47) was generated (Fig. 2). It appears that the number of peaks experiencing chemical shift differences is rather limited and clustered in the 112–115, 139–149, 244–261, and 277–316 regions. Fig. 3A shows the experimental x-ray three-dimensional structure of the FL protein (30) as well as of its isolated domains and linker, exploded to show the regions of contact of the CAT and HPX domains with one another (green) and with the linker (red). Fig. 3B shows the same structures, where the intensity of the colored residues correlates with the size of the mean shift difference experienced by the FL with respect to the isolated domains. It is apparent that most of the chemical shift differences arise from contacts of the CAT and HPX domains with the linker, and very few and with relatively low values are attributable to direct interdomain contacts. It can be concluded that it is likely that the compact structure observed in the x-ray structure of the active FL protein is in equilibrium with other more open structures in solution.
FIGURE 2.

Plot of mean shift differences experienced by FL MMP-1 with respect to the isolated CAT or HPX domains. avg. indicates average.
FIGURE 3.

Mapping of the residues at the interface between the CAT domain, the linker and the HPX domain. A, exploded space fill representation of the experimental three-dimensional x-ray structure of FL-MMP-1 (24) where the regions of contact between CAT and HPX domains are colored in green and the regions of contact of either of the two domains with the linker are colored in red. The arrangement of the two domains and the linker in the crystal structure is visible in the background. B, the same exploded space fill representation where the intensity of the colored residues, from yellow to dark-orange, correlates with the size of the mean shift difference experienced by FL-MMP-1 with respect to the isolated domains. The similarity of the areas experiencing shift changes due to the lack of the linker in B with the contact areas in red, but not with those in green in A, show that most of the shift perturbation arises from the contacts with the linker and not between the two domains.
Relaxation Measurements—Measurements of longitudinal (R1) and transverse (R2) relaxation rates of backbone amide nitrogens at 700-MHz 1H Larmor frequency and 306 K have been performed on 15N-enriched samples of both the isolated CAT domains and the isolated HPX domains as well as of FL-MMP-1. Estimates of R1 and R2 values for these three constructs under the chosen experimental conditions of magnetic field and temperature were obtained using the program HydroNMR (48), and the three-dimensional structures of the constructs were taken from the x-ray structure of the FL protein (30). The experimental and theoretical R1 and R2 values are reported in Fig. 4, A–E), together with the corresponding data for MMP-12 (Fig. 4, F–L) (34) at the same field and 298 K, for comparison purposes. If the experimental data do not agree with the calculated data, outside the experimental error, then they are inconsistent with the structural model. As far as the catalytic domain is concerned (left side), it is clear that the R1 and R2 data for the isolated domain are in excellent agreement with the theoretical expectations. On the contrary, the experimental R1 data for the CAT domain, when it is part of the FL protein, are sizably higher, and the R2 data are sizably lower, with respect to the theoretical values for a rigid FL protein. Higher R1 and lower R2 values, taken together, indicate that the two domains behave as if they belonged to a lower molecular weight protein; that is, the CAT domain possesses some degree of motion, which is independent of the motion of the FL protein as a whole.
FIGURE 4.

Comparison of NMR relaxation data for MMP-1 (data collected at 306 K) and MMP-12 (data collected at 298 K). The calculated (gray bars) and experimental (filled circles) backbone 15NH R1 and R2 values for the isolated CAT and HPX domains of MMP-1 are shown in A and C, and those of MMP-12 are shown in F and H. The 15NH R1 and R2 values for the full-length MMP-1 and MMP-12 are shown in B, D, G, and I, respectively. The experimental NOE values for the full-length proteins are shown in E and L for MMP-1 and MMP-12, respectively. In both proteins, the agreement between experimental and calculated R1 values for the isolated domains is excellent, whereas for the full-length proteins, the experimental R1 values are sizably larger (B and G) than the ones calculated for the rigid x-ray structures. R2 values (D and I) are also in good agreement if a slight tendency of the HPX domain to aggregate is taken into account. The data comparisons are only consistent with interdomain flexibility in MMP-1, which is somewhat less extensive than in MMP-12.
In principle, the same should hold for the HPX domain. Indeed, the R1 and R2 data for the HPX domain in the full-length protein are also higher and lower, respectively, than the theoretical estimate, but here the difference is less marked than for the CAT domain. A plausible explanation lies in the fact that the R1 and R2 data for the isolated HPX domain show the opposite behavior, i.e. the R1 values are lower and the R2 values are higher than expected. This is typically encountered in the presence of self-aggregation. The HPX domain does possess a rather hydrophobic surface, which could allow for the existence of a fraction of dimeric or higher order aggregation species in solution. Partial aggregation brought about by the HPX domain in the FL protein would of course artificially attenuate the effect of interdomain mobility on R1 and R2. The hypothesis of HPX aggregation is easy to check by changing the protein concentration. Indeed, for isolated HPX solutions of higher concentration, the discrepancy with the calculated data sizably increases (data not shown). In conclusion, the HPX data (i) demonstrate the presence of aggregation but also (ii) confirm interdomain mobility in the full-length protein, which is still apparent from R1 and R2 data despite the attenuation of the effect due to aggregation.
A further proof of the interdomain mobility is provided by the very small NOE values of four adjacent residues in the linker region (267, 268, 270, and 271) (Fig. 4E). Comparison with the MMP-12 data (Fig. 4, F–L), collected at lower temperature, shows that interdomain mobility in MMP-1, although certainly present, is less extensive than in MMP-12. This is possibly related (i) to the shorter linker (14 versus 16 residues) and (ii) to the more extensive interdomain contact present in the x-ray structure of active FL-MMP-1 with respect to that observed in the x-ray structure of MMP-12.
A Paramagnetic Probe Monitors Interdomain Surface Exposure—Paramagnetic probes dissolved in protein solutions have been previously used (i) to identify surface residues to help structure determination (49–56), (ii) to determine the orientation of membrane-bound peptides on the membrane surface (57–59), and (iii) to monitor the formation of protein·protein complexes by detecting residues that become shielded from the probe upon complex formation (4, 60, 61). In analogy with the latter strategy, we have used the gadolinium complex Gd(DTPA·BMA) (57) to probe the CAT-HPX interface in the FL protein. An inversion recovery HSQC sequence was used to measure the R1 values of the backbone NH protons of FL-MMP-1 in the absence (R1dia) and in the presence (R1para) of 1.4 mm Gd(DTPA·BMA), as detailed under “Experimental Procedures.” The residues for which the relaxation rate difference R1para – R1dia is higher than a threshold value of 0.7 s–1 are reported in supplemental Table S5. Among them, on the HPX side, residue Leu-314 should be shielded by the CAT domain; on the CAT side, residue Asp-231 should be shielded by the linker, and residues Asp-245, Val-246, and Gln-247 should be shielded by the HPX domain. On the contrary, all of them experience paramagnetic effects particularly strong for Asp-245 (2.7 s–1) and Val-246 (2.4 s–1). These effects are inconsistent with the compact x-ray structure of FL-MMP-1 and confirm that the protein must spend a non-negligible fraction of time in an extended conformation so that the CAT-HPX interface becomes accessible to the probe.
SAXS Measurements—The processed x-ray scattering pattern from FL-MMP-1 is shown in Fig. 5. The experimental radius of gyration Rg is 29 ± 1 Å. This value significantly exceeds that calculated (Rg = 25 Å) from the x-ray structure of active FL-MMP-1 (30). Moreover, the scattering pattern calculated from the active FL-MMP-1 model using CRYSOL (45) fails to fit the experimental data (discrepancy χ = 4.22, Fig. 5, curve 2). Interestingly, the fit to the FL-MMP-12 model is better (discrepancy χ = 1.8, Fig. 5, curve 3). If a distribution of FL-MMP-1 conformations, including compact and extended ones, is taken into consideration, neither individual models nor averaging over the random pool allowed one to fit the SAXS data satisfactorily (χ > 1.9 in both cases). The representative ensembles selected to fit the data give information about the preferable conformations of the protein. To assess the preferable conformations in solution, the EOM method (46) was used. Given a representative pool of (random) structures, the method employs a genetic algorithm to select the ensembles from this pool that best fit the experimental data, as explained under “Experimental Procedures.” Several EOM runs yielded reproducible ensembles neatly fitting the experimental data with discrepancy χ less than 1.2, and a typical fit provided by the ensemble selected by EOM is given in Fig. 5, curve 4. All the fits from different EOM runs are graphically indistinguishable from Fig. 5, curve 4. The Rg distributions of the particles in the initial pool and in the selected ensembles are compared in Fig. 5 (inset). The former distribution is rather broad and covers the Rg range from about 20 to 45 Å, corresponding to extremely compact and completely extended domain configurations, respectively. In contrast, the Rg distribution of the selected ensembles displays a relatively sharp peak around Rg = 25–26 Å, including about 70% of the particles in the selected ensembles. Visual inspection of the models having Rg values in the peak range indicates, not unexpectedly, that their shape is similar to that of the FL-MMP-1 structure in the crystal (30) (although with varying interdomain orientations). However, an extended tail at higher Rg values up to around 40 Å, accounting for about 30% of the particles, is apparent from the multiple EOM reconstructions. These results indicate that the crystallographic conformation of FL-MMP-1 (30) is largely present also in solution, but in addition, the protein experiences noticeable flexibility with a significant amount of extended conformations in equilibrium with the closed one(s). We also tried to explore the possibility of a two-state exchange situation allowing for only two conformations in the mixture. In such a case, two structures with an Rg of around 25 and 32 Å are selected. It is interesting that FL-MMP-1 (30) appears much more compact than FL-MMP-12 (34), as shown by the comparison in Fig. 5, inset, indicating that the two extra residues in the linker region of FL-MMP-12 increase its flexibility.
FIGURE 5.

Experimental x-ray scattering from the NNGH-inhibited, FL-MMP-1 and scattering calculated from the following models: curve 1, experimental data with error bars; curves 2 and 3, computed scattering from the crystallographic models of FL-MMP-1 and FL-MMP-12, respectively; and curve 4, a typical fit by the selected ensemble of structures. The logarithm of the scattering intensity is plotted against the momentum transfer. Inset, the frequency of the models with the given Rg in the initial pool of structures with randomized interdomain linkers (FL-MMP-1 (blue) and FL-MMP-12 (magenta)) and in the selected ensembles (FL-MMP-1 (red) and FL-MMP-12 (green); the latter distribution is obtained by the averaging of several EOM runs. All histograms are normalized to the integral value of unity.
Concluding Remarks and Biological Implications—The present data demonstrate that FL-MMP-1 shows relative mobility of its catalytic and hemopexin-like domains, as recently observed for FL-MMP-12 (34). As in the latter case, the reorientation of the backbone NH vectors with respect to the magnetic field occurs on a time scale that is faster than the rotational time of the whole molecule (62). The amplitude of the motion is probably smaller for FL-MMP-1 than it is for FL-MMP-12, as judged from the SAXS data that suggest that the molecules spend two-thirds of the time in a conformation that is more or less as compact as the solid state structure but is ample enough that the residues at the interface between the two domains are significantly exposed to a paramagnetic probe in solution. Besides MMP-12, relative mobility of the CAT and HPX domains has also been recently suggested for MMP-9, where the linker between the two domains is much longer and constitutes a domain by itself (O-glycosylated domain) (63). In the case of MMP-9, it has been argued (33) that the long O-glycosylated domain may be flexible and may mediate protein-substrate interactions (64, 65). However, the present finding of flexibility in FL-MMP-1 has further biological implications because MMP-1 is a collagenase that is able to hydrolyze intact triple helical type I–III collagen but only in the full-length form, i.e. the presence of the HPX domain is crucial for the function. Notably, the possibility of reorienting the HPX with respect to the CAT domain during catalysis has often been invoked as a means to unwind the collagen triple helix in preparation for the catalytic cleavage (66).
Supplementary Material
Acknowledgments
We acknowledge the support and assistance of the DESY (Hamburg) synchrotron radiation facilities for the SAXS data collection.
This work was supported by grants from the European Commission (Projects MEST-CT-2004-504391, SFMET Number 201640, SPINE2-COMPLEXES Number 031220, and Nano4Drugs Number LSHB-CT-2005-019102), the Ministero dell'Istruzione, dell'Università e della Ricerca (PRIN 2005, Protocollo Number 2005039878, Protocollo RBLA032ZM7, Protocollo RBIP06LSS2), and the Ente Cassa di Risparmio di Firenze.
The on-line version of this article (available at http://www.jbc.org) contains five supplemental tables.
Footnotes
The abbreviations used are: MMP, matrix metalloproteinase; SAXS, small angle x-ray scattering; CAT, catalytic domain; HPX, hemopexin-like domain; FL, full-length; NNGH, N-isobutyl-N-[4-methoxyphenylsulfonyl] glycyl hydroxamic acid; TROSY, transverse relaxation optimized spectroscopy; HSQC, heteronuclear single quantum correlation; NOE, nuclear Overhauser effect; EOM, ensemble optimization method; DTPA, diethylene-triaminepentaacetic acid; BMA, bismethylamide.
References
- 1.Woessner, J. F., Jr., and Nagase, H. (1999) J. Biol. Chem. 274 21491–21494 [DOI] [PubMed] [Google Scholar]
- 2.Brinckerhoff, C. E., and Matrisian, L. M. (2002) Nat. Rev. Mol. Cell Biol. 3 207–214 [DOI] [PubMed] [Google Scholar]
- 3.Page-McCaw, A., Ewald, A. J., and Werb, Z. (2007) Nat. Rev. Mol. Cell Biol. 8 221–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bhaskaran, R., Palmier, M. O., Lauer-Fields, J. L., Fields, G. B., and Van Doren, S. R. (2008) J. Biol. Chem. 283 21779–21788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sternlicht, M. D., and Werb, Z. (2001) Annu. Rev. Cell Dev. Biol. 17 463–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Overall, C. M. (2002) Mol. Biotechnol. 22 51–86 [DOI] [PubMed] [Google Scholar]
- 7.Lauer-Fields, J. L., Juska, D., and Fields, G. B. (2002) Biopolymers 66 19–32 [DOI] [PubMed] [Google Scholar]
- 8.Visse, R., and Nagase, H. (2003) Circ. Res. 92 827–839 [DOI] [PubMed] [Google Scholar]
- 9.Chung, L. D., Dinakarpandian, D., Yoshida, N., Lauer-Fields, J. L., Fields, G. B., Visse, R., and Nagase, H. (2004) EMBO J. 23 3020–3030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gioia, M., Fasciglione, G. F., Marini, S., D'Alessio, S., De Sanctis, G., Diekmann, O., Pieper, M., Politi, V., Tschesche, H., and Coletta, M. (2002) J. Biol. Chem. 277 23123–23130 [DOI] [PubMed] [Google Scholar]
- 11.Gioia, M., Monaco, S., Fasciglione, G. F., Coletti, A., Modesti, A., Marini, S., and Coletta, M. (2007) J. Mol. Biol. 368 1101–1113 [DOI] [PubMed] [Google Scholar]
- 12.Lauer-Fields, J. L., Tuzinski, K. A., Shimokawa, K.-I., Nagase, H., and Fields, G. B. (2000) J. Biol. Chem. 275 13282–13290 [DOI] [PubMed] [Google Scholar]
- 13.Bertini, I., Calderone, V., Fragai, M., Luchinat, C., and Maletta, M. (2006) Angew. Chem. Int. Ed. Engl. 45 7952–7955 [DOI] [PubMed] [Google Scholar]
- 14.Bertini, I., Calderone, V., Fragai, M., Luchinat, C., Mangani, S., and Terni, B. (2003) Angew. Chem. Int. Ed. Engl. 42 2673–2676 [DOI] [PubMed] [Google Scholar]
- 15.Bertini, I., Calderone, V., Cosenza, M., Fragai, M., Lee, Y.-M., Luchinat, C., Mangani, S., Terni, B., and Turano, P. (2005) Proc. Natl. Acad. Sci. U. S. A. 102 5334–5339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rosenblum, G., Meroueh, S., Toth, M., Fisher, J. F., Fridman, R., Mobashery, S., and Sagi, I. (2007) J. Am. Chem. Soc. 129 13566–13574 [DOI] [PubMed] [Google Scholar]
- 17.Gall, A. L., Ruff, M., Kannan, R., Cuniasse, P., Yiotakis, A., Dive, V., Rio, M. C., Basset, P., and Moras, D. (2001) J. Mol. Biol. 307 577–586 [DOI] [PubMed] [Google Scholar]
- 18.Bode, W., Reinemer, P., Huber, R., Kleine, T., Schnierer, S., and Tschesche, H. (1994) EMBO J. 13 1263–1269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cha, J., and Auld, D. S. (1997) Biochemistry 36 16019–16024 [DOI] [PubMed] [Google Scholar]
- 20.Massova, I., Kotra, L. P., Fridman, R., and Mobashery, S. (1998) FASEB J. 12 1075–1095 [PubMed] [Google Scholar]
- 21.Bode, W., and Maskos, K. (2003) Biol. Chem. 384 863–872 [DOI] [PubMed] [Google Scholar]
- 22.Nagase, H., Visse, R., and Murphy, G. (2006) Cardiovasc. Res. 69 562–573 [DOI] [PubMed] [Google Scholar]
- 23.Clark, I. E., and Cawston, T. E. (1989) Biochem. J. 263 201–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Murphy, G., Allan, J. A., Willenbrock, F., Cockett, M. I., O'Connell, J. P., and Docherty, A. J. (1992) J. Biol. Chem. 267 9612–9618 [PubMed] [Google Scholar]
- 25.Knauper, V., Cowell, S., Smith, B., Lopez-Otin, C., O'Shea, M., Morris, H., Zardi, L., and Murphy, G. (1997) J. Biol. Chem. 272 7608–7616 [DOI] [PubMed] [Google Scholar]
- 26.Tam, E. M., Wu, Y. I., Butler, G. S., Stack, M. S., and Overall, C. M. (2002) J. Biol. Chem. 277 39005–39014 [DOI] [PubMed] [Google Scholar]
- 27.Tam, E. M., Moore, T. R., Butler, G. S., and Overall, C. M. (2004) J. Biol. Chem. 279 43336–43344 [DOI] [PubMed] [Google Scholar]
- 28.Minond, D., Lauer-Fields, J. L., Nagase, H., and Fields, G. B. (2004) Biochemistry 43 11474–11481 [DOI] [PubMed] [Google Scholar]
- 29.Jozic, D., Bourenkov, G., Lim, N. H., Visse, R., Nagase, H., Bode, W., and Maskos, K. (2005) J. Biol. Chem. 280 9578–9585 [DOI] [PubMed] [Google Scholar]
- 30.Iyer, S., Visse, R., Nagase, H., and Acharya, K. R. (2006) J. Mol. Biol. 362 78–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li, J., Brick, P., Ohare, M. C., Skarzynski, T., Lloyd, L. F., Curry, V. A., Clark, I. M., Bigg, H. F., Hazleman, B. L., Cawston, T. E., and Blow, D. M. (1995) Structure (Lond.) 3 541–549 [DOI] [PubMed] [Google Scholar]
- 32.Morgunova, E., Tuuttila, A., Bergmann, U., Isupov, M., Lindqvist, Y., Schneider, G., and Tryggvason, K. (1999) Science 284 1667–1670 [DOI] [PubMed] [Google Scholar]
- 33.Rosenblum, G., Van den Steen, P. E., Cohen, S. R., Grossmann, J. G., Frenkel, J., Sertchook, R., Slack, N., Strange, R. W., Opdenakker, G., and Sagi, I. (2007) Structure (Lond.) 15 1227–1236 [DOI] [PubMed] [Google Scholar]
- 34.Bertini, I., Calderone, V., Fragai, M., Jaiswal, R., Luchinat, C., Melikian, M., Mylonas, E., and Svergun, D. (2008) J. Am. Chem. Soc. 130 7011–7021 [DOI] [PubMed] [Google Scholar]
- 35.Andreini, C., Banci, L., Bertini, I., Luchinat, C., and Rosato, A. (2004) J. Proteome Res. 3 21–31 [DOI] [PubMed] [Google Scholar]
- 36.Bertini, I., Fragai, M., Lee, Y.-M., Luchinat, C., and Terni, B. (2004) Angew. Chem. Int. Ed. Engl. 43 2254–2256 [DOI] [PubMed] [Google Scholar]
- 37.Nesi, A., and Fragai, M. (2007) ChemBioChem 8 1367–1369 [DOI] [PubMed] [Google Scholar]
- 38.Keller, R. L. J. (2004) The Computer Aided Resonance Assignment Tutorial, Vol. 1.3, CANTINA Verlag, Goldau, Switzerland
- 39.Kay, L. E., Nicholson, L. K., Delaglio, F., Bax, A., and Torchia, D. A. (1992) J. Magn. Reson. 97 359–375 [Google Scholar]
- 40.Peng, J. W., and Wagner, G. (1994) Methods Enzymol. 239 563–596 [DOI] [PubMed] [Google Scholar]
- 41.Roessle, M. W., Klaering, R., Ristau, U., Robrahn, B., Jahn, D., Gehrmann, T., Konarev, P. V., Round, A., Fiedler, S., Hermes, S., and Svergun, D. I. (2007) J. Appl. Crystallogr. 40 s190–s194 [Google Scholar]
- 42.Konarev, P. V., Volkov, V. V., Sokolova, A. V., Koch, M. H. J., and Svergun, D. I. (2003) J. Appl. Crystallogr. 36 1277–1282 [Google Scholar]
- 43.Guinier, A. (1939) Ann. Phys. 12 161–237 [Google Scholar]
- 44.Svergun, D. I. (1992) J. Appl. Crystallogr. 25 495–503 [Google Scholar]
- 45.Svergun, D. I., Barberato, C., and Koch, M. H. J. (1995) J. Appl. Crystallogr. 28 768–773 [Google Scholar]
- 46.Bernado, P., Mylonas, E., Petoukhov, M. V., Blackledge, M., and Svergun, D. I. (2007) J. Am. Chem. Soc. 129 5656–5664 [DOI] [PubMed] [Google Scholar]
- 47.Grzesiek, S., Bax, A., Clore, G. M., Gronenborn, A. M., Hu, J. S., Kaufman, J., Palmer, I., Stahl, S. J., and Wingfield, P. T. (1996) Nat. Struct. Biol. 3 340–345 [DOI] [PubMed] [Google Scholar]
- 48.de la Torre, J. G., Huertas, M. L., and Carrasco, B. (2000) J. Magn. Reson. 147 138–146 [DOI] [PubMed] [Google Scholar]
- 49.Aime, S., D'Amelio, N., Fragai, M., Lee, Y.-M., Luchinat, C., Terreno, E., and Valensin, G. (2002) J. Biol. Inorg. Chem. 7 617–622 [DOI] [PubMed] [Google Scholar]
- 50.Pintacuda, G., and Otting, G. (2002) J. Am. Chem. Soc. 124 372–373 [DOI] [PubMed] [Google Scholar]
- 51.Scarselli, M., Esposito, G., De Magistris, M. T., Domenighini, M., Rappuoli, R., Burroni, G., Bernini, A., and Niccolai, N. (1998) J. Biochem. 254 313–317 [DOI] [PubMed] [Google Scholar]
- 52.Scarselli, M., Bernini, A., Segoni, C., Molinari, H., Esposito, G., Lesk, A. M., Laschi, F., Temussi, P., and Niccolai, N. (1999) J. Biomol. NMR 15 125–133 [DOI] [PubMed] [Google Scholar]
- 53.Esposito, G., Lesk, A. M., Molinari, H., Motta, A., Niccolai, N., and Pastore, A. (1992) J. Mol. Biol. 224 659–670 [DOI] [PubMed] [Google Scholar]
- 54.Yuan, T., Ouyang, H., and Vogel, H. J. (1999) J. Biol. Chem. 274 8411–8420 [DOI] [PubMed] [Google Scholar]
- 55.Bernini, A., Venditti, V., Spiga, O., Ciutti, A., Prischi, F., Consonni, R., Zetta, L., Arosio, I., Fusi, P., Guagliardi, A., and Niccolai, N. (2008) Biophys. Chem. 137 71–75 [DOI] [PubMed] [Google Scholar]
- 56.Spiga, O., Bernini, A., Scarselli, M., Ciutti, A., Bracci, L., Lozzi, L., Lelli, B., Di Maro, D., Calamandrei, D., and Niccolai, N. (2002) FEBS Lett. 511 33–35 [DOI] [PubMed] [Google Scholar]
- 57.Respondek, M., Madl, T., Göbl, C., Golser, R., and Zangger, K. (2007) J. Am. Chem. Soc. 129 5228–5234 [DOI] [PubMed] [Google Scholar]
- 58.Nanga, R. P., Brender, J. R., Xu, J., Veglia, G., and Ramamoorthy, A. (2008) Biochemistry 47 12689–12697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jarvet, J., Danielsson, J., Damberg, P., Oleszczuk, M., and Graslund, A. (2007) J. Biomol. NMR 39 63–72 [DOI] [PubMed] [Google Scholar]
- 60.Arumugam, S., Hemme, C. L., Yoshida, N., Suzuki, K., Nagase, H., Berjanskii, M., Wu, B., and Van Doren, S. R. (1998) Biochemistry 37 9650–9657 [DOI] [PubMed] [Google Scholar]
- 61.Impagliazzo, A., and Ubbink, M. (2004) J. Am. Chem. Soc. 126 5658–5659 [DOI] [PubMed] [Google Scholar]
- 62.Wang, T. Z., Frederick, K. K., Igumenova, T. I., Wand, A. J., and Zuiderweg, E. R. P. (2005) J. Am. Chem. Soc. 127 828–829 [DOI] [PubMed] [Google Scholar]
- 63.Van den Steen, P. E., Van Aelst, I., Hvidberg, V., Piccard, H., Fiten, P., Jacobsen, C., Moestrup, S. K., Fry, S., Royle, L., Wormald, M. R., Wallis, R., Rudd, P. M., Dwek, R. A., and Opdenakker, G. (2006) J. Biol. Chem. 281 18626–18637 [DOI] [PubMed] [Google Scholar]
- 64.Saffarian, S., Collier, I. E., Marmer, B. L., Elson, E. L., and Goldberg, G. (2004) Science 306 108–111 [DOI] [PubMed] [Google Scholar]
- 65.Overall, C. M., and Butler, G. S. (2007) Structure (Lond.) 15 1159–1161 [DOI] [PubMed] [Google Scholar]
- 66.Ottl, J., Gabriel, D., Murphy, G., Knauper, V., Tominaga, Y., Nagase, H., Kroger, M., Tschesche, H., Bode, W., and Moroder, L. (2000) Chem. Biol. 7 119–132 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.