

Abstract
The human pyruvate dehydrogenase complex (PDC) is a 9.5-megadalton catalytic machine that employs three catalytic components, i.e. pyruvate dehydrogenase (E1p), dihydrolipoyl transacetylase (E2p), and dihydrolipoamide dehydrogenase (E3), to carry out the oxidative decarboxylation of pyruvate. The human PDC is organized around a 60-meric dodecahedral core comprising the C-terminal domains of E2p and a noncatalytic component, E3-binding protein (E3BP), which specifically tethers E3 dimers to the PDC. A central issue concerning the PDC structure is the subunit stoichiometry of the E2p/E3BP core; recent studies have suggested that the core is composed of 48 copies of E2p and 12 copies of E3BP. Here, using an in vitro reconstituted PDC, we provide densitometry, isothermal titration calorimetry, and analytical ultracentrifugation evidence that there are 40 copies of E2p and 20 copies of E3BP in the E2p/E3BP core. Reconstitution with saturating concentrations of E1p and E3 demonstrated 40 copies of E1p heterotetramers and 20 copies of E3 dimers associated with the E2p/E3BP core. To corroborate the 40/20 model of this core, the stoichiometries of E3 and E1p binding to their respective binding domains were reexamined. In these binding studies, the stoichiometries were found to be 1:1, supporting the 40/20 model of the core. The overall maximal stoichiometry of this in vitro assembled PDC for E2p:E3BP:E1p:E3 is 40:20:40:20. These findings contrast a previous report that implicated that two E3-binding domains of E3BP bind simultaneously to a single E3 dimer (Smolle, M., Prior, A. E., Brown, A. E., Cooper, A., Byron, O., and Lindsay, J. G. (2006) J. Biol. Chem. 281, 19772–19780).
The human pyruvate dehydrogenase complex (PDC)3 resides in mitochondria and catalyzes the oxidative decarboxylation of pyruvate to yield acetyl-CoA and reducing equivalents (NADH), serving as a link between glycolysis and the Krebs cycle (1–3). The PDC is a large (∼9.5 MDa) catalytic machine comprising multiple protein components. The three catalytic components are pyruvate dehydrogenase (E1p), dihydrolipoyl transacetylase (E2p), and dihydrolipoamide dehydrogenase (E3), with E3 being a common component between different α-keto acid dehydrogenase complexes. The two regulatory enzymes in the PDC are the isoforms of pyruvate dehydrogenase kinase and pyruvate dehydrogenase phosphatase.
The PDC is organized around a structural core, which includes the C-terminal domains of E2p and a noncatalytic component that specifically binds E3, i.e. the E3-binding protein (E3BP). To this E2p/E3BP core, multiple copies of the other PDC components are tethered through noncovalent interactions. Each E2p subunit contains two consecutive N-terminal lipoic acid-bearing domains (LBDs), termed L1 and L2, followed by the E1p-binding domain (E1pBD) and the C-terminal inner-core/catalytic domain, with these independent domains connected by unstructured linkers. Similarly, each E3BP subunit consists of a single N-terminal LBD (referred to as L3), the E3-binding domain (E3BD), and the noncatalytic inner core domain. Together, the inner core domains of E2p and E3BP assemble to form the dodecahedral 60-meric E2p/E3BP core. The role of the E1pBD and E3BD domains is to tether E1p and E3, respectively, to the periphery of the E2p/E3BP core. It is presumed that the LBDs (L1, L2, and L3) shuttle between the active sites of the three catalytic components of the PDC during the oxidative decarboxylation cycle (4). The eukaryotic PDC is unique among α-keto acid dehydrogenase complexes in its requirement for E3BP; prokaryotic PDCs employ the single subunit-binding domain to secure either E1p or E3 to the complex (5).
Using a “divide-and-conquer” approach, a wealth of structural information on the PDC has been accumulated recently. High-resolution crystal structures are available for the human E1p (6–8) and E3 components (9). A model for the human E2p has been constructed based on an 8.8-Å electron density map available from cryo-electron microscopy (10). Additionally, solution and crystal structures of the L1 and L2 domains of E2p have been determined (11–13), and the high-resolution crystal structures of the E3BD (14, 15), pyruvate dehydrogenase kinase isoforms 1–4 (12, 16–18), and pyruvate dehydrogenase phosphatase isoform 1 (19) are known. Therefore, atomic models are available for almost all components and domains of the mammalian PDC.
With the successes of the above structural approach, attention has turned to the overall structure of the PDC. There are two outstanding questions as follows. What are the subunit and overall catalytic component stoichiometries? What are the positions and orientations of the components in this large catalytic machine? Yu et al. (10) recently determined the cryo-EM structure of a PDC core comprising only human E2p subunits. Like yeast E2p, human E2p adopts a dodecahedral structure composed of 60 E2p proteins; each face of the dodecahedron has a large gap. Although this structure is highly informative, the composition of this core deviates substantially from that of the native PDC, because no E3BP subunits are present in the core structure. Based on the similar structure of the dodecahedral yeast PDC, a hypothesis was formed that, in human PDC, 12 copies of E3BP bind in the 12 gaps, which is termed the “60/12” model (20). Biophysical studies on complexes of E2p and E3BP later negated the 60/12 model; Hiromasa et al. (21) therefore posited an alternative, the “48/12” model, in which the dodecahedral core includes 48 E2p subunits and 12 E3BP proteins. A further source of conjecture is how many E1p and E3 components bind to the periphery of the PDC. If one binding domain binds to one peripheral catalytic component, a maximally occupied 60/12 PDC would harbor 60 E1p heterotetramers and 12 E3 dimers (or 48 E1ps and 12 E3s in the 48/12 model). The notion of such 1:1 binding is supported by the preponderance of available biophysical evidence. Specifically, two crystal structures, site-directed mutagenesis, and calorimetric measurements describe a 1:1 interaction between E3BD and E3 (14, 15). Also, although no structures are available for the human E1p-E1pBD complex, a crystal structure of the homologs of these proteins from Bacillus stearothermophilus also demonstrates a 1:1 interaction between the E1pBD of E2p and the E1p heterotetramer (22). In addition, ITC experiments performed on the bacterial E1p and the cognate subunit-binding domain indicate a 1:1 association (23). At variance with the above observations, a different subunit stoichiometry has been proposed by Smolle et al. (24, 25). Their evidence suggests that two binding domains bind for every peripheral component; such an arrangement potentially yields a PDC with half as many peripheral components bound.
This study was undertaken to ascertain the subunit and component stoichiometries of the human PDC, particularly with regard to interactions between the E3BD and the E3 dimer. We show that quantification of bands on an SDS-polyacrylamide gel of a PDC reconstituted at saturating E1p and E3 concentrations supports neither the 60/12 nor the 48/12 model. Instead, a “40/20” model is proposed, and subsequent ITC and analytical ultracentrifugation (AUC) data corroborate this new model. In addition, results from electrophoretic mobility shift assays, ITC, and AUC presented here uniformly show a 1:1 interaction between E3BD and the E3 dimer as well as between E1pBD and the E1p heterotetramer. The implications of this 1:1 binding stoichiometry for the macromolecular assembly of the PDC are discussed.
EXPERIMENTAL PROCEDURES
Protein Expression and Purification—Recombinant human E3 (9, 14) and E1p (7) were expressed and purified as described previously. The recombinant fusion protein LBDb-E3BD was constructed, expressed, and purified as described before. Descriptions and schematic representations of the LBDb-E1pBD, LBDb-E3BD, L3-E3BD, and XDD1 constructs in the pET28b expression vector can be found under “Results and Discussion” (see Fig. 2). The expression plasmids for human E1p and the full-length human E2p/E3BP core were generous gifts from Dr. Kirill Popov, University of Alabama Medical Center, Birmingham, AL (26). For use as a protein standard, the E2p expression construct was generated by the deletion of a SalI fragment containing the mature E3BP coding sequence from the E2p/E3BP plasmid. The E3BP construct was prepared by amplifying the coding sequence for mature E3BP from the E2p/E3BP plasmid; the amplified DNA fragment was also cloned into the pET28b expression vector.
FIGURE 2.

E1pBD- and E3BD-containing constructs used or discussed in this study. A, five protein constructs are represented schematically. Residue numbers are those of the E1pBD and E3BP of the human PDC, based on mature sequence numbering. The cyan-colored boxes in the LBDb-E1pBD and LBDb-E3BD constructs depict the LBDb from the E2b component of the human BCKDC. The blue boxes represent the L3 domain from E3BP. The tan box is the TEV site; excluding E1pBD, the magenta boxes (residues 121–129 and 170–175) are the sequences flanking E3BD (residues 130–170) depicted by the red box. The green boxes in all constructs except XDD represent the 8 amino acids from the vector sequence, including the His6 tag. The XDD1 protein contains the same C-terminal truncation of 7 amino acids in E3BD as the XDD construct (nonprocessed sequence residues 59–216, or mature sequence residues 6–163) reported previously (25). The number of N-terminal amino acids from the pET14b vector (green box) in the XDD construct is unknown. B, secondary structure and amino sequence of human E3BD (hE3BD). The amino acid residues in hE3BD that directly interact with residues from both subunits of the E3 dimer are annotated by asterisks. The solid lines represent the linker regions connecting the two helices in hE3BD.
All E3BD-containing proteins were expressed in Escherichia coli BL21 DE3 pLys S cells (Novagen, San Diego, CA). Lipoic acid was added to the growth media to a final concentration of 0.2 mm before induction with 0.5 mm isopropyl 1-thio-β-d-galactopyranoside. After overnight growth at 30 °C, harvested cells were suspended in the lysis buffer (100 mm potassium phosphate, pH 8.0, 500 mm NaCl, 1% (v/v) Triton X-100, 0.5% (v/v) Tween 20, 10% (v/v) glycerol, 1 mm benzamidine, 1 mm phenylmethylsulfonyl fluoride, and 20 mm β-mercaptoethanol) and disrupted by the addition of lysozyme and sonication. Cleared cell extract was treated with nickel-nitrilotriacetic acid resin (Qiagen, Valencia, CA), and the eluted protein was further purified on a Superdex 200 26/60 column (GE Healthcare) that had been equilibrated with 50 mm potassium phosphate, pH 7.5, 150 mm KCl, 5% (v/v) glycerol, and 20 mm β-mercaptoethanol. The E3BD protein (E3BP residues 121–175) was prepared by incubating LBDb-E3BD protein with the TEV protease (100:1 (w/w)) for 24 h at 4 °C, followed by nickel-nitrilotriacetic acid extraction and purification on a Sephacryl S-100 column equilibrated in the same buffer. The recombinant E2p/E3BP core and E2p core proteins were expressed and purified according to the method described by Hiromasa et al. (21); a key modification was that the E2p/E3BP core and E2p core proteins in the absence of detergent were purified from the pellet of the first 8% (w/v) polyethylene glycol 8000 precipitation. The individually expressed E3BP protein was purified from the pellet of the second 8% polyethylene glycol 8000 precipitation, followed by purification on gel filtration and FPLC Resource Q column.
Protein Concentration Determination—At least two different measurements were used to determine the concentrations of the individual proteins. For E3, the absorbance of the flavin chromophore at 450 nm (ε450 nm = 10,325 m-1 cm-1) was monitored. Alternatively, E3 was precipitated using trichloroacetic acid, washed with trichloroacetic acid, and then dissolved in guanidinium hydrochloride to remove the bound FAD; the absorbance of E3 at 278 nm (ε278 nm = 23,971 m-1 cm-1) was used to determine the concentration of E3. The concentrations of LBDb-E1pBD, LBDb-E3BD, L3-E3BD, and XDD1 were determined by using the calculated extinction coefficients (ε280 nm) of 14,000, 14,435, 11,000, and 11,000 m-1 cm-1, respectively, and colorimetrically by the advanced protein determination assay (Cytoskeleton, Inc., Denver, CO). Because the E3BD sequence does not contain any UV-absorbing aromatic amino acid residues, a colorimetric assay was used to determine its protein concentration. The subunit-binding domain from E2b of the BCKDC was used as a standard in the advanced protein determination assay used to determine the E3BD concentration. Deviations between protein measurements were generally within 10%. Final protein concentrations were taken as the average of the spectral and colorimetric values. The ε278 nm of the 60-meric E2p/E3BP core was calculated to be 29,596 m-1 cm-1 assuming a subunit ratio of E2p:E3BP = 40:20 (this study). The ε278 nm values of individual E2p and E3BP proteins were 33,920 and 20,970 m-1 cm-1, respectively. The ε278 nm value for the E1p heterotetramer was 143,438 m-1 cm-1.
In Vitro Assembly of the Human PDC—The purified recombinant E2p/E3BP core (0.45 nmol) in 50 mm potassium phosphate, pH 7.5, 150 mm KCl, and 5% (v/v) glycerol was combined with purified His6-tagged E3 dimers (17.5 nmol) in a final volume of 2.6 ml, or with purified E1p heterotetramers (32 nmol), or with both E3 and E1p. The assembly mixtures were passed through an FPLC Superdex S-200 column to remove excess unbound E3 and/or E1p proteins from the E2p/E3BP core. To determine subunit stoichiometries, the purified reconstituted PDC was subjected to SDS-PAGE and stained with Coomassie Brilliant Blue. All sample gels and individual protein standard gels were run simultaneously and stained and destained together. The separated bands were quantified by both staining density alone and by comparing with the standard curves obtained with individual protein standards. The molar extinction coefficients described above were used. Results from the two methods are in good agreement except for the E1β subunit, which stained weaker with the dye than other subunits. The substoichiometric amount of E1β subunit obtained by direct staining intensity was corrected by comparing with the standard curve generated for this subunit.
Native PAGE Binding Assay—Before electrophoresis, 180 pmol of E3 were mixed with varying concentrations of the E3BD-containing proteins in 50 mm potassium phosphate, pH 7.5, 50 mm KCl, 5% (v/v) glycerol, and 5 mm dithiothreitol in a final volume of 15 μl. Half of each sample was loaded onto Tris-glycine polyacrylamide gels (5% (w/v) stacking gel with 7% (w/v) resolving gel). After electrophoresis at 100 V for 2.5 h, the gels were stained with Coomassie Brilliant Blue R-250 for 20 min and destained in 10% (v/v) methanol and 10% (v/v) acetic acid.
Isothermal Titration Calorimetry—ITC measurements were performed in a VP-ITC microcalorimeter (MicroCal, Northampton, MA). Titrations were carried out in 50 mm potassium phosphate, pH 7.5, 50 mm KCl, 10 mm β-mercaptoethanol, and 0.01% (v/v) Tween 20 at 20 °C. Prior to the titration, both proteins had been exhaustively dialyzed against this buffer. In a typical measurement for E3BD binding, 30 injections (at 3-min intervals) of 8 μl each of the E3BD-containing protein (200 μm) or LBDb-E1pBD (186 μm) were made into 1.4271 ml of the E3 dimer (20 μm) or the E1p heterotetramer (18 μm), respectively, in the cell. To measure E3 or E1p binding to the E2p/E3BP core, 210 μm of the E3 dimer or 150 μm of the E1p heterotetramer in the syringe was injected into the cell containing 14 or 50 μm (based on monomer concentration), respectively, of the E2p/E3BP core. The binding isotherms calculated by integration of the thermograms were used to derive the three nonlinear parameters for the interactions: the association constant (Ka), the molar ratio (n), and the enthalpy change (ΔH). Ka was converted to the dissociation constant (Kd) using the relationship Kd = 1/Ka. Curve fitting and the derivation of thermodynamic parameters were carried out using the nonlinear parameter optimization protocols implemented in the ORIGIN version 7.0 software package provided by MicroCal. The concentrations of the proteins were determined as described above.
Analytical Ultracentrifugation—All analytical ultracentrifugation sedimentation velocity (SV) experiments were carried out in a model XL-I (Beckman-Coulter, Fullerton, CA) centrifuge. Experiments with LBDb-E3BD were carried out in a buffer composed of 50 mm potassium phosphate, pH 7.5, 12 mm KCl. These studies were performed at 20 °C in an An60-Ti rotor (Beckman-Coulter) using a rotor speed of 50,000 rpm. The buffer for the experiments with L3-E3BD and XDD1 included 20 mm Tris-HCl, pH 7.5. They were performed using a rotor speed of 45,000 rpm at 4 °C in an An50-Ti rotor (Beckman-Coulter). AUC studies featuring reconstituted PDC with either E1p or E3 were done at 20 °C, with rotor speeds of 15,000 or 20,000 rpm, respectively, in the An50Ti rotor. All AUC experiments were carried out in dual-sector charcoal-filled Epon centerpieces fitted with sapphire windows; all sample volumes were ∼390 μl. Both the on-board laser interferometer and UV-visible spectrophotometer were used to monitor the sedimentation of the proteins. The partial specific volumes of the proteins, the buffer viscosity, and the buffer density were estimated using the freeware program SEDNTERP (27). The initial qualities of the experiments were analyzed by performing simple c(s) analyses, in which the raw data are directly modeled as a continuous distribution of noninteracting, ideally sedimenting species. The freeware program SEDFIT (28, 29) was used for such analyses.
Although the concentrations of the proteins included in all SV experiments were estimated using UV spectrophotometry, the laser interferometer in the XL-I centrifuge was used as an independent, unbiased means for determining protein concentration. The raw data from the interferometer can be expressed in terms of the “number of fringes displaced,” termed ΔJ. This quantity is related concentration of the protein by an equation that is similar to the familiar Beer-Lambert law as shown in Equation 1,
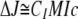 |
(Eq. 1) |
where c is concentration (expressed in mol/liter); M is the molar mass of the species; l is the optical path length (1.2 cm in this case), and CI is a constant with a value of 2.75 fringes·liter·g-1·cm-1. The term CIM assumes the place of the conventional extinction coefficient in this Beer-Lambert-like formulation; this term should be applicable for all proteins that do not have significant post-translational modifications, such as glycosylation. This term throughout this paper is referred to as “εIF”, making Equation 1 take the form shown in Equation 2,
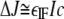 |
(Eq. 2) |
In practice, εIF was calculated using Equation 3,
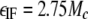 |
(Eq. 3) |
where Mc is the predicted molar mass of the protein based on its amino acid sequence. The mass of the FAD cofactor was not considered in calculating the εIF of E3. In an SV experiment with only one protein, ΔJ is observed; l is known, and εIF is easily calculated using Equation 3. Therefore, the only unknown in Equation 2 is c, allowing its calculation from the experimentally observed ΔJ.
In the SV experiments presented in this work, there are usually two boundaries: a leading boundary, representing the co-sedimenting complexes of the larger and smaller proteins, and a lagging boundary that represents excess free smaller protein. For most of these experiments, three methods were used to determine the concentrations of the smaller protein that cosedimented with the larger. In the first, called the “lagging” method, the concentration of lagging material was quantified using Equations 2 and 3. Because a known concentration of lagging protein was input, the lagging concentration was subtracted from the input concentration, and this value was taken as the amount of lagging protein in the complex. For the second method, ΔJ for the leading boundary was determined. From this value, the input ΔJ of the larger protein was subtracted. This latter value was taken as the signal because of the cosedimenting smaller protein. Using Equations 2 and 3, the concentration of the cosedimenting smaller protein can be deduced. The third method is the multisignal method (30–32), which utilizes the ability of the data acquisition software to collect concentration profile data from both on-board detection systems. In parallel with the interferometric (IF) data, absorbance (ABS) profiles of the centrifugation cells were also obtained. Because c is known from the IF data, this value can be fixed, and the molar extinction coefficient for the absorbance optics, εABS, may be determined using the Beer-Lambert law shown in Equation 4,
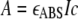 |
(Eq. 4) |
where A is the absorbance. The freeware program SEDPHAT takes this approach in globally analyzing SV data acquired from the two different optical systems (32); εABS is treated as a “floating” nonlinear parameter that is optimized along with sample menisci, frictional ratio, etc. By performing SV experiments with the individual proteins, the εABS may be established for each protein in isolation. The relationship between εIF and εABS for a particular protein defines a spectral signature for that protein. When two of the components are mixed and a multisignal SV experiment is carried out, it has been shown that these signatures may be used to deconvolute the c(s) distribution into two distributions, one for each component (32). Such distributions are called ck(s) distributions. In the current context, two ck(s) distributions may be calculated, one representing the population of the smaller protein and the other of the larger protein (or complex). Such distributions may be integrated to yield the concentrations of the components at defined s20,w values. In this way, the concentrations and thus stoichiometry of the individual components of a cosedimenting protein complex can be determined. For experiments performed with the core complex, the concentration derived was that of a subunit of the complex assuming one of two models. For the 40/20 model, the subunit was a heterotrimer comprising two copies of E2p and one of E3BP. For the 48/12 model, the subunit was a heteropentamer made up of four copies of E2p and one of E3BP.
The c(s) and ck(s)
distributions in this study were regularized using the maximum entropy and
Tikhonov-Phillips protocols
(28), respectively, such that
ratio of the goodness-of-fit statistics of the regularized distribution to the
best fit distribution
(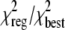 )
corresponded to a confidence level of 0.683. In this way, the statistical
quality of the fitted distributions with regularization was not allowed to
deviate from that of the best distributions by more than 1σ. For the SV
experiments performed at 4 °C, the sample meniscus for the IF data refined
to unrealistic values, likely indicating that some convection was present in
the sample. In these cases, the meniscus was fixed at a position that
corresponded to an optical artifact in the data that has reliably represented
the meniscus in past experiments.
)
corresponded to a confidence level of 0.683. In this way, the statistical
quality of the fitted distributions with regularization was not allowed to
deviate from that of the best distributions by more than 1σ. For the SV
experiments performed at 4 °C, the sample meniscus for the IF data refined
to unrealistic values, likely indicating that some convection was present in
the sample. In these cases, the meniscus was fixed at a position that
corresponded to an optical artifact in the data that has reliably represented
the meniscus in past experiments.
RESULTS AND DISCUSSION
Subunit and Molar Stoichiometries of the Human PDC Reconstituted in Vitro—The organization of the human PDC was investigated by in vitro assembly using individually purified recombinant E1p, E3, and the E2p/E3BP core proteins. The 60-meric E2p/E3BP core was incubated with molar excesses of E3 dimers and/or E1p heterotetramers. The assembly mixture was passed through an FPLC Superdex 200 column to remove excess unbound E3 and/or E1p components. The purified reconstituted PDC was subjected to SDS-PAGE and stained with Coomassie Brilliant Blue (Fig. 1A). Subunit stoichiometries of the assembled PDC were estimated by comparing the staining intensity of each band and by estimating the amount of each subunit from the standard curve obtained with individual protein standards. The subunit:component ratio of E2p:E3BP: E1p heterotetramer:E3 dimer is 43 ± 2.4:20:36.9 ± 5.3:18.6 ± 1.1 by direct comparison of staining intensity and 48.7 ± 2.2: 20:38.2 ± 4.3:16.1 ± 0.8 by comparing to the standard curves, with nine experiments for both methods. The component ratios are indexed to the E3BP subunit. These results support a stoichiometry of E2p:E3BP:E1p:E3 = 40:20:40:20.
FIGURE 1.

Subunit and catalytic component stoichiometries and solution behavior of an in vitro reconstituted PDC. A, in vitro assembly of the human pyruvate dehydrogenase complex. The E2p/E3BP core (0.45 nmol) in a potassium phosphate buffer, pH 7.5, was mixed with E3 (17.5 nmol) in a final volume of 2.6 ml (lane 1); the same sample of E2p/E3BP core was mixed with E1p (32 nmol) (lane 2); or the same E2p/E3BP core sample was mixed with E3 (17.5 nmol) and E1p (32 nmol) (lane 3). The protein mixtures were passed through a Sephacryl S-400 column to remove the excess unbound E3 or E1p. The stoichiometries of protein subunits were estimated by both Coomassie Blue staining intensity directly and by comparing to the standard curves obtained with individual protein standards. The subunit stoichiometry of E2p:E3BP in the 60-meric E2p/E3BP core is 40:20. The molar ratio of E2p/E3BP 60-mer:E1p heterotetramer:E3 dimer in the reconstituted PDC is 1:37:18 based on the staining intensity method normalized for the E1β subunit by comparison with the standard curve for this subunit. B, SV studies on PDC and its complexes with E3 and E1p. The solid line shows the c(s) distribution of PDC core alone, and the dashed line shows the distribution when a saturating concentration of E3 is added, and the dotted line shows the distribution when a saturating concentration of E1p is added. The large peaks on the left side of the distributions are because of excess E3 or E1p. C, ITC measurements of E3 binding to the reconstituted PDC core. The top panel is the thermogram that results from titrating the E3 dimer into a solution of E2p/E3BP. The solid varying line depicts enthalpy changes, and the flat line is the base line, as calculated by ORIGIN 7.0. The squares in the bottom panel are the integrated heats from each injection as derived from the data in the top panel, and the solid line is the fit to these data using a model that considers only a single type of binding sites. The molar ratio for E3 dimer:E2p/E3BP core is 18.8:1, based on the 40/20 E2p/E3BP model.
The present subunit ratio of E2p:E3BP = 40:20, estimated by staining intensity or by comparison with individual standard curves, is different from that of 48:12 reported previously by Roche and co-workers (21). Our E2p:E3BP subunit ratio also disagrees with the 6–12 copies of E3BP in the PDC estimated previously (20, 33). The 40:20 subunit ratio that we propose would be consistent with a dodecahedral arrangement of 20 heterotrimers, with each E2p/E3BP heterotrimer harboring two E2p subunits and one E3BP subunit. In contrast, based on the ratio of E2p:E3BP = 48:12 reported previously, a fraction of trimers in the E2p/E3BP core would be devoid of the E3BP subunit. The molecular ratio of E1p:E3:E2p/E3BP = 40:20:1 obtained by examination of the present in vitro assembly also differs from that of the 30:6:1 reported previously for the native bovine heart PDC (20, 34). However, the present ratio of E1p: E3:E2p/E3BP = 40:20:1 was achieved through in vitro assembly at saturating E1p and E3 concentrations. The results suggest that the E1p- and E3-binding sites in the E2p/E3BP core in the native PDC complex are not fully occupied by these two catalytic components. This raises the possibility that the copy numbers of E1p and E3 components may play a role in regulating PDC activity, as proposed for the E1b component of mammalian BCKDC (35).
Sedimentation velocity (SV) AUC experiments were used to examine the hydrodynamic behavior of the E2p/E3BP core in the absence and presence of E3. The s20,w of core alone is 30.4 ± 0.2 S (Fig. 1B). In the presence of saturating concentrations of E3, the s20,w increases to 43.3 ± 0.4 S, indicating a substantial increase in size (Fig. 1B). For these analyses, three different means of calculating the concentration of cosedimenting E3 were employed (see “Experimental Procedures”). If the concentration of the core complex is calculated using the putative heterotrimer of two E2ps and one E3BP, the molar ratio of heterotrimer to E3 dimer was found to be close to 1:1. Indeed, if all three methods are combined to yield a single average, the molar ratio of heterotrimer to E3 is 1.0 ± 0.2. This ratio translates into an average of 20 ± 4 E3s bound per core complex. The concentration of the core complex can also be calculated on the basis of the 48/12 model; in this case, the core can be conceptualized as comprising 12 heteropentamers of four E2ps and one E3BP. When calculated based on the 48/12 model, the molar ratio of E3 to heteropentamer is 1.7 ± 0.4, corresponding to 21 ± 5E3 dimers cosedimenting with the complex.
The hydrodynamic properties of the interactions between E1p and the E2p/E3BP core were also studied. In a preliminary experiment, the core was incubated in the presence of a molar excess of E1p heterotetramers. When subjected to SV, the resultant complex had a sedimentation coefficient of about 70 S (Fig. 1B). Using the “Leading” method described under “Experimental Procedures,” the E1p co-sedimenting with the E2p/E3BP core was quantified. Assuming a 40/20 core, the result indicates that 40 E1p tetramers bind to each 60-meric E2p/E3BP core. Assuming a 48/12 model, the result is nearly identical; about 40 E1p heterotetramers cosediment with the E2p/E3BP core. It is noteworthy, however, that this model features 48 potential binding sites for E1p, meaning that eight sites remain unoccupied despite the large molar excess of E1p (∼100 E1ps are present for every E2p/E3BP core in the AUC experiments).
ITC was also used to characterize the interaction between E3 and the E2p/E3BP core. When the E3 dimer was titrated into the reaction cell containing the E2p/E3BP core, a binding isotherm is obtained (Fig. 1C) that can be easily fit to obtain an association constant and a binding stoichiometry (n). First, the core concentration was calculated on the basis of the 40/20 complex; the Kd of the E3:E2p-E3BP interaction was 102 ± 7.5 nm, with an n value of 18.8 ± 0.94. When calculating the core concentration based on the 48/12 model, the Kd and n values are 102 nm and 17, respectively.
The binding of E1p to the E2p/E3BP core was also deciphered by ITC (supplemental Fig. S1A). The titration of E1p into the E2p/E3BP core produced an isotherm that could be fit with a Kd of 218 ± 8.6 nm in three independent experiments. Significantly, the n value is 40.5 ± 3.6 based on the E2/E3BP stoichiometry of 40:20. The data indicate that the E2p/E3BP core maximally binds 40 copies of E1p heterotetramers at saturating E1p concentrations. The n value is 38 ± 3.4, if the protein concentration is based on the 48/12 E2p/E3BP model, again indicating unused binding potential if the 48/12 model is correct.
E1pBD- and E3BD-containing Constructs Studied—The data presented above, when combined with the assumption that one binding domain (either E1pBD or E3BD) binds to one peripheral catalytic component (E1p heterotetramer or E3 dimer, respectively), provide strong support for the 40/20 model of the E2p/E3BP core postulated herein. Although the preponderance of evidence supports the 1:1 binding stoichiometry (5, 14, 15), this supposition has been challenged in the literature recently with respect to the E3BP-E3 interactions (24, 25). To provide additional empirical grounds for the 40/20 model, studies on the E1p- and E3-binding domains of E2p and E3BP, respectively, were carried out and are described below.
To facilitate its expression, the E1pBD domain (mature E2p residues 260–315) was N-terminally linked to the LBDb of the E2b component of the BCKDC. The resulting LBDb-E1pBD construct harbored a TEV protease-recognition site immediately proximal to the E1pBD sequence and a C-terminal His6 tag (Fig. 2A). As for E3 binding studies, the ideal means of determining how many E3 dimers may bind to an E3BP monomer would be to overexpress and purify E3BP. However, the E3BP protein, when overexpressed in E. coli, is not suitable for biophysical characterization, because it forms unstable putative dimers that aggregate at higher concentrations (data not shown). Therefore, soluble versions of E3BP were constructed; these proteins harbor LBDs and E3BDs but not the C-terminal domain of E3BP (Fig. 2). As also shown in Fig. 2A, the LBDb-E3BD construct includes residues 121–175 (the mature sequence numbering) of human E3BP (26), which included the full-length E3BD (residues 130–170) (Fig. 2B) based on the E3BD structure (14). The L3-E3BD construct included an N-terminal methionine, residues 1–175 of mature E3BP. The XDD1 protein (mature sequence residues 1–163) was used to mimic the XDD construct (nonprocessed sequence residues 59–216 or mature sequence residues 6–163) that had been studied by Smolle et al. (25). The XDD1 construct contains the same C-terminal truncation of seven amino acid residues in the E3BD as reported for the XDD vector (Fig. 2A). However, unlike the XDD construct, XDD1 does not contain the N-terminal deletion of five amino acids in the L3 domain; the latter domain does not interact with the E3 dimer. It is important to note that both XDD1 and XDD feature only half of the H2 helix of the E3BD structure (14), as a result of the C-terminal truncation (Fig. 2B).
Analytical Ultracentrifugation Studies of E3 Binding to E3BD-containing Proteins—SV was used to study the behavior of all three E3BD-containing proteins in the presence of E3 (Fig. 3). For E3 alone, there is a dominant peak at 5.64 ± 0.03 S in a c(s) distribution (Fig. 3). When combined with the refined frictional ratio, these s values can be used to estimate the mass of the sedimenting species using Svedberg's equation; the mass values thus obtained in these experiments (115 ± 4 kDa) conform to the expectation that E3 is a dimer (calculated dimeric mass = 105 kDa). Often, a second, minor peak appears at ∼8S, indicating a high molecular weight contaminant or oligomerization of E3. Using the program HYDROPRO (36), it is possible to use bead modeling to predict the hydrodynamic properties of a protein from the coordinates of its crystal structure. Such modeling using the crystal structure of E3 (9) resulted in a predicted sedimentation coefficient of 5.678 S for the E3 dimer, which agrees with the experimental observation. The observed s20,w values for LBDb-E3BD, L3-E3BD, or XDD1 were 1.7,4 1.6 ± 0.1, and 1.4 ± 0.1 S, respectively. The refined frictional ratios (f/f0) of these three proteins, 1.5, 1.7 ± 0.1, and 1.7 ± 0.1, respectively, indicate that the proteins adopt an elongated conformation. The masses for all three proteins, when estimated as delineated above (19.2, 23.0 ± 2.2, and 19.6 ± 1.4 kDa, respectively), are consistent with the calculated monomeric molecular masses of the proteins (19.4, 20.0, and 18.7 kDa, respectively). When LBDb-E3BD (17.1 μm) is included in solution with E3 (3.1 μm), there are two peaks present in a c(s) distribution: one at 1.75 S, representing free LBDb-E3BD; and one at 5.97 S, presumably representing the complex between the two proteins (Fig. 3A). The sedimentation of L3-E3BD in the presence of E3 is similar, with peaks at 1.62 ± 0.07 S and at 5.81 ± 0.03 S (Fig. 3B). Finally, XDD1 and E3 together in solution exhibited two peaks at 1.44 ± 0.08 S and 5.81 ± 0.02 S (Fig. 3C). As expected, the complex of E3 and an E3BD-containing protein therefore sediments faster than either protein alone (p < 0.0001 for both the L3-E3BD:E3 complex and the XDD1-E3 complex).
FIGURE 3.

Sedimentation velocity experiments showing c(s) distributions for E3, LBDb-E3BD, L3-E3BD, and XDD1. Each panel represents a separate sedimentation velocity experiment. In each experiment, the E3 dimer and the E3BD-containing protein were sedimented alone, and in a third cell, they were sedimented together after being incubated with one another for at least 2 h. In all panels, the c(s) distributions for E3 alone are shown as dotted lines, the distributions for the E3BD-containing protein are represented as dashed lines, and the distributions describing the mixture are shown as a solid line. A, LBDb-E3BD + E3; B, L3-E3BD + E3; C, XDD1 + E3.
As with the PDC core complexes with E3, the SV experiments with the E3BD-containing proteins were evaluated to determine the stoichiometries of the cosedimenting complexes. The results are shown in Tables 1, 2, 3; samplings of the multisignal SV results are shown in Figs. 4 and 5 and supplemental Fig. S2. All three methods of examining the concentrations of cosedimenting E3 and E3BD-containing protein indicate that there is a 1:1 complex between the E3 dimer and any E3BD-containing proteins. Even under conditions that would most favor the binding of a second E3BD-binding protein, i.e. a large molar excess of L3-E3BD or XDD1, there is no indication that a second protein binds to the E3 dimer.
TABLE 1.
Molar ratios derived from AUC of E3 and LBDb-E3BD
|
Input molar ratio (LBDb-E3BD/E3)
|
Molar ratio of interacting LBDb-E38D/E3
|
||
|---|---|---|---|
| MWLa | Laggingb | Leadingc | |
| 6.1 | 1.1 | 0.7 | 1.3 |
| Overall average | 1.0 ± 0.3 | ||
This value was calculated using the concentrations obtained by integrating the peak at ∼6 S of the ck(s) distributions. MWL indicates multiwavelength
This value was measured by subtracting the observed concentration of LBDb-E38D from the input concentration
This value was measured by subtracting the input signal of E3 from the signal observed at ∼6 S and then calculating the concentration of cosedimenting LBDb-E3BD
TABLE 2.
Molar ratios derived from AUC of E3 and L3-E3BD
|
Input molar ratio (L3-E3BD/E3)
|
Molar ratio of interacting L3-E3BD/E3
|
||
|---|---|---|---|
| MWLa | Lagging | Leading | |
| 1.3 | 1.1 | 0.9 | 0.8 |
| 2.6 | 0.9 | 1.3 | 1.0 |
| 4.2 | 0.6 | 1.5 | 0.9 |
| 6.7 | 0.8 | 1.0 | 1.2 |
| 13 | 1.1 | 1.2 | 1.0 |
| Averages | 0.9 ± 0.2 | 1.2 ± 0.2 | 1.0 ± 0.1 |
| Overall average | 1.0 ± 0.2 | ||
Calculation methods are as outlined in Table 1. MWL indicates multiwavelength
TABLE 3.
Molar ratios derived from AUC of E3 and XDD1
|
Input molar ratio (XDD1/E3)
|
Molar ratio of interacting XDD1/E3
|
||
|---|---|---|---|
| MWLa | Lagging | Leading | |
| 1.6 | 1.3 | 0.6 | 1.1 |
| 3.4 | 1.1 | 1.1 | 1.3 |
| 5.0 | 1.1 | 0.6 | 1.6 |
| 8.3 | 1.2 | 1.3 | 1.5 |
| 17 | 1.1 | 1.4 | 1.4 |
| Averages | 1.1 ± 0.1 | 1.0 ± 0.4 | 1.4 ± 0.2 |
| Overall Average | 1.2 ± 0.3 | ||
Calculation methods are as outlined in Table 1. MWL indicates multiwavelength
FIGURE 4.

ck(s) distributions obtained from multisignal sedimentation velocity experiments with L3-E3BD and E3. A, data and fit to the data for the interference optical system. In the top panel, circles represent individual data points. Radially invariant (IF data) and time-invariant (both IF and ABS data) noise features have been subtracted from these data, and all SV data presented in this paper. Lines denote the calculated sedimentation velocity concentration profiles obtained by fitting the data to the ck(s) model. The lines are color-coded, with violet indicating early scan times, and indigo, blue, green, yellow, orange, and red denoting progressively later scan times. For clarity, only every sixth data point and only every third scan used in the analysis are shown. In the bottom panel, the residuals between the data and the calculated profiles are shown. The residuals for a particular scan are connected by a line whose color corresponds to the color assigned in the top panel. B, data and fit to the data for the absorbance optical system. The wavelength of detection was 276 nm. The selection of shown data points and scans as well as the color-coding is the same as in A, except that every third data point is shown. C, ck(s) distributions. The green line is the distribution because of L3-E3BD. The red line is that due to E3. For this distribution, the areas beneath the peaks at ∼ 6 S are 3.5 μm (red, E3) and 3.0 μm (green, L3-E3BD).
FIGURE 5.

ck(s) distributions obtained from multisignal sedimentation velocity experiments with XDD1 and E3. The scheme for color, symbols, and for shown data is the same as in Fig. 6. A, data and fit to the data for the IF optical system. B, data and fit to the data for the ABS (276 nm) optical data. C, ck(s) distributions. For this distribution, the areas beneath the peaks at ∼6 S are 3.2 μm (red, E3) and 3.4 μm (green, XDD1).
ITC Measurements of E3 and E1p Binding to E3BD- and E1pBD-containing Proteins—The thermodynamic characteristics of E3 binding to E3BD-containing proteins were studied using ITC. As reported previously (14), the association of E3 with nonfused E3BD alone is exothermic and has a high association constant (i.e. a low Kd, Fig. 6A). Indeed, the sudden saturation of E3 by E3BD precludes accurate determination of Kd, but the stoichiometry of the interaction should be accurate (37).5 This experiment, which was performed in quadruplicate, yielded an average n value of 0.9 ± 0.1, i.e. one E3BD binds per E3 dimer. A similar result was obtained when titrating LBDb-E3BD into E3 (Fig. 6B); the n value was 1.0 ± 0.1. L3-E3BD and XDD1 have lower affinities for E3, and therefore the Kd values could be reliably calculated from the integrated heat changes upon injection. They are 37 ± 5 and 200 ± 30 nm, respectively (Fig. 6, C and D). Nevertheless, the n values in both the L3-E3BD and XDD1 experiments were nearly 1:0.96 ± 0.05 and 0.95 ± 0.06, respectively. No evidence for a second binding event could be detected in any of the ITC experiments. Significantly, the Kd of E3 binding to XDD1 is 6-fold higher than that for E3 binding to the equivalent XDD construct reported previously (25). In addition, the latter study showed that two XDD molecules apparently bind one E3 dimer. The differences in buffer systems used and in the temperature of the experiments likely account for the divergent Kd values, but it is unlikely that the titration conditions explain the differing binding stoichiometries.
FIGURE 6.

ITC measurements of E3 binding to E3BD-containing proteins. The protein constructs titrated into E3 were: A, E3BD; B, LBDb-E3BD; C, L3-E3BD; D, XDD1; and E, E2p/E3BP core. See the “Experimental Procedures” for the ITC titration conditions. The Kd values from three determinations are: A, 2.25 ± 1.25 nm; B, 2.75 ± 0.96 nm; C,37 ± 5 nm; and D, 200 ± 30 nm. The n values are as follows: A, 0.9 ± 0.1; B, 1.0 ± 0.1; C, 0.96 ± 0.05; and D, 0.95 ± 0.06.
In parallel ITC studies, an E1pBD-containing construct LBDb-E1pBD was titrated into the E1p in the reaction cell (supplemental Fig. S1B). The binding isotherm shows a Kd of 103 ± 6.2 nm and an n value of 0.96 ± 0.09 in three independent experiments. The n value indicates a 1:1 stoichiometry for the binding of LBDb-E1pBD to the E1p heterotetramer. Taken together, the ITC results establish a 1:1 stoichiometry for E1p and E3 interactions with their respective binding domains.
Native Gel Analysis of E3 Binding to E3BD-containing Proteins—Native PAGE was used to further establish the binding stoichiometries between the E3 dimer and E3BD-containing proteins. A fixed concentration of the E3 dimer was titrated with increasing concentrations of E3BD-containing proteins, and the reaction mixtures were analyzed by native PAGE (Fig. 7). When substoichiometric quantities of LBDb-E3BD were added to E3, two bands are observed (Fig. 7A); the lower bands represent the LBDb-E3BD:E3 complex, and the upper bands represent free E3 dimers. The E3BD-containing proteins studied here (LBDb-E3BD, L3-E3BD, and XDD1) are more negatively charged (calculated pI = 5.32, 6.0, and 5.58, respectively) than the E3 dimer (calculated pI = 6.66), but they add relatively little mass to the complex. Therefore, the binding of these proteins to E3 is expected to result in an increased mobility for the complex in native PAGE. In the 7% polyacrylamide gels, LBDb-E3BD migrated with the dye front off the gel and is therefore not visible. When the two proteins are present at a 1:1 ratio, the upper band completely disappears, indicating that all of the E3 is present as a complex with LBDb-E3BD. No enhancement or migration change of the lower band is evident upon further additions of LBDb-E3BD (data not shown). For L3-E3BD comprising the first two domains of E3BP of PDC, the same pattern is observed (Fig. 7B). When one molecule of L3-E3BD is present per E3 dimer, only the lower band (the L3-E3BD-E3 complex) is present. When greater than 1:1 stoichiometric quantities of L3-E3BD are added relative to the E3 dimer, the excess L3-E3BD starts to appear as the faster moving band below the complex. The constant faint band below the L3-E3BD is a minor contaminant from the E3 sample.
FIGURE 7.

Native gel electrophoresis shows 1:1 binding stoichiometry between full-length E3BD-containing proteins and the E3 dimer. A, titration of LBDb-E3BD with the concentration of the E3 dimer (12 μm) held constant. Molar ratios of LBDb-E3BD:E3 dimer are indicated, with the rightmost lane having E3 alone. The excess free LBDb-E3BD migrated off the gel. B, titration of L3-E3BD into E3. The rightmost and leftmost lanes contain L3-E3BD and E3 alone, respectively. The L3-E3BD:E3 dimer ratio is indicated above the gel. The constant faint band below the L3-E3BD is a minor contaminant from the E3 sample. C, titration of XDD1 into E3. The E3 and the XDD1:E3 complex overlap at increasing XDD1:E3 ratios. The minor band below the XDD1 is a contaminant from the XDD1 sample.
Anomalous Native Gel Profiles with the C-terminally Truncated XDD1 Construct—The binding of XDD1, which mimics the XDD construct reported by Smolle et al. (25), to E3 was also studied using native PAGE. Fig. 7C shows that with increasing XDD1:E3 dimer ratios (from the left to the right side of the gel), there is a subtle increase of the faster moving band than E3, which presumably corresponds to the XDD1-E3 complex band. However, free XDD1 is present even at XDD1:E3 ratios below 1:1. At the 1:1 stoichiometry of XDD1:E3 dimer, a significant amount of the free XDD1 band is still present. The minor band below the XDD1 band is a contaminant of the XDD1 protein. The similar pattern persists at the XDD1:E3 dimer ratios of 1.5:1 and 2:1, accompanied by the increasing intensity of the free XDD1 band. We interpret the data to indicate that, unlike its full-length counterpart (L3-E3BD), the C-terminal truncated XDD1 construct binds the E3 dimer weakly (see ITC-binding results in Fig. 6D) under the conditions of the electrophoretic experiment. Also, the fact that the upper gel band adopts a presumably intermediate position between the free E3 and XDD1:E3 complex band suggests that this system behaves like those described by Gilbert and Jenkins (38, 39); the koff of the interaction is likely to be instantaneous on the time scale of the electrophoretic experiment. Accordingly, the binding stoichiometry between XDD1 and the E3 dimer cannot be determined by the native PAGE method.
Although the 1:1 stoichiometry of E3BD-containing proteins associating with E3 dimers presented above lends credence to previous work (5, 14, 40), it is in stark contrast to that reported by Smolle et al. (25). Their studies suggested that two copies of their E3BD-containing XDD construct bound to one E3 dimer. These authors hypothesize that the two copies of XDD bind to the E3 dimer near the 2-fold axis of the larger protein. This arrangement seems unlikely, given that two independently obtained crystal structures (14, 15) of the E3-E3BD complex showed that two copies of E3BD could not simultaneously bind to the E3 dimer. This situation arises because the two binding sites for E3BD on E3 overlap, crossing the local 2-fold axis of the dimeric protein; therefore, the binding of one E3BD sterically precludes the binding of a second (Fig. 8A). Solution binding evidence in conjunction with site-directed mutagenesis indicated that the crystallographically observed E3-E3BD interface was correct (14). Smolle et al. (25) further suggest that E2p can be cross-linked in the periphery of the PDC by virtue of two E3BDs interacting with a single E3. Our data offer no evidence for such cross-links. It is important to note that active site couplings through transient cross-links between LBDs do exist in the PDC (41, 42). For example, an LBD with a reduced lipoyl moiety from E2p could interact with the active site of E3. During the time that this LBD resides on E3, E2 and E3BP are noncovalently cross-linked via their mutual interaction (at different sites) with E3 (Fig. 8B). Once E3 has catalyzed the oxidation of the lipoic acid moiety bound at its active site, it is likely that the LBD will be released, breaking the cross-link. The extensive interaction network between LBDs has been suggested as a mechanism for catalytic rate enhancement in the E. coli PDC (4, 41, 42).
FIGURE 8.

Protein-protein interactions in the native human PDC. A, close-up view of E3BD (ribbons representation) bound to E3 (surface) (14). One monomer of E3 is colored orange, and the other is blue. The approximate position of the dyad axis of the E3 dimer is shown by the black symbol. Most of E3BD is colored green, but those residues with atoms that would clash with a second bound E3BD are shown in purple. B, schematic model of the native human PDC. The dodecahedral 60-meric core of the human PDC is modeled using the structure of the catalytic domain of B. stearothermophilus E2 (44). The E2p polypeptides are colored magenta, with E3BP polypeptides colored green. The E3 dimers are shown in blue and orange, with a single E3BD bound per dimer of E3 (14), as indicated by the data. In this model, it is possible for 20 E3 dimers to bind; only 7 are shown for clarity. A single E1p heterotetramer docked to the E1pBD of E2p is shown, with the α subunits shown in tan and the β subunits in cyan. The structure of the human versions of E1p bound to E1pBD is unknown; shown here is the structure from B. stearothermophilus (45). The circled E3 has an LBD of E. coli E2p docked to the active site. E2p and E3BD are therefore noncovalently cross-linked via their mutual interaction with E3. C, possible arrangement of E2p and E3BP components in a 40/20 core. Shown is a dodecahedral arrangement of 20 heterotrimers composed of 2 E2p proteins (purple) and one E3BP (green).
The results of hydrodynamic and E3-binding properties that we obtained here with the XDD1 construct differ significantly from those reported for the XDD by Smolle et al. (25). Both constructs contain the C-terminal truncation of seven amino acids in the helix 2 of E3BD (Fig. 2A). Yet, XDD has an s20,w of 2.0 S, whereas that of XDD1 is 1.4 S (Fig. 3C); the former suggests a compact, globular structure, whereas the latter indicates an elongated structure that would be expected of a protein that consists of two domains tethered by a flexible peptide linker. Additionally, their putative (XDD)2-E3 complex has the same sedimentation coefficient as E3 (indicating a significantly elongated complex), whereas the XDD1-E3 complex has a greater sedimentation coefficient than E3 alone (Fig. 3C). Furthermore, in their titration experiments monitored by native PAGE, no 1:1 intermediate complex was observed, implying that there is significant cooperativity in the XDD-E3 interaction; however, no cooperativity was mentioned in the analysis of their ITC data (25). XDD1, conversely, bound only very poorly under the conditions of the electrophoretic assay, and the weaker binding is supported by the ITC data (Fig. 6D). The causes of the significant differences between the two proteins are not immediately evident, because the exact vector construction and amino acid composition of XDD were not described (25). Regardless, claims of the superstoichiometric binding of E3BD to E3 are not supported by our data.
Implications for the Overall Architecture of the PDC—The model studies presented above clearly demonstrate that a single E3BD-containing protein interacts with a single E3 dimer. We also show that ∼20 E3 dimers associate with the E2p-E3BP core complex. The model data therefore firmly establish that there must be 20 copies of E3BP in the core. Moreover, we have demonstrated that 40 E1p heterotetramers bind to the core complex, and that the binding stoichiometry of E1pBD to E1p is 1:1. These facts allow the conclusion that there are 40 copies of E2p in the core. Therefore, the evidence points to a 40/20 model for the reconstituted E2p/E3BP core. A hypothetical arrangement for such a core is shown in Fig. 8C. All 20 vertices of the dodecahedral core include a heterotrimer of two E2ps and one E3BP. The protein-protein contacts at the midpoints between vertices (a local 2-fold) are made only by like subunits. The 40/20 model presented here is a possible configuration of the PDC core that has some appealing features. Unlike the 48/12 model, the 40/20 model features an identical building block at all 20 vertices of the dodecahedron, a heterotrimer of two E2p subunits and one E3BP subunit. The 40/20 arrangement also allows for more E3 subunits to bind, which could result in the more efficient functioning of the PDC.
The results described in this study are incompatible with an earlier model of the core posited by Hiromasa et al. (21). Using methods similar to those employed here, these authors concluded that the PDC core includes 48 copies of E2p and 12 of E3BP. Their conclusions were largely based on titrating either E1p or E3 into solutions of E2p/E3BP core and quantifying the concentration of the smaller protein that cosediments with the core. They found that ∼48 copies of E1p and 12 copies of E3 bound to a reconstituted core. Using the supposition of 1:1 ratios of the binding domains to the peripheral subunits, it was concluded that the stoichiometry of the E2p/E3BP core is 48:12. However, their estimation of E3 subunits may have been low. One means of quantification of E3 was performed by extrapolation of the linear part of a graph of E3:core ratio versus apparent sedimentation coefficient (and radius of gyration using small angle x-ray scattering). However, the maximal values obtained by these methods were not obtained until the E3:core ratio was greater than 20 (21). Another means used by Hiromasa et al. (21) to quantify the number of E3 subunits was similar to the leading methodology employed in this study (see “Experimental Procedures”). The concentration of E3 used in the previously reported experiment was such that only 20 E3 dimers were present for every core, a situation under which the core would not be saturated in our model.
Supplementary Material
Acknowledgments
We are indebted to Masato Kato for the discussion and help in producing the graphics of the E2p and E3BP subunit arrangement in the 40/20 model.
This work was supported, in whole or in part, by National Institutes of Health Grants DK26758 and DK62306. This work was also supported by Grant I-1286 from the Welch Foundation.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1 and S2.
Footnotes
The abbreviations used are: PDC, pyruvate dehydrogenase complex; E1p, pyruvate dehydrogenase; E2p, dihydrolipoyl transacetylase; E3, dihydrolipoamide dehydrogenase; E3BP, E3-binding protein; E3BD, E3-binding domain; LBD, lipoyl-bearing domain; L3, LBD from E3-binding protein; BCKDC, branched-chain α-keto acid dehydrogenase complex; E2b, dihydrolipoyl transacylase; ITC, isothermal titration calorimetry; AUC, analytical ultracentrifugation; SV, sedimentation velocity; TEV, tobacco-etched virus; IF interference; ABS, absorbance; FPLC, fast protein liquid chromatography.
No error estimates were given for the values associated with LBD-E3BD because only one experiment was performed with this protein.
References
- 1.Reed, L. J. (2001) J. Biol. Chem. 276 38329-38336 [DOI] [PubMed] [Google Scholar]
- 2.Reed, L. J., Damuni, Z., and Merryfield, M. L. (1985) Curr. Top. Cell Regul. 27 41-49 [DOI] [PubMed] [Google Scholar]
- 3.Patel, M. S., and Roche, T. E. (1990) FASEB J. 4 3224-3233 [DOI] [PubMed] [Google Scholar]
- 4.Perham, R. N. (2000) Annu. Rev. Biochem. 69 961-1004 [DOI] [PubMed] [Google Scholar]
- 5.Fries, M., Stott, K. M., Reynolds, S., and Perham, R. N. (2007) J. Mol. Biol. 366 132-139 [DOI] [PubMed] [Google Scholar]
- 6.Ciszak, E. M., Korotchkina, L. G., Dominiak, P. M., Sidhu, S., and Patel, M. S. (2002) J. Biol. Chem. 278 21240-21246 [DOI] [PubMed] [Google Scholar]
- 7.Kato, M., Wynn, R. M., Chuang, J. L., Tso, S.-C., Machius, M., Li, J., and Chuang, D. T. (2008) Structure (Lond.) 16 1849-1859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Seifert, F., Ciszak, E., Korotchkina, L., Golbik, R., Spinka, M., Dominiak, P., Sidhu, S., Brauer, J., Patel, M. S., and Tittmann, K. (2007) Biochemistry 46 6277-6287 [DOI] [PubMed] [Google Scholar]
- 9.Brautigam, C. A., Chuang, J. L., Tomchick, D. R., Machius, M., and Chuang, D. T. (2005) J. Mol. Biol. 350 543-552 [DOI] [PubMed] [Google Scholar]
- 10.Yu, X., Hiromasa, Y., Tsen, H., Stoops, J. K., Roche, T. E., and Zhou, Z. H. (2008) Structure (Lond.) 16 104-114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Howard, M. J., Fuller, C., Broadhurst, R. W., Perham, R. N., Tang, J.-G., Quinn, J., Diamond, A. G., and Yeaman, S. J. (1998) Gastroenterology 115 139-146 [DOI] [PubMed] [Google Scholar]
- 12.Kato, M., Chuang, J. L., Tso, S.-C., Wynn, R. M., and Chuang, D. T. (2005) EMBO J. 24 1763-1774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ruhul Momen, A. Z. M., Hirota, H., Hayashi, F., and Yokoyama, S. (2006) Protein Data Bank 10.2210/pdb2dne/pdb [DOI]
- 14.Brautigam, C. A., Wynn, R. M., Chuang, J. L., Machius, M., Tomchick, D. R., and Chuang, D. T. (2006) Structure (Lond.) 14 611-621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ciszak, E. M., Makal, A., Hong, Y. S., Vettaikkorumakankauv, A. K., Korotchkina, L. G., and Patel, M. S. (2006) J. Biol. Chem. 281 648-655 [DOI] [PubMed] [Google Scholar]
- 16.Kato, M., Li, J., Chuang, J. L., and Chuang, D. T. (2007) Structure (Lond.) 15 992-1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Steussy, C. N., Popov, K. M., Bowker-Kinley, M. M., Sloan, R. B., Jr., Harris, R. A., and Hamilton, J. A. (2001) J. Biol. Chem. 276 37443-37450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wynn, R. M., Kato, M., Chuang, J. L., Tso, S.-C., Li, J., and Chuang, D. T. (2008) J. Biol. Chem. 283 25305-25315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vassylyev, D. G., and Symerski, J. (2007) J. Mol. Biol. 370 417-426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sanderson, S. J., Miller, C., and Lindsay, J. G. (1996) Eur. J. Biochem. 236 68-77 [DOI] [PubMed] [Google Scholar]
- 21.Hiromasa, Y., Fujisawa, T., Aso, Y., and Roche, T. E. (2004) J. Biol. Chem. 279 6921-6933 [DOI] [PubMed] [Google Scholar]
- 22.Frank, R. A. W., Pratap, J. V., Pei, X. Y., Perham, R. N., and Luisi, B. F. (2005) Structure (Lond.) 13 1119-1130 [DOI] [PubMed] [Google Scholar]
- 23.Jung, H.-I., Cooper, A., and Perham, R. N. (2003) Eur. J. Biochem. 270 4488-4496 [DOI] [PubMed] [Google Scholar]
- 24.Smolle, M., and Lindsay, J. G. (2006) Biochem. Soc. Trans. 34 815-818 [DOI] [PubMed] [Google Scholar]
- 25.Smolle, M., Prior, A. E., Brown, A. E., Cooper, A., Byron, O., and Lindsay, J. G. (2006) J. Biol. Chem. 281 19772-19780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Harris, R. A., Bowker-Kinley, M. M., Wu, P., Jeng, J., and Popov, K. M. (1997) J. Biol. Chem. 272 19746-19751 [DOI] [PubMed] [Google Scholar]
- 27.Laue, T. M., Shah, B. D., Ridgeway, R. M., and Pelletier, S. L. (1992) in Analytical Ultracentrifugation in Biochemistry and Polymer Science (Harding, S. E., Rowe, A. J., and Horton, J. C., eds) pp. 90-125, The Royal Society of Chemistry, Cambridge, UK
- 28.Schuck, P. (2000) Biophys. J. 78 1606-1619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schuck, P., Perugini, M. A., Gonzales, N. R., Howlett, G. J., and Schubert, D. (2002) Biophys. J. 82 1096-1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Deka, R. K., Brautigam, C. A., Tomson, F. L., Lumpkins, S. B., Tomchick, D. R., Machius, M., and Norgard, M. V. (2007) J. Biol. Chem. 282 5944-5958 [DOI] [PubMed] [Google Scholar]
- 31.Houtman, J. C. D., Yamaguchi, H., Barda-Saad, M., Braiman, A., Bowden, B., Appella, E., Schuck, P., and Samelson, L. E. (2006) Nat. Struct. Mol. Biol. 13 798-805 [DOI] [PubMed] [Google Scholar]
- 32.Balbo, A., Minor, K. H., Velikovsky, C. A., Mariuzza, R. A., Peterson, C. B., and Schuck, P. (2005) Proc. Natl. Acad. Sci. U. S. A. 102 81-86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jilka, J. M., Rahmatullah, M., Kazemi, M., and Roche, T. E. (1986) J. Biol. Chem. 261 1858-1867 [PubMed] [Google Scholar]
- 34.Barrera, C. R., Namihira, G., Hamilton, L., Munk, P., Eley, M. H., Linn, T. C., and Reed, L. J. (1972) Arch. Biochem. Biophys. 148 343-358 [DOI] [PubMed] [Google Scholar]
- 35.Harris, R. A., Hawes, J. W., Popov, K. M., Zhao, Y., Shimomura, Y., Sato, J., Jaskiewicz, J., and Hurley, T. D. (1997) Adv. Enzyme Regul. 37 271-293 [DOI] [PubMed] [Google Scholar]
- 36.de la Torre, J. G., Huertas, M. L., and Carrasco, B. (2000) Biophys. J. 78 719-730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Velazquez-Campoy, A., Leavitt, S., and Freire, E. (2004) in Methods in Molecular Biology (Fu, H., ed) pp. 35-54, Humana Press, Inc., Totowa, NJ [DOI] [PubMed]
- 38.Gilbert, G. A., and Jenkins, R. C. L. (1956) Nature 177 853-854 [DOI] [PubMed] [Google Scholar]
- 39.Gilbert, G. A., and Jenkins, R. C. L. (1959) Proc. R. Soc. Lond. Ser. A 253 420-437 [Google Scholar]
- 40.Mande, S. S., Sarfaty, S., Allen, M. D., Perham, R. N., and Hol, W. G. J. (1995) Structure (Lond.) 4 277-286 [DOI] [PubMed] [Google Scholar]
- 41.Danson, M. J., Fersht, A. R., and Perham, R. N. (1978) Proc. Natl. Acad. Sci. U. S. A. 75 5386-5390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hackert, M. L., Oliver, R. M., and Reed, L. J. (1983) Proc. Natl. Acad. Sci. U. S. A. 80 2226-2230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sigurskjold, B. W. (2000) Anal. Biochem. 277 260-266 [DOI] [PubMed] [Google Scholar]
- 44.Izard, T., Aevarsson, A., Allen, M. D., Westphal, A. H., Perham, R. N., de Kok, A., and Hol, W. G. J. (1999) Proc. Natl. Acad. Sci. U. S. A. 96 1240-1245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Frank, R. A. W., Titman, C. M., Pratap, J. V., Luisi, B. F., and Perham, R. N. (2004) Science 306 872-876 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.