

Summary
UV-light induced pyrimidine photodimers are repaired by the nucleotide excision repair pathway. Photolesions have biophysical parameters closely resembling undamaged DNA, impeding discovery through damage surveillance proteins. The DDB1-DDB2 complex has the highest known affinity for photodimers in metazoan cells, and serves in the initial detection of UV-lesions in vivo. The structure of the DDB1-DDB2 complex bound to a 6-4 pyrimidine-pyrimidone photodimer (6-4PP) shows that the lesion is held exclusively by the WD40 domain of DDB2. A DDB2 hairpin inserts into the minor groove, extrudes the photodimer into a binding pocket and kinks the duplex by ∼40°. The tightly localized probing of the photolesions, combined with proofreading in the photodimer pocket enables DDB2 to detect lesions refractory to detection by other damage surveillance proteins. The structure provides insights into damage recognition in chromatin, and suggests a mechanism by which the DDB1-associated CUL4 ubiquitin ligase targets proteins surrounding the site of damage.
Introduction
Nucleotide excision repair (NER) is a central pathway for the removal of structurally and chemically diverse lesions (Friedberg et al., 2006; Gillet and Scharer, 2006). These lesions range from UV-induced cyclobutane pyrimidine dimers (CPD) and 6-4 pyrimidine-pyrimidone photoproducts (6-4PP) to a variety of bulky adducts formed by environmental carcinogens. Mutations in NER are associated with rare autosomal recessive syndromes such as xeroderma pigmentosum (XP) characterized by heightened UV-sensitivity, neurological abnormalities and an increased propensity to develop skin neoplasms (Cleaver, 2005).
In the global-genome branch of NER (GG-NER), the DNA is initially surveyed for lesions by XPC-RAD23B (Sugasawa et al., 1998) and the UV DNA damage binding (UV-DDB) complex (Fitch et al., 2003; Moser et al., 2005; Sugasawa et al., 2005). Arrival of XPC-RAD23B at the site of damage triggers the recruitment of TFIIH, with its associated XPB and XPD helicases, followed by XPA, the single strand DNA binding protein RPA and the two nucleases XPG and XPF-ERCC1 (reviewed in Gillet and Scharer, 2006). Once assembled, the NER repairosome excises a 24-32nt ssDNA fragment containing the damage, and this is followed by gap re-synthesis (Aboussekhra et al., 1995).
The UV-DDB complex contains two principal subunits DDB1 (p127) and DDB2 (p48) (Dualan et al., 1995; Feldberg and Grossman, 1976; Takao et al., 1993). Mutations in DDB2 give rise to XP complementation group E (reviewed in Tang and Chu, 2002). Several lines of evidence indicate that UV-DDB is the GG-NER factor specialized for the detection of UV-induced lesions in chromatin. In vitro, UV-DDB binds to pyrimidine dimers including isomers of CPD and 6-4PP with the highest reported affinity and specificity of all NER proteins (Fujiwara et al., 1999; Payne and Chu, 1994; Reardon et al., 1993; Treiber et al., 1992; Wittschieben et al., 2005). XPC, by contrast, has substantially lower affinity and specificity for UV-lesions (Batty et al., 2000; Sugasawa et al., 1998). In vivo, DDB2 localizes ahead of XPC to CPD and 6-4PP lesions (Fitch et al., 2003; Moser et al., 2005; Yasuda et al., 2007). In the absence of DDB2, XPC still localizes to 6-4PP and to a lesser extent to CPDs, although with substantially delayed kinetics (Moser et al., 2005; Wakasugi et al., 2002; Yasuda et al., 2007). In DDB2-deficient XPE cells, CPD repair is largely abolished, while 6-4PP repair is affected to a lesser extent (Hwang et al., 1998; Moser et al., 2005; Tang et al., 2000).
The DDB1 subunit associates tightly with the CUL4-RBX1 complex and forms a cullin family ubiquitin ligase (Groisman et al., 2003; Shiyanov et al., 1999). Following UV exposure, the DDB1-DDB2-CUL4A-RBX1 complex (DDBCUL4) localizes to the site of damage, and ubiquitinates XPC and DDB2 (El-Mahdy et al., 2006; Sugasawa et al., 2005). Polyubiquitination of DDB2 reduces its affinity for damage while XPC remains unaffected, and this is thought to facilitate the handover of the lesion to XPC (Sugasawa et al., 2005). Additional DDBCUL4 substrates include histones H2A, H3 and H4 around the site of damage (Kapetanaki et al., 2006; Wang et al., 2006). H3 and H4 ubiquitination has been shown to loosen nucleosome binding in vitro (Wang et al., 2006) providing a pathway to assemble the NER repairosome in the otherwise inaccessible chromatin environment.
Here we present the structures of the DDB1-DDB2 complex alone and bound to DNA containing either a 6-4PP lesion or an abasic site. The structures reveal the molecular mechanism underlying high affinity recognition of photo-lesions that are refractory to detection by XPC. The structures also suggest a mechanism for the assembly of the DDBCUL4 ubiquitin ligase in chromatin and provide a framework for understanding the ubiquitination of proteins proximal to damage.
Results
The overall structure of the DDB1-DDB2 complex
The crystal structures of the human DDB1-DDB2 (DDBhs) complex and that of human DDB1 bound to the zebrafish DDB2 ortholog (DDBdr) were determined at 3.3 Å and 2.3 Å resolution, respectively (Supplemental Tables S1, S2). A 2.8 Å refined structure of DDBdr with UV-damaged DNA was obtained with a 14 base pair (bp) DNA duplex containing a synthetic 6-4 pyrimidine-pyrimidone (6-4PP) dimer located opposite two adenine bases (thereafter DDBdr–DNA6-4PP). Additionally, co-crystal structures with DNA duplexes containing a single base tetrahydrofuran (THF) abasic site mimic, which has been shown to be recognized by UV-DDB in vitro (Fujiwara et al., 1999; Wittschieben and Wood, 2003), were obtained for DDBdr (16 bp DNA; 2.6 Å; thereafter DDBdr–DNATHF) and DDBhs (31 bp DNA; 4.0 Å; thereafter DDBhs–DNATHF). The overall sequence conservation of zebrafish DDB2 versus its human ortholog is 74% (similarity) and 51% (identity) for the constructs crystallized (see Supplemental Figure S1). The overall Cα-backbone rmsd of the human and zebrafish DDB2 structures is 1.1 Å, with 48 % similarity in the DDB1-DDB2 interface. As the protein-DNA interactions in DDBhs-DNATHF are more than 80% identical to those in the DDBdr–DNA complexes, our discussion will focus on the high resolution zebrafish DDBdr structures.
The DDB2 structure is composed of an N-terminal helix-loop-helix segment (residues 101 to 136) followed by a 7-bladed WD40 β-propeller domain (residues 137 to 455; Figures 1, 2B, C; Supplemental Figure S1). The DDB1 structure was previously shown to consist of three WD40 β-propeller domains (BPA, BPB & BPC) and a C-terminal helical domain (CTD) (Angers et al., 2006; Li et al., 2006). DDB2 binds to an interface between the DDB1 BPA and BPC propellers, where its helix-loop-helix motif (helices h1 and h2) inserts into a cavity formed by the narrow ends of BPA and BPC (Figure 1; Supplemental Figure S3A). The wide end of DDB2, which anchors the helix-loop-helix motif, contains a large hydrophobic surface patch (∼3900 Å2) that is buried at the DDB1-DDB2 interface upon complex formation.
Figure 1. Overall structure of the DDB1-DDB2-DNA complex.
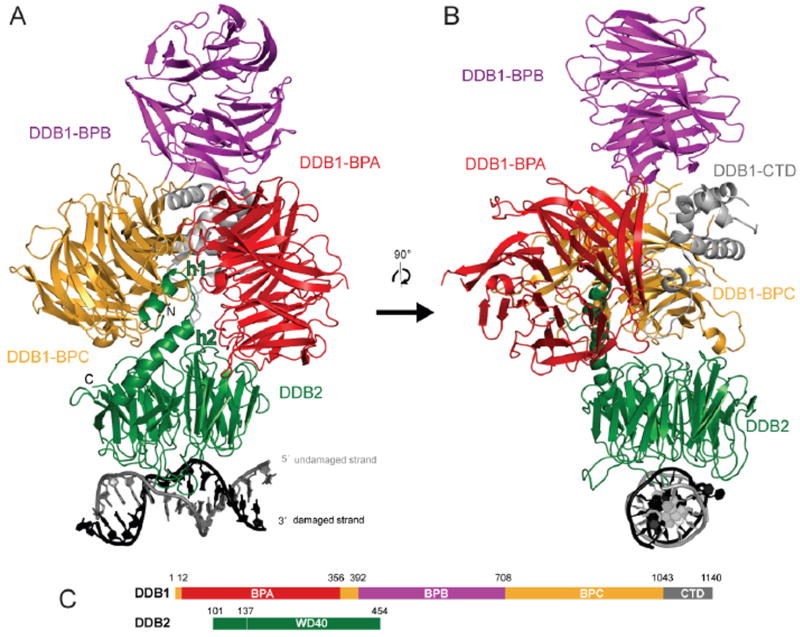
(A) Ribbon representation of the DDBdr–DNA6-4PP complex: DDB2, green; DDB1-BPA, red; DDB1-BPB, magenta; DDB1-BPC, yellow; DDB1-CTD, gray; DNA6-4PP damaged/undamaged strand in black/gray.
(B) Ribbon representation of the DDBdr–DNA6-4PP complex rotated by 90° about the vertical axis relative to (A).
(C) Schematic representation of hsDDB1 and drDDB2 with domain boundaries.
Figure 2. DDB1-DDB2 induced DNA-kinking upon complex formation and location of DNA-binding residues.

(A) Superposition of the DDBdr–DNA6-4PP and DDBdr–DNATHF complex structures, highlighting the induced kink of ∼40° at the site of damage. DDB2, gray; DNA6-4PP, blue; DNATHF, red.
(B) Cartoon representation of the DDB2dr-WD40 β-propeller with DNA6-4PP. Hairpin residues contacting the DNA are shown as yellow spheres; residues contacting the DNA-backbone are shown in orange. Bound DNA is shown in black/gray (carbon atoms) with phosphates in orange/purple for the damaged/undamaged strand, respectively. 2Fo-Fc electron density (contoured at 1.0 σ) for the DNA is shown in blue.
(C) Cartoon representation of DDB2dr in rainbow colors depicting the WD40 domain nomenclature. Blades are labelled 1-7 with the individual β-strands of blade 4 labelled with A-D. The backbone of the damaged and undamaged DNA strands is shown in black and gray, respectively, with the damaged 6-4 photoproduct shown as stick model.
The DDB2 WD40 propeller exclusively binds the damage containing DNA duplex
All DDB1-DDB2-DNA structures (DDBdr–DNA6-4PP, DDBdr–DNATHF and DDBhs–DNATHF) show that DNA binding is carried out exclusively by the DDB2 subunit (Figures 1, 2). DDB2 binds DNA with the narrow end of its β-propeller, opposite from where the DDB1-binding site is located. The DNA runs across the full diameter of the DDB2 β-propeller, slightly offset towards one side (WD40 repeats 4-7; Figure 2C). The overall DDB2-DNA binding interface comprises seven base pairs: two directly at, three 5′ and two 3′ to the lesion. The surface buried upon DNA-DDB2 complex formation is on the order of ∼2100 Å2. To our knowledge, this is the first direct indication of a β-propeller involved in nucleic acid binding.
The 14 bp 6-4PP DNA structure consists of two double-stranded B-DNA segments (6 bp and 5 bp) that flank a central 2 bp distortion where the two strands are separated. The 2 bp distortion forms over the 6-4PP dinucleotide, and the separation of the damaged and undamaged strands results in a gap in the duplex (dimensions 10Å × 9.2 Å) (Figure 3). The DNA is kinked by ∼40° around the site of the lesion (Figure 2). The 6-4PP photodimer is flipped-out of the duplex, and is held near the central cavity of the DDB2 β-propeller by a shallow pocket formed by residues from the loops connecting strands D1-A2, B2-C2 and D2-A3 (Figure 3). The DDB2-DNA interactions include charge-stabilized hydrogen bonds to the phosphodiester backbone distributed equally among the two DNA strands. These involve 3 arginine (Arg148, Arg369, Arg404) and 2 lysine residues (Lys168, Lys280), as well as the side chains of Tyr393 and Gln345 and the backbone amide group of Ile428 (Figures 3B, E). An additional set of eleven mostly conserved arginine, lysine and histidine residues located more distally to the DNA backbone (> 5.5 Å) line the path of the DNA phosphate backbone along the propeller (Figure 6A).
Figure 3. Mechanism of 6-4 photodimer recognition.
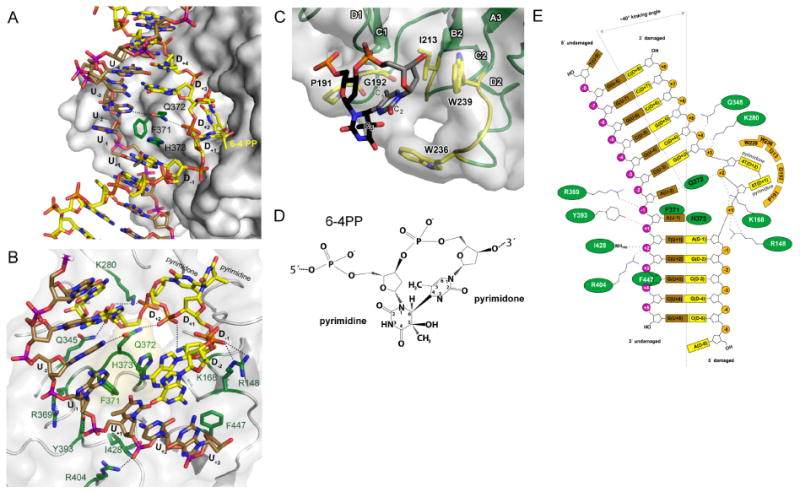
(A) Close-up of the DDB2 hairpin insertion (green) at the lesion with the damaged and undamaged strand depicted in yellow and brown, respectively.
(B) Interaction of DDB2 with the DNA6-4PP-backbone. The backbone of both strands is contacted by an array of positively charged residues crucial for the stabilization of the phosphate backbone compression at the damaged site (D+1, D+2). Parts of the DNA are omitted for clarity.
(C) Close-up of the photodimer binding pocket stabilizing the flipped-out dinucleotide. Contacting residues are shown as stick models in yellow. The pyrimidine ring D+1 and the pyrimidone ring D+2 are shown in black and gray, respectively. Parts of the DNA have been omitted for clarity.
(D) Chemical structure of the 6-4 pyrimidine-pyrimidone dimer.
(E) Schematic representation of interactions between drDDB2 and DNA6-4PP (with colors as in (A) and (B)).
Figure 6. The DDB2-surface and hairpin are important structural features of the damage recognition process.
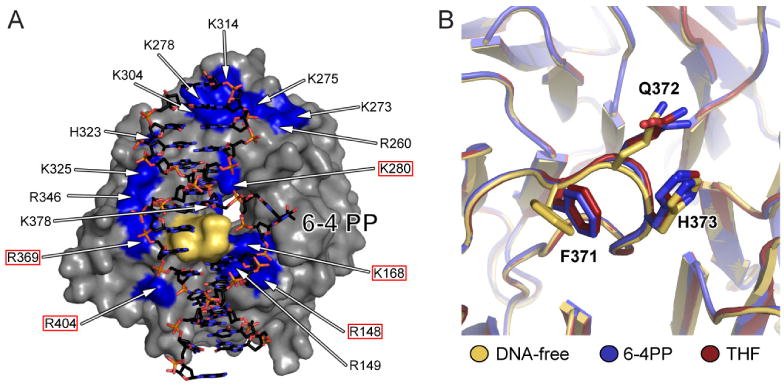
(A) Conserved basic residues mapped onto the DDB2 surface (gray). Residues framed with a red box are in direct contact with the DNA phosphate backbone. The remaining 11 residues, located more distally (> 5.5 Å), form a second layer of arginines, lysines and histidines potentially guiding and pre-orienting the DNA towards DDB2 through long-range charged interaction. The 371FQH373-hairpin motif is shown in yellow.
(B) Overlay of the DDB2 hairpin residues 371FQH373 from DNA-free (yellow) the DNA6-4PP (blue) and DNATHF (red) bound complex structures indicating that the hairpin acts as a rigid unit.
DDB2 mediated damage detection involves insertion of an invariant hairpin at the lesion combined with photodimer flipping
The separation of the damaged and undamaged strands at the lesion is triggered by the insertion of a 3-residue DDB2 hairpin (residues 371 to 373) that occurs in the connection between blades 5 and 6. The side chains of Phe371, Gln372 and His373, all of which are strictly conserved amongst DDB2 orthologs, insert part way into the DNA duplex, on the minor groove side. This insertion is associated with the minor groove widening from 13 Å to 18 Å and causes unwinding of the DNA around the lesion by ∼23°. The major groove remains largely unaffected (∼18 Å; Supplemental Tables S3, S4). The 6-4PP nucleotides flip out in an extra-helical conformation, orphaning the opposing adenine bases (U-1, U-2). Upon photodimer flipping, U-1 and U-2 are juxtaposed to hairpin residues His373 and Gln372, respectively (Figures 3A, B) such that the His373 imidazole ring and Gln372 carboxamide group are approximately co-planar with the opposing bases. The bases of the undamaged strand largely retain their stacking interactions with adjacent bases within the duplex. The one exception is the step from the U-1 base to the U+1 base. This base step exhibits diminished stacking owing to a large shift value of -5.8 Å (calculated with 3DNA (Lu and Olson, 2008)), with U-1 moving away from DDB2. Although both base planes remain parallel and in a helical conformation, they make only a small number of van der Waals contacts. The gap in the duplex that results from the extra-helical 6-4PP conformation of the damaged strand is stabilized through Gln372 π-stacking with the base 3′ to the lesion (D+3), and also through His373 stacking against base D-1 on the 5′ side. The hairpin residues Gln372 and His373 thereby substitute for the flipped-out 6-4PP photodimer by stabilizing the orphaned bases on the undamaged strand (Figure 3A), as well as the flanking bases on the damaged strand that have lost their stacking partners due to photodimer flipping.
The photodimer binding pocket restricts the size of base adducts bound by DDB2
The 6-4 pyrimidine-pyrimidone photodimer binds to a shallow DDB2 pocket lined by Pro191, Gly192, Ile213, Trp236 and Trp239 (Figure 3C, E). Approximately half of the surface area of the photodimer becomes buried in the complex. The 3′-pyrimidone (position D+2) makes more extensive contacts compared to the 5′-pyrimidine (position D+1). The pyrimidone ribose group makes van der Waals contacts to Trp239, while the edge of the base group that contains the C5-methyl group packs with hydrophobic groups of Gly192 and Ile213. The opposing edge of the pyrimidone base containing the carbonyl group (O2) is solvent exposed. The 5′-pyrimidine base has one edge (C6 carbon, and methyl and hydroxyl groups of the C5 carbon) packing loosely (∼4.2 Å) with Trp236 and Pro191. The opposite edge of the pyrimidine base (O2, N3 and O4) is fully solvent exposed (Figure 3C). Overall, the complementarity between the photodimer surface and the pocket is partial, consistent with the pocket accommodating the CPD lesion as well (Figure 5). However, the shape and composition of the pocket would limit the size and the chemical nature of the lesion that can be accommodated. Larger substitutes including 4-nitroquinolone (4NQ) and nitrogen mustard, both of which are weak substrates (Payne and Chu, 1994) can only be accommodated on the solvent exposed edge of the D+1 position.
Figure 5. Predicted binding mode of dewar 6-4PP and CPD in the damage binding pocket.
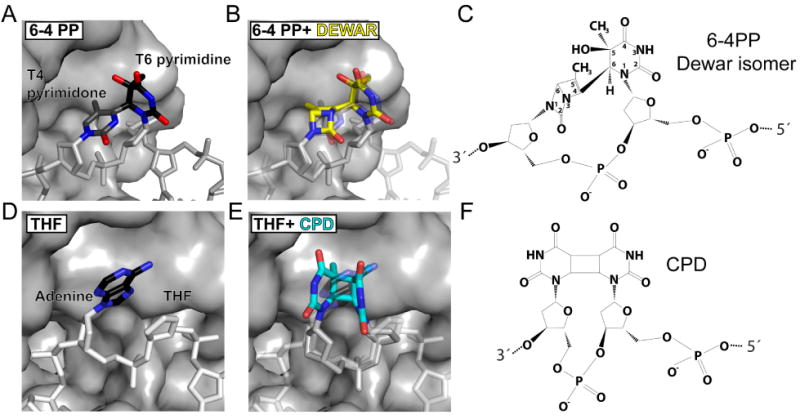
(A) The 6-4 photodimer in the damage binding pocket with the pyrimidine carbon atoms shown in black and the pyrimidone carbon atoms in gray.
(B) Overlay of the 6-4 PP (shown faded) with the Dewar isomer shown in yellow.
(C) Chemical structure of the Dewar isomer.
(D) The abasic site mimic (THF) with the neighboring Adenine in the damage binding pocket.
(E) Overlay of the abasic site damage (shown faded) with a CPD shown in cyan. The covalently linked bases of the CPD can easily be accommodated in the damage binding pocket without sterical clashes.
(F) Chemical structure of the CPD.
The 6-4 photoproduct (D+1-D+2) exhibits a compression of the phosphate backbone from 7.0 Å to <6.0 Å (Supplemental Tables S5, S6). This conformation is stabilized by Arg148 and Lys168, the latter being positioned in-between the two phosphates of the photodimer (Figures 3B, E). Additional stabilization of the lesion backbone is contributed by Gln372 (hairpin) which hydrogen bonds to the 5′ phosphate group of the pyrimidone (position D+2). The observed compression in the D+1-D+2 backbone is expected to favor the binding of intra-strand crosslinks, which in solution are often pre-compressed (reviewed in (Lukin and de Los Santos, 2006). Accordingly, cis-Pt modified duplexes having a compressed phosphate backbone (5′-3′→ cis-PT: 6.31 Å, 5.95 Å (Gelasco and Lippard, 1998)) are an in vitro DDB2 substrate.
The observed kink in the damage duplex is induced by DDB2
The kinking of the DNA around the 6-4PP is caused by DDB2 interactions with both the damaged and undamaged strands. On the damaged strand, the flipping out of the 6-4PP dinucleotide and their replacement by the smaller Gln372 and His373 side chains allows for kinking towards the major groove, with the DDB2 contacts to the phosphate groups flanking the 6-4PP serving as pivot points. On the undamaged strand, the Phe371 side chain from the hairpin pushes the U-1 base away from the DNA axis facilitating the compression of the major groove and changing the direction of the 5′ segment of the DNA strand. The kink in the 3′ direction of the undamaged strand end is caused by Phe447 pushing the U+3 ribose group away from DDB2 (Figures 3B, E).
The DDB2 hairpin displaces two nucleotides irregardless of the presence of a single nucleotide THF lesion or a dinucleotide photoproduct
The tetrahydrofuran moiety is a single base pair lesion that is bound tightly in vitro (Fujiwara et al., 1999; Wittschieben and Wood, 2003). The DNATHF conformation in the 2.6 Å DDBdr-DNATHF structure is surprisingly similar to the di-nucleotide crosslink seen in the DDBdr-DNA6-4PP structure. As in DDBdr-DNA6-4PP, DDBdr-DNATHF has two nucleotides flipped out, with the THF occupying position D+1 and the unmodified adenine base 3′ to the THF at position D+2 (Figure 4; Supplemental Figure S4). In addition, the overall DNA-backbone conformations and the DNA paths along the surface of DDB2 are essentially identical, despite different lesion types and DNA sequences (6-4PP vs. THF rmsd of 0.39 Å for the protein; 1.7 Å for DNA backbone over 10 bp). The kinking angle observed in THF and 6-4PP is indistinguishable within experimental error with slight variations (±5°) likely due to differences in crystal packing of the very ends, 3′ to the damage (Figure 2A). The low resolution DDBhs-DNATHF structure exhibits an essentially identical kinking angle and DNA path (Supplemental Figure S5).
Figure 4. Mechanism of abasic site recognition.

(A) Interaction of DDB2 with the DNATHF-backbone showing essentially identical interactions despite of a different sequence and type of damage. Parts of the DNA are omitted for clarity.
(B) Sequence of DNATHF used for co-crystallization. Disordered bases are shown faded.
(C) Chemical structure of the abasic site mimic (THF) and the neighboring Adenosine.
DDB2 is a DNA sequence independent damage detection protein
In the THF and 6-4PP structures, DDB2 contacts the lesion, their unstacked neighbors (D+3, D-1), the two orphaned undamaged bases (U-1, U-2) and the phosphate backbone. The only DNA sequence-specific hydrogen bond is between Gln372 and the orphaned U-2 base. Purines (adenine in DDBdr-DNA6-4PPvia atom C2) as well as pyrimidines (thymine in DDBdr-DNATHFvia atom N3) function as hydrogen bond donor to the Gln372 carboxamide oxygen. In principle, analogous hydrogen bonds can be made with guanine (via N1) and cytosine (via N4), making DDB2 a sequence-independent damage-detection protein.
The DDB2 β-propeller uses canonical WD40 features to serve as a DNA-binding platform
Seven out of the twelve DDB2 residues in direct contact with the DNA occur at positions frequently used by other WD40 domain proteins for binding to phosphopeptides and other ligands (Wu et al., 2003). These positions include the residue immediately preceding strand A, the innermost strand of the WD40 blade, where the DNA-contacting residues Arg148, Lys280 and Ile428 of DDB2 map to (see Figure 2C for nomenclature). The other position is the residue immediately following strand B, which contains four more DNA-contacting DDB2 residues (Lys168, Gln345, Tyr393 and Phe447). The third WD40 position, the second residue in strand A, is not used by DDB2 in ligand binding (see also Supplemental Figure S1). In addition to DDB2, several other proteins involved in chromatin and nucleic acid-associated processes contain WD40 domains. The high versatility of the β-propeller fold and the relatively few changes required for nucleic acid binding suggest these proteins may use WD40 domains for protein-DNA interactions as well. A likely example is the kelch type β-propeller protein RAG2, which is involved in V(D)J recombination. Biochemical studies point at the β-propeller propeller domain as being directly involved in DNA binding (Fugmann and Schatz, 2001).
Discussion
The mechanism of DNA damage recognition by DDB2
Damaged-DNA recognition by DDB2 involves the insertion of the hairpin into the minor groove, the stabilization of the flipped lesion and of the orphaned bases, and the kinking of the DNA. Due to the large footprint of the hairpin, minor groove insertion can proceed only upon flipping of the damage-strand bases out of the duplex. Since the footprint spans two bases, a dinucleotide flip is required, regardless of the presence of a single base THF or a two base 6-4PP lesion. The 16 basic side chains on the DDB2 surface likely guide and pre-orient the DNA towards DDB2, increasing the probability that the intermolecular encounter will result in productive complex formation (Figure 6A).
The hairpin structure, which is stabilized by an i+3 backbone hydrogen bond, does not change on DNA binding. The three Cα atoms of the hairpin in the DDBdr–DNA6-4PP can be superimposed on those in the DDBdr–DNATHF and the apo-DDBdr complexes with rmsd values of 0.18 Å and 0.56 Å, respectively (Figure 6B).
The DNA structure, on the other hand, changes substantially on DDB2 binding. Isolated 6-4PP and CPD DNA molecules exhibit overall double helical structures in solution, and the lesions or the opposing bases are not flipped out of the duplex (reviewed in Lukin and de Los Santos, 2006). However, these lesions destabilize the double helical DNA structure, presumably due to suboptimal stacking and lack of base pairing at the lesion. (Jing et al., 1998; Lukin and de Los Santos, 2006). This destabilization may well be associated with conformational flexibility at the lesion, and indeed molecular dynamics (MD) simulations suggest an increased propensity of the lesions to flip out, combined with an increased mobility of the bases in direct proximity of the lesion (Barsky et al., 2000; Maillard et al., 2007; O'Neil et al., 2007). This suggests that the isolated lesion may sample a conformational space populated with structures that share aspects of the flipped dinucleotide conformation of the DDB2-bound DNA. DDB2 could then pre-select those DNA conformations that most resemble the bound-DNA state. We presume that the sampling of the lesion's conformational space by DDB2 could be aided by an induced-fit mechanism whereby the binding energy from initial DDB2-DNA interactions may help distort the DNA structure further. Such initial DDB2-DNA interactions may involve a subset of the interactions seen in the final specific complex, but they could also involve the large number of basic side chains on the DDB2 surface (Figure 6A).
All these binding mechanisms are based on the diminished intra-strand stacking and inter-strand hydrogen bonding interactions around the lesion allowing the insertion of the hairpin and the flipping out of a dinucleotide segment. In the absence of damage, however, the intact base stacking and hydrogen bonding interactions would represent an energy barrier that precludes the insertion of the hairpin.
Additional specificity toward the photoproducts is provided by (i) the DDB2 lesion binding pocket, which would limit the size of the damaged base at the D+2 position and the nature of the covalent modifications at the D+1 position, and (ii) the interactions with the lesion's phosphodiester backbone, the compression of which would be stabilized by the 6-4PP or CPD intra-strand crosslink.
By probing the photoproduct directly, in a tightly localized fashion, DDB2 detects lesions refractory to binding by Rad4/XPC
The difficulty in detecting CPD lesions, as opposed to other photodimers, lies in the comparatively small structural and thermodynamic perturbation caused by lesions of this type. Structures of 6-4PP containing duplexes in solution show a greater extent of helix distortion than identical duplexes containing a CPD. In the context of a 12 bp DNA, a 6-4PP has been reported to thermodynamically destabilize the duplex by ∼6 kcal/mol, whereas a CPD resulted in ∼1.5 kcal/mol destabilization (Jing et al., 1998). Structural and biochemical studies have indicated that Rad4/XPC detects the lesion-induced thermodynamic destabilization of the Watson Crick duplex by flipping the lesion-containing dinucleotide as well as the two opposing, undamaged nucleotides (Min and Pavletich, 2007). In the structure of Rad4/XPC bound to a CPD-containing DNA, the damaged bases and their phosphodiester backbone are disordered in a fully-solvent exposed area of the complex, allowing Rad4/XPC to accommodate a wide range of bulky lesions. From a thermodynamic point of view, however, Rad4/XPC pays a penalty for flipping the undamaged nucleotides opposing the lesion. Solution NMR studies of CPD-containing duplexes have shown that the bases opposing the lesion are largely stacked within the duplex, although they are likely to be more flexible and have a higher probability of flipping out. Consequently, Rad4/XPC has only limited specificity towards CPD lesions, but it can recognize a wide range of lesions that destabilize the duplex substantially. DDB2, on the other hand, more directly probes for the limited perturbations caused by the photolesion. It binds to the lesion's phosphodiester backbone in a compressed conformation, essentially recognizing a DNA structural feature induced by the lesion. DDB2 also binds to the damaged bases through its surface pocket, stabilizing the flipped-out conformation of the lesion. In addition, because DDB2 does not flip out the undamaged bases, it can sense lesions that destabilize the duplex minimally. These features provide DDB2 with a heightened specificity towards photodimers, but they also limit the range of lesions that it can recognize compared to Rad4/XPC.
DDB2 mediated recognition of nucleosome embedded UV-lesions
The DDB1-DDB2 complex localizes to chromatin following UV-irradiation (Rapic Otrin et al., 1998), and it remains tightly associated with mononucleosomes when chromatin from UV-irradiated cells is solubilized by micrococcal nuclease digestion (Groisman et al., 2003). The DNA duplexes in DDBTHF and DDB6-4PP are remarkably similar in backbone conformation to that of the DNA wrapped around the nucleosome core particle. In fact DDBhs-DNATHF and the nucleosomal DNA can be superimposed with an rmsd of ∼2.4 Å over 23 bp (including the lesion and its two flanking 11 bp segments), while DDBdr-DNA6-4PP can be superimposed with a rmsd of ∼3.4 Å over 12 bp (including the lesion and its two flanking 5 bp segments) (Davey and Richmond, 2002). Upon superposition the ∼40° kinking angle around the lesion mimicks the nucleosomal DNA curvature. The best fit with the nucleosomal DNA is obtained when placing the lesion where the minor groove faces outwards, and thus points away from the core particle. In such configuration, DDB2 would, in principle, be able to read out the photodimer from the solvent without having to interfere with the remainder of the nucleosome structure (Supplemental Figure S6). Interestingly, CPDs isolated from irradiated human cells do not distribute randomly, but rather cluster at these exact locations, with the minor groove pointing outwards, thus potentially facilitating CPD detection by DDB2 in vivo. XPC, in contrast, does not bind well to nucleosomal substrates (Yasuda et al., 2005).
A model for DNA mediated XPC-recruitment by the DDB1-DDB2 complex
Comparison of our DDB2-DNA complex with the structure of the yeast XPC homolog Rad4 bound to damaged DNA (Min and Pavletich, 2007) indicates that the two proteins cannot bind to the same lesion simultaneously, as the DDB2 hairpin would clash extensively with a Rad4 β-hairpin that also inserts through the DNA duplex. However, because XPC/Rad4 binds to DNA in a bipartite manner, utilizing separate domains for binding to the lesion-containing DNA segment and to the adjacent undamaged dsDNA segment, it is possible that XPC can bind to the DDB2-damaged DNA complex by interacting only with the undamaged dsDNA 3′ to the lesion (see Supplemental Figure S7). Further biochemical and structural studies are required to address the possibility of a DDB2-DNA-XPC complex.
Mutations in DDB2 found in xeroderma pigmentosum complementation group E impair DNA and DDB1 binding
The DDB1-DDB2-DNA structure indicates that most XPE mutations affect residues or structural elements involved in binding to either DNA or DDB1. Mutations that affect DNA binding include Lys244Glu and Arg273His (Tang and Chu, 2002; Wittschieben and Wood, 2003). In the structure, Lys244 (Lys280 in drDDB2) makes a direct contact to the phosphodiester backbone of the DNA. Arg273 (Arg309 in drDDB2) does not contact the DNA directly, but is involved in intramolecular interactions that may have a role in stabilizing the structure of blade 4, part of which forms the DNA-binding surface of DDB2. The DDB1-DDB2 interface is affected in XPE patient GM01389 with a Leu350Pro mutation combined with the deletion of Asn349 (Nichols et al., 2000; Rapic-Otrin et al., 2003) (Leu387 and Asp386 in drDDB2, respectively). The XPE phenotype of these DDB1-DDB2 interface mutations suggests that the lack of DDB1 and its associated ubiquitin ligase activity would result in a defective NER response. It is also possible that lack of DDB1 binding may reduce the amount of functional DDB2 available in the cell, because, in vitro, we find that the solubility of DDB2 is highly dependent on the presence of DDB1 (A. Scrima & N. Thoma, unpublished). Other XPE mutations such as Asp307Tyr may affect the structural integrity of the entire DDB2 WD40 domain, indirectly interfering with the binding to both DNA and DDB1 (see Supplemental Figure S1 for a detailed discussion of XPE mutations).
The DDBCUL4 ubiquitin ligase complex establishes a spatially defined ubiquitination zone around the lesion
The DDB1-DDB2 structure provides the first insights into the DDB1-DCAF ubiquitin ligase architecture (O'Connell and Harper, 2007) (Implications for DCAF complexes architecture outside DNA repair are discussed in Supplemental Figure S3). In vivo the DDB1-DDB2-CUL4-RBX1 ubiquitin ligase complex (DDBCUL4) has been shown to ubiquitinate DDB2, XPC (El-Mahdy et al., 2006; Sugasawa et al., 2005), and histones (Wang et al., 2006). Although the role of DNA binding in the ubiquitin ligase activity of DDBCUL4 is not well understood, several lines of evidence suggest that DNA binding may facilitate ubiquitination. In vivo, DDBCUL4 is associated with the COP9 signalosome (Luijsterburg et al., 2007) that inhibits CUL4 ubiquitin ligase activity (Groisman et al., 2003), and chromatin localization leads to the release of COP9 (Groisman et al., 2003; Luijsterburg et al., 2007), activating DDBCUL4. In vitro, the ubiquination of XPC by DDBCUL4 is accelerated through direct binding of DDB1-DDB2 to damaged DNA (Sugasawa et al., 2005). Ubiquitination of XPC and DDB2 have been suggested to mediate an ubiquitin-dependent handover of the lesion from DDB2 to XPC (Sugasawa et al., 2005). The ubiquitination of histones surrounding the lesions, on the other hand, has been suggested to loosen the nucleosome structure, thereby facilitating the access of the NER repair machinery to the site of damage (Wang et al., 2006). The complete assembly of the NER repairosome requires around 100 bp, and this likely necessitates the displacement of at least one nucleosome (reviewed in Thoma, 2005).
A model of the DDBCUL4 ubiquitin ligase, constructed by superimposing the DDB1 structures from our DDB1-DDB2-DNA and the previously reported DDB1-CUL4-RBX1 (Angers et al., 2006) complexes, shows that the ubiquitin ligase can reach DNA-bound proteins in the vicinity of the lesion (Figure 7 and Supplemental Figure S6C). The DDB1 BPB-domain, which is the attachment site for CUL4A (Figures 1 and 7A) has rotational flexibility relative to the rest of DDB1-DDB2 (this work and Angers et al., 2006; Li et al., 2006). Because of this, the RBX1-bound ubiquitin conjugating enzyme (E2) would be able to sweep an active zone of ∼120° to maximally 180° within a distance of ∼35 to 60 Å from the photodimer (Figures 7B-D). Because Cullin-based ubiquitination is determined in part by the proximity of the substrate lysine(s) to the E2 active site (Wu et al., 2003), our DNA-bound DDBCUL4 model suggests that the lesion-bound nucleosome would be an efficient substrate (Supplemental Figure S6). Ubiquitination of DNA-bound DDB2 and other NER factors may be less efficient however, as the geometry of DDBCUL4 results in a “blind spot” in the immediate vicinity of the lesion that can not be reached by the ligase (Figures 7C, D). We note, however, that unstructured segments such as the approximately 55-residue hsDDB2 N-terminus could, in principle, reach the “active zone” of the ligase. The exact lysine(s) targeted in DDB2 has not been determined, but in vitro studies have suggested that more than six lysine residues are modified in the course of the reaction (Sugasawa et al., 2005). While the precise timecourse of ubiquitination events at sites of UV-damage has to await further studies, our structure suggests that DDB1-DDB2 may facilitate the localization of XPC to the lesion by ubiquitinating histones H3-H4 and destabilizing the nucleosome structure in the immediate vicinity of the lesion.
Figure 7. Implications for the role of UV-DDB mediated damage recognition in the context of chromatin.

In structures of DDB1, DDB1-CUL4A/RBX1 and that of the DDB1-DDB2 complexes, the B-domain, which serves as the main attachment site for CUL4, is found in 8 different orientations (1. PDB:2HYE; 2. PDB: 2B5L; 3. DDBhs–DNAfree; 4. DDBhs–DNATHF (Mol. A); 5. DDBhs–DNATHF (Mol. C); 6. DDBdr-DNAfree; 7. DDBdr–DNATHF/6-4PP; 8. PDB:2B5M). The position of the E2 enzyme (UbcH5A; PDB: 2C4P) in relation to CUL4A/RBX1 was modeled using the c-Cbl-UbcH7 structure (PDB: 1FBV) as template.
(A) Model of the DDB1-DDB2-DNA-CUL4A/RBX1-E2 complex (CUL4A/RBX1: lightgray; E2; darkgray). Numbers along the dotted path depict positions of the active site of the E2 (red spheres) based on the BPB-domain orientation in 8 different DDB1-structures.
(B) As in (A) but rotated by 90° horizontally.
(C-D) Different inclination angles of the BPB-domain tilt the CUL4 ligase by ∼20°, and confer a rotational movement of ∼ 120°. The CUL4/E2 samples ∼120° in a zone of ∼35-60 Å from the damage.
In conclusion, the structures of the DDB1-DDB2-DNA complexes provide a structural basis for understanding the high affinity detection of lesions refractory to recognition by XPC. It also provides a framework for understanding damage recognition in chromatin and the role of ubiquitination in the NER response to damage.
Experimental Procedures
Protein expression & purification
hsDDB1 (1-1140), hsDDB2 (10-427) and drDDB2 (94-457) were cloned into a pAC-derived plasmid (BD biosciences Pharmingen, San Jose, CA, USA). Recombinant DDB1-DDB2 baculoviruses were prepared according to the manufacturer's instruction. Proteins were co-expressed as N-terminal (His)6 tagged fusion proteins in High Five insect cells (Invitrogen).
For the DDB1-DDB2 purification, cells were resuspended in lysis buffer (50 mM Tris pH 8.0; 200 mM NaCl; 5 mM β-ME; 1 mM PMSF; 0.1 % Triton X-100) and lysed by sonication. Supernatant was harvested and protein was purified by sequential HisTrap affinity chromatography (Sigma) and Source15Q ion exchange chromatography (GE Healthcare). The purification was completed by size exclusion chromatography (Superdex 200; GE Healthcare) in 50 mM Hepes 7.4, 200 mM NaCl and 5 mM DTT. The purified complex was concentrated to ∼10 mg/ml, flash-frozen in liquid N2 and stored at -80°C.
Oligonucleotides used for crystallization
Single stranded DNA-oligonucleotides containing the tetrahydrofuran (THF) lesion were ordered from Sigma Genosys Switzerland. The oligonucleotides containing the 6-4 PP were synthesized and purified in the laboratory of S. Iwai (Iwai et al., 1996). The complementary oligos (containing no lesion) were synthesized by GeneLink and Sigma Genosys with further purification on a Microsorb 300-5 PureDNA HPLC column (Varian, Inc., USA). Oligos were annealed in 10 mM Hepes 7.4; 50 mM NaCl and subsequently lyophilized and stored at -20°C.
Crystallization
Crystals of the DDBdr-DNA complexes as well as the DDBdr DNA-free complex were grown at 20-25°C using the hanging drop diffusion method. For protein DNA-complexes a 1.2 molar excess of DNA was added to the protein solution. The 16mer THF complex and the DNA free complex were crystallized after mixing the protein solution in a 1:1 ratio with reservoir containing 100 mM Ca-Acetate; 100 mM MES pH 5.7; 12-14 % PEG 400. Diffraction grade 6-4 PP crystals were obtained under identical conditions with the help of streak seeding. Crystals were transferred into cryo-solution (100 mM Ca-Acetate; 100 mM MES pH 5.7; 12 % PEG 400; 20 % Ethylene glycol) and flash frozen in liquid N2 for data collection. Data sets were collected at the Swiss Light Source, beamline X10SA, Paul Scherrer Institut, Villigen, Switzerland. Collected data were processed with XDS (Kabsch, 1993).
The DDBhs-DNATHF complex as well as the DDBhs DNA-free complex was grown at 20-25°C using the hanging drop diffusion method. For the protein DNA-complex a 1.2 molar excess of 31mer THF-DNA (damaged strand: 5′ AAGTCCTGAATGAAT(THF)AAGCAGGCGTTGAAG 3′; undamaged strand: 5′ CTTCAACGCCTGCTTTATTCATTCAGGACTT 3′) was added to the protein solution. The protein solution was mixed in a 1:1 ratio with reservoir solution. DDBhs-DNATHF crystallized in 100 mM (NH4)2SO4; 18% PEG 4000; 200 mM Ammoniumformate; 100 mM Na-Citrate pH 5.6. The DNA-free crystals were grown in 200 mM (NH4)2SO4; 800 mM LiSO2; 100 mM Na-Citrate pH 5.6. Crystals were transferred into cryo-solution (DDBhs-DNATHF: mother liquor + 15 % Ethyleneglycol; DNA-free: mother liquor + 30 % Glycerol) and flash frozen in liquid N2 for data collection. Data sets were collected at the 8BM and 17IDB beamlines of the Advanced Photon Source, Argonne, USA. Reflection data were indexed, integrated, and scaled using the HKL2000 package (Otwinowski and Minor, 1997).
Structure solution and model building
Crystals of DNA-free DDBhs contain three complexes in the asymmetric unit. The structure was solved by the MIRAS method using KAu(CN)2 and thimerosal derivates. Initial heavy atom sites were localized using ShelxD (Sheldrick, 2008). Phases were calculated with the program SHARP (delaFortelle and Bricogne, 1997), and were improved using solvent flattening and three-fold averaging with the program DM (Collaborative Computational Project, 1994). Sequence assignment was confirmed using the anomalous signal from SeMet-derived crystals (data not shown). The structure of the DDBhs-DNATHF crystals was solved by molecular replacement with Phaser (Mccoy et al., 2007) using the individual molecules of the DNA-free structure as search models. The DDBhs-DNATHF crystals contain two complexes in the asymmetric unit.
Structures of DDBdr were solved by molecular replacement with Molrep (Vagin and Teplyakov, 1997) using the structure of hsDDB1 as a search model. The position of DDB1 was fixed and an additional round of Molrep with hsDDB2 as search model resulted in the full hsDDB1-drDDB2 complex. In the DNA containing complexes positive density appearing in the difference map was clearly identifiable as bound DNA, which was manually build into the density.
All structures were built using Coot (Emsley and Cowtan, 2004) and refined with Refmac5 (Murshudov et al., 1997). Tight NCS restraints were applied during the refinement of the DNA-free DDBhs and DDBhs-DNATHF complexes. Figures were generated with PyMol (DeLano Scientific; http://www.pymol.org).
Supplementary Material
The Supplemental Data include six tables and seven figures and can be found with this article online at xxx.
Acknowledgments
We thank Norman Kairies for crystallographic support. We would like to thank Fritz Thoma, Hanspeter Naegeli and Susan Gasser for discussions relating to DNA repair in the context of chromatin. We thank Hans Widmer for help and support. Part of this work was performed at the Swiss Light Source at the Paul Scherrer Institute, Villigen, Switzerland. Y.K and S.I. acknowledge funding from the Human Frontier Science Program (HFSP). Funding in the laboratory of N.H.T. is provided by the Swiss National Fond (SNF;#31-120205) and the Novartis Research Foundation. A.S. gratefully acknowledges funding from the European Molecular Biology Organization (EMBO).
Footnotes
Accession numbers: The models and structure factors have been deposited in the Protein Data Bank with the PDB accession codes 3EI1 (DDBdr-DNA6-4PP), 3EI2 (DDBdr-DNATHF), 3EI3 (DDBdr-DNAfree) and 3EI4 (DDBhs-DNAfree).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aboussekhra A, Biggerstaff M, Shivji MK, Vilpo JA, Moncollin V, Podust VN, Protic M, Hubscher U, Egly JM, Wood RD. Mammalian DNA nucleotide excision repair reconstituted with purified protein components. Cell. 1995;80:859–868. doi: 10.1016/0092-8674(95)90289-9. [DOI] [PubMed] [Google Scholar]
- Angers S, Li T, Yi X, MacCoss MJ, Moon RT, Zheng N. Molecular architecture and assembly of the DDB1-CUL4A ubiquitin ligase machinery. Nature. 2006;443:590–593. doi: 10.1038/nature05175. [DOI] [PubMed] [Google Scholar]
- Barsky D, Foloppe N, Ahmadia S, Wilson DM, 3rd, MacKerell AD., Jr New insights into the structure of abasic DNA from molecular dynamics simulations. Nucleic acids research. 2000;28:2613–2626. doi: 10.1093/nar/28.13.2613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batty D, Rapic'-Otrin V, Levine AS, Wood RD. Stable binding of human XPC complex to irradiated DNA confers strong discrimination for damaged sites. J Mol Biol. 2000;300:275–290. doi: 10.1006/jmbi.2000.3857. [DOI] [PubMed] [Google Scholar]
- Cleaver JE. Cancer in xeroderma pigmentosum and related disorders of DNA repair. Nat Rev Cancer. 2005;5:564–573. doi: 10.1038/nrc1652. [DOI] [PubMed] [Google Scholar]
- Collaborative Computational Project, N. The CCP4 suite: programs for protein crystallography. Acta crystallographica. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- Davey CA, Richmond TJ. DNA-dependent divalent cation binding in the nucleosome core particle. Proceedings of the National Academy of Sciences of the United States of America; 2002. pp. 11169–11174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- delaFortelle E, Bricogne G. Maximum-likelihood heavy-atom parameter refinement for multiple isomorphous replacement and multiwavelength anomalous diffraction methods. Macromolecular Crystallography. 1997;276(Pt A):472–494. doi: 10.1016/S0076-6879(97)76073-7. [DOI] [PubMed] [Google Scholar]
- Dualan R, Brody T, Keeney S, Nichols AF, Admon A, Linn S. Chromosomal localization and cDNA cloning of the genes (DDB1 and DDB2) for the p127 and p48 subunits of a human damage-specific DNA binding protein. Genomics. 1995;29:62–69. doi: 10.1006/geno.1995.1215. [DOI] [PubMed] [Google Scholar]
- El-Mahdy MA, Zhu Q, Wang QE, Wani G, Praetorius-Ibba M, Wani AA. Cullin 4A-mediated proteolysis of DDB2 protein at DNA damage sites regulates in vivo lesion recognition by XPC. The Journal of biological chemistry. 2006;281:13404–13411. doi: 10.1074/jbc.M511834200. [DOI] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallographica Section D-Biological Crystallography. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Feldberg RS, Grossman L. A DNA binding protein from human placenta specific for ultraviolet damaged DNA. Biochemistry. 1976;15:2402–2408. doi: 10.1021/bi00656a024. [DOI] [PubMed] [Google Scholar]
- Fitch ME, Nakajima S, Yasui A, Ford JM. In vivo recruitment of XPC to UV-induced cyclobutane pyrimidine dimers by the DDB2 gene product. The Journal of biological chemistry. 2003;278:46906–46910. doi: 10.1074/jbc.M307254200. [DOI] [PubMed] [Google Scholar]
- Friedberg EC, Walker GC, Siede RD, Wood RD, Schultz RA, Ellenberger T. DNA repair and mutagenesis. 2nd. Washington, DC: ASM Press; 2006. [Google Scholar]
- Fugmann SD, Schatz DG. Identification of basic residues in RAG2 critical for DNA binding by the RAG1-RAG2 complex. Molecular cell. 2001;8:899–910. doi: 10.1016/s1097-2765(01)00352-5. [DOI] [PubMed] [Google Scholar]
- Fujiwara Y, Masutani C, Mizukoshi T, Kondo J, Hanaoka F, Iwai S. Characterization of DNA recognition by the human UV-damaged DNA-binding protein. The Journal of biological chemistry. 1999;274:20027–20033. doi: 10.1074/jbc.274.28.20027. [DOI] [PubMed] [Google Scholar]
- Gelasco A, Lippard SJ. NMR solution structure of a DNA dodecamer duplex containing a cis-diammineplatinum(II) d(GpG) intrastrand cross-link, the major adduct of the anticancer drug cisplatin. Biochemistry. 1998;37:9230–9239. doi: 10.1021/bi973176v. [DOI] [PubMed] [Google Scholar]
- Gillet LC, Scharer OD. Molecular mechanisms of mammalian global genome nucleotide excision repair. Chemical reviews. 2006;106:253–276. doi: 10.1021/cr040483f. [DOI] [PubMed] [Google Scholar]
- Groisman R, Polanowska J, Kuraoka I, Sawada J, Saijo M, Drapkin R, Kisselev AF, Tanaka K, Nakatani Y. The ubiquitin ligase activity in the DDB2 and CSA complexes is differentially regulated by the COP9 signalosome in response to DNA damage. Cell. 2003;113:357–367. doi: 10.1016/s0092-8674(03)00316-7. [DOI] [PubMed] [Google Scholar]
- Hwang BJ, Toering S, Francke U, Chu G. p48 Activates a UV-damaged-DNA binding factor and is defective in xeroderma pigmentosum group E cells that lack binding activity. Molecular and cellular biology. 1998;18:4391–4399. doi: 10.1128/mcb.18.7.4391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwai S, Shimizu M, Kamiya H, Ohtsuka E. Synthesis of a Phosphoramidite Coupling Unit of the Pyrimidine (6-4) Pyrimidone Photoproduct and Its Incorporation into Oligodeoxynucleotides. J Amer Chem Soc. 1996;118:7642–7643. [Google Scholar]
- Jing Y, Kao JF, Taylor JS. Thermodynamic and base-pairing studies of matched and mismatched DNA dodecamer duplexes containing cis-syn, (6-4) and Dewar photoproducts of TT. Nucleic acids research. 1998;26:3845–3853. doi: 10.1093/nar/26.16.3845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W. Automatic Processing of Rotation Diffraction Data from Crystals of Initially Unknown Symmetry and Cell Constants. Journal of Applied Crystallography. 1993;26:795–800. [Google Scholar]
- Kapetanaki MG, Guerrero-Santoro J, Bisi DC, Hsieh CL, Rapic-Otrin V, Levine AS. The DDB1-CUL4ADDB2 ubiquitin ligase is deficient in xeroderma pigmentosum group E and targets histone H2A at UV-damaged DNA sites. Proceedings of the National Academy of Sciences of the United States of America; 2006. pp. 2588–2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Chen X, Garbutt KC, Zhou P, Zheng N. Structure of DDB1 in complex with a paramyxovirus V protein: viral hijack of a propeller cluster in ubiquitin ligase. Cell. 2006;124:105–117. doi: 10.1016/j.cell.2005.10.033. [DOI] [PubMed] [Google Scholar]
- Lu XJ, Olson WK. 3DNA: a versatile, integrated software system for the analysis, rebuilding and visualization of three-dimensional nucleic-acid structures. Nature protocols. 2008;3:1213–1227. doi: 10.1038/nprot.2008.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luijsterburg MS, Goedhart J, Moser J, Kool H, Geverts B, Houtsmuller AB, Mullenders LH, Vermeulen W, van Driel R. Dynamic in vivo interaction of DDB2 E3 ubiquitin ligase with UV-damaged DNA is independent of damage-recognition protein XPC. J Cell Sci. 2007;120:2706–2716. doi: 10.1242/jcs.008367. [DOI] [PubMed] [Google Scholar]
- Lukin M, de Los Santos C. NMR structures of damaged DNA. Chemical reviews. 2006;106:607–686. doi: 10.1021/cr0404646. [DOI] [PubMed] [Google Scholar]
- Maillard O, Camenisch U, Clement FC, Blagoev KB, Naegeli H. DNA repair triggered by sensors of helical dynamics. Trends Biochem Sci. 2007;32:494–499. doi: 10.1016/j.tibs.2007.08.008. [DOI] [PubMed] [Google Scholar]
- Mccoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. Journal of Applied Crystallography. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min JH, Pavletich NP. Recognition of DNA damage by the Rad4 nucleotide excision repair protein. Nature. 2007;449:570–575. doi: 10.1038/nature06155. [DOI] [PubMed] [Google Scholar]
- Moser J, Volker M, Kool H, Alekseev S, Vrieling H, Yasui A, van Zeeland AA, Mullenders LH. The UV-damaged DNA binding protein mediates efficient targeting of the nucleotide excision repair complex to UV-induced photo lesions. DNA repair. 2005;4:571–582. doi: 10.1016/j.dnarep.2005.01.001. [DOI] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallographica Section D-Biological Crystallography. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- Nichols AF, Itoh T, Graham JA, Liu W, Yamaizumi M, Linn S. Human damage-specific DNA-binding protein p48. Characterization of XPE mutations and regulation following UV irradiation. The Journal of biological chemistry. 2000;275:21422–21428. doi: 10.1074/jbc.M000960200. [DOI] [PubMed] [Google Scholar]
- O'Connell BC, Harper JW. Ubiquitin proteasome system (UPS): what can chromatin do for you? Curr Opin Cell Biol. 2007;19:206–214. doi: 10.1016/j.ceb.2007.02.014. [DOI] [PubMed] [Google Scholar]
- O'Neil LL, Grossfield A, Wiest O. Base flipping of the thymine dimer in duplex DNA. J Phys Chem B. 2007;111:11843–11849. doi: 10.1021/jp074043e. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Macromolecular Crystallography. 1997;276(Pt A):307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- Payne A, Chu G. Xeroderma pigmentosum group E binding factor recognizes a broad spectrum of DNA damage. Mutat Res. 1994;310:89–102. doi: 10.1016/0027-5107(94)90012-4. [DOI] [PubMed] [Google Scholar]
- Rapic-Otrin V, Navazza V, Nardo T, Botta E, McLenigan M, Bisi DC, Levine AS, Stefanini M. True XP group E patients have a defective UV-damaged DNA binding protein complex and mutations in DDB2 which reveal the functional domains of its p48 product. Human molecular genetics. 2003;12:1507–1522. doi: 10.1093/hmg/ddg174. [DOI] [PubMed] [Google Scholar]
- Rapic Otrin V, Kuraoka I, Nardo T, McLenigan M, Eker AP, Stefanini M, Levine AS, Wood RD. Relationship of the xeroderma pigmentosum group E DNA repair defect to the chromatin and DNA binding proteins UV-DDB and replication protein A. Molecular and cellular biology. 1998;18:3182–3190. doi: 10.1128/mcb.18.6.3182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reardon JT, Nichols AF, Keeney S, Smith CA, Taylor JS, Linn S, Sancar A. Comparative analysis of binding of human damaged DNA-binding protein (XPE) and Escherichia coli damage recognition protein (UvrA) to the major ultraviolet photoproducts: T[c,s]T, T[t,s]T, T[6-4]T, and T[Dewar]T. The Journal of biological chemistry. 1993;268:21301–21308. [PubMed] [Google Scholar]
- Sheldrick GM. A short history of SHELX. Acta Crystallographica Section A. 2008;64:112–122. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- Shiyanov P, Nag A, Raychaudhuri P. Cullin 4A associates with the UV-damaged DNA-binding protein DDB. The Journal of biological chemistry. 1999;274:35309–35312. doi: 10.1074/jbc.274.50.35309. [DOI] [PubMed] [Google Scholar]
- Sugasawa K, Ng JM, Masutani C, Iwai S, van der Spek PJ, Eker AP, Hanaoka F, Bootsma D, Hoeijmakers JH. Xeroderma pigmentosum group C protein complex is the initiator of global genome nucleotide excision repair. Molecular cell. 1998;2:223–232. doi: 10.1016/s1097-2765(00)80132-x. [DOI] [PubMed] [Google Scholar]
- Sugasawa K, Okuda Y, Saijo M, Nishi R, Matsuda N, Chu G, Mori T, Iwai S, Tanaka K, Tanaka K, et al. UV-induced ubiquitylation of XPC protein mediated by UV-DDB-ubiquitin ligase complex. Cell. 2005;121:387–400. doi: 10.1016/j.cell.2005.02.035. [DOI] [PubMed] [Google Scholar]
- Takao M, Abramic M, Moos M, Jr, Otrin VR, Wootton JC, McLenigan M, Levine AS, Protic M. A 127 kDa component of a UV-damaged DNA-binding complex, which is defective in some xeroderma pigmentosum group E patients, is homologous to a slime mold protein. Nucleic acids research. 1993;21:4111–4118. doi: 10.1093/nar/21.17.4111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang J, Chu G. Xeroderma pigmentosum complementation group E and UV-damaged DNA-binding protein. DNA repair. 2002;1:601–616. doi: 10.1016/s1568-7864(02)00052-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang JY, Hwang BJ, Ford JM, Hanawalt PC, Chu G. Xeroderma pigmentosum p48 gene enhances global genomic repair and suppresses UV-induced mutagenesis. Molecular cell. 2000;5:737–744. doi: 10.1016/s1097-2765(00)80252-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoma F. Repair of UV lesions in nucleosomes--intrinsic properties and remodeling. DNA Repair (Amst) 2005;4:855–869. doi: 10.1016/j.dnarep.2005.04.005. [DOI] [PubMed] [Google Scholar]
- Treiber DK, Chen Z, Essigmann JM. An ultraviolet light-damaged DNA recognition protein absent in xeroderma pigmentosum group E cells binds selectively to pyrimidine (6-4) pyrimidone photoproducts. Nucleic acids research. 1992;20:5805–5810. doi: 10.1093/nar/20.21.5805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagin A, Teplyakov A. MOLREP: an automated program for molecular replacement. Journal of Applied Crystallography. 1997;30:1022–1025. [Google Scholar]
- Wakasugi M, Kawashima A, Morioka H, Linn S, Sancar A, Mori T, Nikaido O, Matsunaga T. DDB accumulates at DNA damage sites immediately after UV irradiation and directly stimulates nucleotide excision repair. The Journal of biological chemistry. 2002;277:1637–1640. doi: 10.1074/jbc.C100610200. [DOI] [PubMed] [Google Scholar]
- Wang H, Zhai L, Xu J, Joo HY, Jackson S, Erdjument-Bromage H, Tempst P, Xiong Y, Zhang Y. Histone H3 and H4 ubiquitylation by the CUL4-DDB-ROC1 ubiquitin ligase facilitates cellular response to DNA damage. Molecular cell. 2006;22:383–394. doi: 10.1016/j.molcel.2006.03.035. [DOI] [PubMed] [Google Scholar]
- Wittschieben BO, Iwai S, Wood RD. DDB1-DDB2 (xeroderma pigmentosum group E) protein complex recognizes a cyclobutane pyrimidine dimer, mismatches, apurinic/apyrimidinic sites, and compound lesions in DNA. The Journal of biological chemistry. 2005;280:39982–39989. doi: 10.1074/jbc.M507854200. [DOI] [PubMed] [Google Scholar]
- Wittschieben BO, Wood RD. DDB complexities. DNA repair. 2003;2:1065–1069. doi: 10.1016/s1568-7864(03)00113-7. [DOI] [PubMed] [Google Scholar]
- Wu G, Xu G, Schulman BA, Jeffrey PD, Harper JW, Pavletich NP. Structure of a beta-TrCP1-Skp1-beta-catenin complex: destruction motif binding and lysine specificity of the SCF(beta-TrCP1) ubiquitin ligase. Molecular cell. 2003;11:1445–1456. doi: 10.1016/s1097-2765(03)00234-x. [DOI] [PubMed] [Google Scholar]
- Yasuda G, Nishi R, Watanabe E, Mori T, Iwai S, Orioli D, Stefanini M, Hanaoka F, Sugasawa K. In vivo destabilization and functional defects of the xeroderma pigmentosum C protein caused by a pathogenic missense mutation. Molecular and cellular biology. 2007;27:6606–6614. doi: 10.1128/MCB.02166-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda T, Sugasawa K, Shimizu Y, Iwai S, Shiomi T, Hanaoka F. Nucleosomal structure of undamaged DNA regions suppresses the non-specific DNA binding of the XPC complex. DNA Repair (Amst) 2005;4:389–395. doi: 10.1016/j.dnarep.2004.10.008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The Supplemental Data include six tables and seven figures and can be found with this article online at xxx.