

Abstract
Previously, IL-1β secretion from Type 2 diabetic patients has been shown to be increased compared with controls. In this study, we aimed to delineate the mechanism of IL-1β induction under high-glucose (HG) conditions in human monocytes. THP-1 cells cultured in normal glucose were treated with increasing concentrations of D-glucose (10–25 mM) for 6–72 h. IL-1β and IL-1 receptor antagonist levels were measured by ELISA and Western blots, whereas mRNA was quantitated by RT-PCR. Specific inhibitors and small interfering RNAs of PKC, p38, ERK1/2, NF-κB, and NADPH oxidase were used to determine the mediators in parallel experiments under HG conditions. IL-1β-secreted protein, cellular protein, and mRNA increase under HG conditions is time and dose dependent, with maximum increase at 15 mM (48 h; P < 0.05). IL-1 receptor antagonist release was time and dose dependent, similar to IL-1β expression pattern; however, the molar ratio of IL-1β to IL-1RA was increased. Data from inhibitor and small interfering RNA experiments indicate that IL-1β release under HG is mediated by PKC-α, via phosphorylation of p38 MAPK, and ERK1/2 leading to NF-κB activation, resulting in increased mRNA and protein for IL-1β. At the same time, it appears that NADPH oxidase via p47phox activates NF-κB, resulting in increased IL-1β secretion. Data suggest that, under HG conditions, monocytes release significantly higher amounts of IL-1β through multiple mechanisms, further compounding the disease progression. Targeting signaling pathways mediating IL-1β release could result in the amelioration of inflammation and possibly diabetic vasculopathies.
Keywords: protein kinase C, diabetes, p38 phosphorylation, interleukin-1β
Clinical evidence and experimental evidence support a central role for inflammation in diabetes, as shown by the importance of adhesion molecules, cytokines, chemokines, and so forth (7, 15, 17). The monocyte-macrophage is a crucial and the most readily accessible cell in the artery wall, secreting several proinflammatory, proatherogenic cytokines, such as IL-1β, IL-6, and TNF-α, which are increased in diabetes (18, 19, 25).
IL-1β is a multipotent, proinflammatory cytokine that affects most cell types and cooperates with other cytokines, chemokines, and a variety of cellular mediators (12). Production of IL-1β is tightly controlled by a variety of molecules, including surface receptors, such as IL-1 receptor antagonist (IL-1ra), a specific receptor antagonist. IL-1ra binds with equal affinity to both IL-1α and IL-1β. In an increasing number of infectious and noninfectious clinical and experimental situations, data indicate that IL-1ra may be essential for the host defense against excessive, deleterious inflammation (3), with corresponding higher IL-1β-to-IL-1ra molar ratios. An imbalance between IL-1ra and IL-1β may predispose individuals to the development of diabetes. Previously, it has been shown that high levels of IL-1ra are required to block IL-1β-mediated deleterious effects on pancreatic β-cell function (39). A recent study by Kowluru and Odenbach (26) suggests a major role for IL-1β in the pathogenesis of diabetic retinopathies and also in pancreatic β-cell toxicity.
Convincing evidence exists for a relationship between microvascular complications and high glucose (HG), with several other studies suggesting a similar relationship for macrovascular complications (13, 28, 35, 46). Studies from our laboratory show increased IL-1β levels in basal and LPS-activated monocytes in type 2 diabetes compared with that shown for controls (11).
Hyperglycemia is one of the key factors that contribute to diabetic complications (9, 32, 48). Shanmugam et al. (43) have shown that HG treatment activates monocytes and induces an increase in TNF-α, IL-1β, and monocyte chemoattractant protein-1 gene expression in human THP-1 monocytic cells (43). However, it is not clear how HG affects IL-1β and IL-1ra expression in monocytes. The present study was designed to examine the mechanism of HG on IL-1β expression in a human monocytic cell line (THP-1).
MATERIALS AND METHODS
Reagents
THP-1 human monocytic cell line was obtained from American Type Culture Collection (Manassas, VA). Endotoxin-free, glucose-free RPMI 1640 medium and FBS were purchased from GIBCO BRL (Carlsbad, CA). Antibiotics, glutamine, PMSF, endotoxin-free D-glucose/mannitol, HEPES, protease inhibitor cocktail, Triton X-100, and DTT were from Sigma (St. Louis, MO). Calphostin-C (10 μmol/l), SB-203580 (5 μmol/l), caffeic acid phenethyl ester (CAPE; 5 μmol/l), BAY-11–7085 (10 μmol/l), 2,2′,3,3′,4,4′-hexa-hydroxy-1,1′-biphenyl-6,6′-dimethanol dimethyl ether (HBDDE; 50 μmol/l), apocynin (30 μmol/l), diphenyleneiodonium chloride (DPI; 10 μmol/l), and LJNKI1 (5 μmol/l) were obtained from Calbiochem. Eli Lilly kindly provided LY-379196 (30 nmol/l). U-0126 (10 μmol/l) was purchased from Cell Signaling (Boston, MA). Phosphorylated p38 antibody (catalog no. sc-7973), phosphorylated ERK1/2 (catalog no. sc-7976), total p38 (catalog no. sc-7149), total ERK (catalog no. sc-94), and IL-1β (catalog no. sc-7884) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Polyvinylidene difluoride membranes and Tris-glycine gels were from Invitrogen (Carlsbad, CA). Bicinchoninic acid kit was obtained from Pierce (Rockford, IL). Prevalidated PKC-α (GenBank NM_002737), PKC-β (GenBank NM_002738), NF-κB (GenBank NM_003998), and p47phox (GenBank NM_000265) small interfering RNAs (siRNAs) and siPORT amine transfection reagent were purchased from Ambion (Austin, TX). siRNA SMART pool ERK1/2 or MAPK1 (GenBank NM_002745) and siGENOME SMART pool p38 MAPK (GenBank NM_138957) were obtained from Dharmacon (Chicago, IL). Enhanced chemiluminescence (ECL) kits were purchased from Amersham Pharmacia (Piscataway, NJ). The concentrations of the various inhibitors and siRNAs used in the present study were cautiously titrated and determined in pilot studies, and some were reported previously (10, 47).
Cell culture
THP-1 cells were subcultured in endotoxin-free RPMI 1640 containing 5.5 mmol/l glucose, 50 μmol/l mercaptoethanol, 10% FBS, 2 mmol/l glutamine, 1 mmol/l sodium pyruvate, and 10 mmol/l HEPES supplemented with 10% FBS and antibiotics at 37°C and 5% CO2 and used between passages 3 and 6 with 90% confluency for all the experiments (47). We monitored the endotoxin levels in the culture medium reagents (glucose, mannitol, etc.) using limulus amoebocyte lysate assay (Cambrex, Milwaukee, WI), and the average endotoxin level was <100 endotoxin unit/ml consistently in all of the experiments, as any endotoxin contamination interferes with accurate IL-1β and IL-1ra quantitation.
Treatments
Cells were cultured (1 × 106 cells/ml) for 3 days in either 5.5 mmol/l glucose [normal glucose (NG)] or for indicated time points with 10–25 mmol/l glucose (HG); as an osmotic control, 9.5–14.5 mmol/l mannitol was added along with NG with daily changes in medium. Cell viability, as determined by trypan blue exclusion, was >92%. In addition, cells were also treated with inhibitors for 24 h with NG and HG (15 mM), with daily changes in medium for 48 h (47). Cell supernatants, lysates, and RNA were collected for ELISA, Western blotting, and RT-PCR, respectively.
IL-1β and IL-1ra ELISA
The release of IL-1β and IL-1ra were measured in the supernatants of THP-1 cells treated with NG or HG (10–25 mM) by highly sensitive ELISAs (R&D Systems, Minneapolis, MN), as reported previously (10). IL-1β and IL-1ra concentrations were expressed as picograms per milligram protein. The intra-and interassay coefficient of variation was determined to be <10%.
Western blots
At the end of treatments, cells were lysed, and total protein was isolated as reported previously (47). Total protein (20–30 μg) was resolved in 10% Tris-glycine gel, and the proteins on the gel were transferred onto polyvinylidene difluoride membrane. Blots were blocked with 5% nonfat milk and then incubated with specific phosphoantibody and anti-rabbit or anti-mouse IgG conjugated with horse-radish peroxidase, and the protein bands were detected with ECL detection reagents from Amersham Biosciences. The membrane was then stripped with the use of Restore Western blot stripping buffer (Pierce), and the membranes were incubated with nonphosphoantibody or β-actin and detected by ECL detection reagents. The results are representative of a minimum of three experiments using three different batches of THP-1 cells treated with HG for different times.
RNA extraction and real-time RT-PCR
THP-1 cells were treated as indicated above, and total RNA was obtained using TRI reagent (Invitrogen). The first strand of cDNA was synthesized with the use of total 1 μg RNA. cDNA (50–100 ng) was amplified by Taqman primers (Applied Biosystems, Foster City, CA) specific for IL-1β and GAPDH, following manufacturer’s cycling parameters and using an ABI 7700 sequence detection system (Applied Biosystems). GAPDH was used as an endogenous reference to correct for differences in the amount of total RNA added to the reaction and to compensate for different levels of inhibition during reverse transcription of RNA and during PCR. Data are calculated by use of the 2−ΔΔCT method (where CT is cycle threshold) and are presented as multiples of induction of transcripts for IL-1β gene normalized to GAPDH in cells treated with HG (31).
siRNA transfection assays
Prevalidated siRNAs were obtained from Ambion and Dharmacon. THP-1 cells in 12-well plates were transiently transfected with 20 μmol/l siRNAs and siPORT amine reagent following manufacturer’s instructions, with suitable vehicle and scrambled siRNA controls, and subsequently treated with HG (15 mM) for 48 h with daily changes of medium. Cell supernatants were collected for IL-1β assays, and cell lysates were utilized to determine the proximal signaling mediator using phosphospecific antibodies to p38 and ERK/MAPK. Transfection rates of 70–80% of cells were accepted for all the experiments. Knock-down efficiency of the siRNAs is indicated via 1) Western blot (for downstream mediators of the target sequence) and 2) the measured end product (IL-1β) of the target gene.
PKC activity assays
PKC activity in THP-1 cells was determined by nonradioactive immunoassay (Stressgen, Ann Arbor, MI). It is based on the analysis of PKC activity in the solution phase and on ELISA that utilizes a synthetic peptide as a substrate for PKC and a polyclonal antibody that recognizes the phosphorylated form of the substrate. Briefly, readily phosphorylated PKC (substrate) was pre-coated on the wells of microtiter plates. Cellular extract (10 μg) were added to the appropriate wells, followed by the addition of ATP to initiate the reaction. The kinase reaction was terminated, and a phospho-specific substrate antibody was added to the wells that specifically recognizes the phosphorylated peptide substrate bound by a peroxidase-conjugated secondary antibody. Color was developed with tetramethylbenzidine substrate and was proportional to PKC phosphotransferase activity. The reaction was stopped with acid stop solution, and the intensity of the color was measured in a microplate reader at 450 nm. Relative PKC activity in the cell lysates was calculated and expressed as absorbance per milligram protein. The intra- and interassay coefficient of variation was <10%. This assay is specific for all isoforms of PKC.
NF-κB p65 activity assays
We examined NF-κB p65 activity in nuclear extracts of THP-1 cells treated with HG. Nuclear extracts were prepared as described previously (42), and NF-κB p65 activity was determined using reagents from Active Motif (Carlsbad, CA). Briefly, 5 μg of nuclear extract were added to each NF-κB consensus-binding site oligonucleotide-precoated well. A primary antibody specific for an epitope (p65) on the bound and active form of the transcription factor was then added, followed by subsequent incubation with secondary antibody. Color was developed with the use of developing solution and stopped. Absorbance was read at 450 nm and normalized to milligram cell protein. The intra- and interassay coefficient of variation for these assays was <7%.
Mechanistic studies were confirmed in monocytes from healthy controls, after obtaining informed consent, on a University of California Davis Institutional Review Board-approved protocol.
Data analysis
All experiments were performed on at least five occasions in duplicate or triplicate. Experimental results are presented as means ± SD. Paired t-tests and ANOVA were used for data analyses, and significance was defined as P < 0.05.
RESULTS
IL-1β secreted protein, cellular protein, and mRNA increases under HG conditions are time and dose dependent
THP-1 cells secreted significant amounts of IL-1β in the presence of HG (5.5 ± 0.9 pg/mg protein at 15 mM, 7.4 ± 1 pg/mg protein at 25 mM) compared with in the presence of NG (2.5 ± 0.1 pg/mg protein; P < 0.05). This increase in IL-1β secretion was time dependent, with significant levels secreted after 48 h at both 15 and 25 mM glucose (Fig. 1, A and B). Addition of 9.5 mM mannitol to NG did not result in increased IL-1β secretion, suggesting that the glucose-induced increase was not an osmotic effect.
Fig. 1.
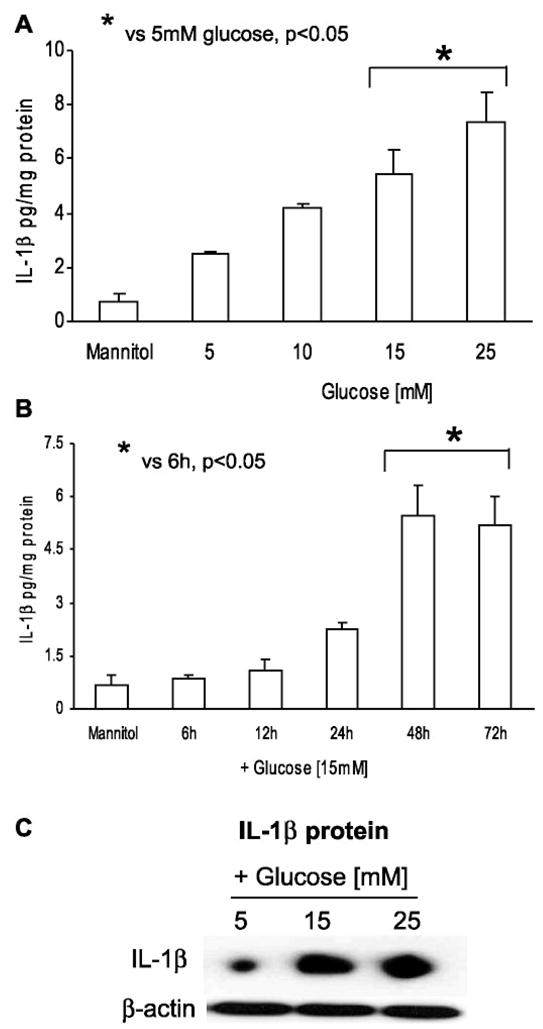
A: dose-dependent effect of high glucose on IL-1β release from THP-1 cells. Cells were challenged with increasing glucose concentration (10–25 mmol/l) for 48 h, and IL-1β release was measured as described in MATERIALS AND METHODS. As a control, 9.5 mmol/l mannitol was added with normal glucose (NG) in simultaneous wells (n = 5). *P < 0.05 vs. 5 mM glucose. B: time-dependent effect of high glucose on IL-1β release from THP-1 cells. Cells were challenged with 15 mmol/l glucose for 6–72 h, and IL-1β release was measured as described in MATERIALS AND METHODS. As a control, 9.5 mmol/l mannitol was added with NG in simultaneous wells (n = 5). *P < 0.05 vs. 6 h. C: effect of high glucose on cellular IL-1β protein. Total cell lysates of THP-1 cells were run on Tris-glycine gels and then blotted for IL-1β protein (n = 3).
To further confirm the stimulation of IL-1β levels under HG conditions, intracellular IL-1β protein was quantitated by Western blot. Figure 1C shows a significant increase in IL-1β protein level under HG conditions with no change observed in β-actin levels.
Real-time RT-PCR was used to quantitate the IL-1β mRNA. The level of the IL-1β mRNA was normalized to GAPDH. Figure 2A shows that IL-1β-to-GAPDH mRNA ratio increased with both 15 and 25 mM glucose compared with that shown at 5 mM (2.7 ± 0.3, 3 ± 0.2 vs. 1.6 ± 0.1; P < 0.05). Incubation of THP-1 cells in 15 mM glucose resulted in significant increase in IL-1β mRNA expression, with maximum increase at 24 h (Fig. 2B). Because of the maximal increase in IL-1β expression at 48-h incubation with HG, all subsequent experiments were conducted for this duration.
Fig. 2.

A: dose-dependent effect of high glucose on IL-1β mRNA from THP-1 cells. Cells were challenged with increasing glucose concentration (10–25 mmol/l) for 48 h, and real-time RT-PCR analysis was performed to detect IL-1β and GAPDH mRNA, using sequence-specific primers as described in MATERIALS AND METHODS. As a control, 9.5 mmol/l mannitol was added with NG in simultaneous wells (n = 5). *P < 0.05 vs. 5 mM glucose. B: time-dependent effect of high glucose on IL-1β mRNA from THP-1 cells. Cells were challenged with 15 mmol/l glucose for 6–72 h, and real-time RT-PCR analysis was performed to detect IL-1β and GAPDH mRNA, using sequence-specific primers as described in MATERIALS AND METHODS. As a control, 9.5 mmol/l mannitol was added with NG in simultaneous wells (n = 5). *P < 0.05 vs. 5 mM glucose.
IL-1ra secretion in human monocytes under HG conditions is time and dose dependent
Incubation of THP-1 cells in glucose (5–25 mM) resulted in significant increase in IL-1ra levels. THP-1 cells secreted significant amounts of IL-1ra in the presence of HG (15 and 25 mM) compared with that shown in NG (37 ± 2 and 36 ± 4 vs. 14 ± 1 pg/mg protein, respectively; P < 0.05; Fig. 3A). This increase in IL-1ra release with HG (15 and 25 mM) was time dependent, similar to the IL-1β expression pattern, with a considerable decrease at 72 h; however, no significant changes in IL-1ra levels were noticed at 6, 12, and 24 h (Fig. 3B).
Fig. 3.

A: dose-dependent effect of high glucose on IL-1 receptor antagonist (IL-1ra) release from THP-1 cells. Cells were challenged with increasing glucose concentration (10–25 mmol/l) for 48 h, and IL-1ra release was measured as described in MATERIALS AND METHODS. As a control, 9.5 mmol/l mannitol was added with NG in simultaneous wells (n = 5). *P < 0.05 vs. 5 mM glucose B: time-dependent effect of high glucose on IL-1ra release from THP-1 cells. Cells were challenged with 15 mmol/l glucose for 6–72 h, and IL-1ra release was measured as described in MATERIALS AND METHODS. As a control, 9.5 mmol/l mannitol was added with NG in simultaneous wells (n = 5). *P < 0.05 vs. 6 h. C: effect of high glucose on IL-1β-to-IL-1ra molar ratio in THP-1 cells. Cells incubated with high-glucose concentration show a higher IL-1β-to-IL-1ra molar ratio than those incubated with NG (n = 5).
IL-1β-to-IL-1ra ratio is increased under HG conditions in human monocytes
Because IL-1β is proinflammatory and IL-1ra is anti-inflammatory, we calculated the IL-1β-to-IL-1ra molar ratio for THP-1 cells treated with HG (15 and 25 mM) at 48 h in the supernatants. Thus high molar IL-1β/IL-1ra would indicate inflammation. THP-1 cells incubated with HG showed higher IL-1β/IL-1ra (0.3 ± 0.01 and 0.23 ± 0.02) with 15 and 25 mM compared with NG (0.1 ± 0.01 with 5 mM glucose; P < 0.05; Fig. 3C). Data suggested that HG caused significant production of IL-1β and resulted in an imbalance in the IL-1β/IL-1ra, leading to increased inflammation.
HG-induced IL-1β protein release is attenuated with PKC inhibition
Next, to investigate the HG-mediated mechanistic events in THP-1 cells, we used two strategies: 1) specific pharmacological inhibitors and 2) siRNA technology.
First, we examined the role of PKC in IL-1β release from THP-1 cells under HG. The pan-specific PKC inhibitor calphostin C resulted in significant decrease in IL-1β (from 6.9 ± 0.9 to 1.8 ± 0.1 pg/mg protein at 15 mM; P < 0.05) release from monocytes (Fig. 4A). Next, we determined which isoform of PKC mediates IL-1β release from human monocytes under HG, using isotype selective PKC-α/γ inhibitor HBDDE and the specific PKC-βII inhibitor LY-379196. HBBDE significantly decreased IL-1β release induced with HG (from 6.9 ± 0.9 to 0.6 ± 0.1 pg/mg protein at 15 mM; P < 0.05), whereas LY-379196 did not have an effect (from 6.9 ± 0.9 to 5.5 ± 1 pg/mg protein at 15 mM) (Fig. 4A).
Fig. 4.

A: effect of PKC isoform inhibitors on IL-1β release from THP-1 cells under high glucose. Cells were cultured as described in Fig. 1 except for exposure to calphostin C (10 μmol/l), 2,2′,3,3′,4,4′-hexahydroxy-1,1′-biphenyl-6,6′-dimethanol dimethyl ether (HBDDE; 50 μmol/l), and LY-379196 (30 nmol/l) for 2 h followed by 15 mmol/l glucose challenge for 48 h. IL-1β release was then measured as described in MATERIALS AND METHODS (n = 5). *P < 0.05 vs. control (15 mmol/l). B: effect of small interfering RNAs (siRNAs) to PKC isoforms on IL-1β release from THP-1 cells under high glucose. Cells were transfected with PKC-α, PKC-β, and scrambled (S) siRNAs. After 48 h, cells were challenged with 15 mmol/l glucose. IL-1β release was then measured as described in MATERIALS AND METHODS (n = 5). *P < 0.05 vs. control (scrambled siRNA + 15 mmol/l glucose).
Second, to further clarify the role of PKC-α and PKC-β, we used sequence-specific siRNAs to confirm the inhibitor data. As shown in Fig. 4B, siRNA to PKC-α significantly decreased HG-induced IL-β release compared with scrambled siRNA controls (at 15 mM, 5.9 ± 1.7 to 2.8 ± 1.1 pg/mg protein; P < 0.05) from monocytes under HG. However, the addition of siRNA to PKC-β did not have any effect on IL-β (at 15 mM, 5.9 ± 1.7 to 6.8 ± 3.7 pg/mg protein).
HG-induced IL-1β protein release is abrogated with p38 and ERK1/2 MAPK inhibition
To assess the involvement of p38 and ERK1/2 in HG stimulation of IL-1β expression and protein release, we used the same two strategies.
First, cells were incubated with a specific inhibitor of p38 MAPK (SB-203580; 10 μmol/l) or ERK1/2 (U-0126; 10 μmol/l). SB-203580 is a selective inhibitor of p38, and U-0126 is a potent inhibitor of dual-specificity MEK immediately upstream from ERK1/2. Western blot analyses confirmed increased phosphorylation of p38 and ERK1/2 in THP-1 cell lysates under HG conditions (Fig. 5, A and B), and addition of inhibitors abrogated HG-enhanced p38 and ERK1/2 phosphorylation (Fig. 5, C and D).
Fig. 5.

A and B: effect of high glucose on p38 MAPK and ERK phosphorylation in THP-1 cells. After cells were cultured with high glucose, cells were lysed, and cell lysates were blotted for total and phosphorylated p38 (pp38) and total and phosphorylated ERK (pERK) (n = 3). C and D: effect of inhibitors (SB-203580 and U-0126) on p38 and ERK phosphorylation in THP-1 cells. After cells were cultured with inhibitors and high glucose, cells were lysed, and cell lysates were blotted for total and phosphorylated p38 and total and phosphorylated ERK (n = 3).
We also found that both p38 and ERK inhibitors significantly decreased IL-1β release from THP-1 cells (Fig. 6A), but the JNK inhibitor (LJNKI1; 5 μmol/l) did not have any significant effect (6.9 ± 0.8 to 8.2 ± 1 pg/mg protein at 15 mM).
Fig. 6.

A: effect of MAPK inhibitors on IL-1β release from THP-1 cells under high glucose. Cells were cultured as described in Fig. 1 except for exposure to SB-203580 (5 μmol/l), U-0126 (10 μmol/l), and LJNKI1 (JNK; 5 μmol/l) for 2 h followed by 15 mmol/l glucose challenge for 48 h. IL-1β release was then measured as described in MATERIALS AND METHODS (n =5). *P < 0.05 vs. control (15 mmol/l). B: effect of siRNAs to p38 and ERK1/2 on IL-1β release from THP-1 cells under high glucose. Cells were transfected with p38, ERK1/2, and scrambled siRNAs. After 48 h, cells were challenged with 15 mmol/l glucose. IL-1β release was then measured as described in MATERIALS AND METHODS (n = 5). *P < 0.05 vs. control (scrambled siRNA + 15 mmol/l glucose).
Second, using siRNA smart pools for p38 and ERK1/2 (Dharmacon), we suppressed p38 and ERK1/2 activity (data not shown) and measured IL-1β release from THP-1 cells under HG conditions. Figure 6B shows a significant decrease in IL-1β release from THP-1 cells, suggesting that both p38 and ERK1/2 activation is required.
IL-1β release under HG conditions is abolished with NF-κB inhibition
Transcription factor NF-κB is important for IL-1β gene expression. We examined the role of NF-κB in HG stimulation of IL-1β protein production in THP-1 cells preincubated with NF-κB activation inhibitors CAPE and BAY 11-7085. CAPE (5 μmol/l) is a potent and specific inhibitor of NF-κB activation that prevents translocation of the p65 subunit of NF-κB and its binding to the DNA. BAY 11-7085 (5 μmol/l) is an inhibitor of IκBα phosphorylation. Both inhibitors significantly blocked IL-1β protein compared with 15 mM glucose (6.9 ± 0.8 to 1.9 ± 0.1 pg/mg protein with CAPE and 2 ± 0.3 pg/mg protein for BAY 11-7085; P < 0.05; Fig. 7A).
We used NF-κB siRNA to further validate the inhibitor data. Figure 7B shows a significant decrease in IL-1β secretion at 15 mM glucose compared with scrambled control (P < 0.05).
Fig. 7.

A: effect of NF-κB inhibitors on IL-1β release from THP-1 cells under high glucose. Cells were cultured as described in Fig. 1 except for exposure to caffeic acid phenethyl ester (CAPE; 5 μmol/l) and BAY-11-7085 (10 μmol/l) for 2 h followed by 15 mmol/l glucose challenge for 48 h. IL-1β release was then measured as described in MATERIALS AND METHODS (n = 5). *P < 0.05 vs. control (15 mmol/l). B: effect of siRNAs to NF-κB on IL-1β release from THP-1 cells under high glucose. Cells were transfected with NF-κB and scrambled siRNAs. After 48 h, cells were challenged with 15 mmol/l glucose. IL-1β release was then measured as described in MATERIALS AND METHODS (n = 5). *P < 0.05 vs. control (scrambled siRNA + 15 mmol/l glucose).
NAPDPH oxidase inhibitors and p47phox siRNA decrease IL-1β secretion in THP-1 cells under HG conditions
We have previously shown that monocytes release superoxide under hyperglycemic conditions because of activation of NADPH oxidase by PKC-α (10, 47). Thus we examined whether the increased superoxide anion released under HG could result in increased IL-1β secretion. To delineate the NADPH oxidase activation in IL-1β release, we used NADPH oxidase inhibitors apocyanin (specific inhibitor of the plasma membrane NADPH oxidase; 30 μmol/l) and DPI (inhibitors of flavin enzymes that reduce activities of NADPH-dependent oxidase; 10 μmol/l). Both inhibitors significantly decreased IL-1β (Fig. 8A) compared with 15 mM glucose alone.
To further validate the role of NADPH oxidase, we investigated the effect of inhibiting p47phox (an essential cytosolic subunit of NADPH oxidase that translocates to membrane and results in its activation) (2) using specific siRNAs. IL-1β secretion decreased significantly under HG conditions in cells transfected with p47phox siRNA compared with scrambled siRNA-transfected controls (at 15 mM, 5.9 ± 1.7 to 0.5 ± 0.7 pg/mg protein; P < 0.05; Fig. 8B).
Fig. 8.
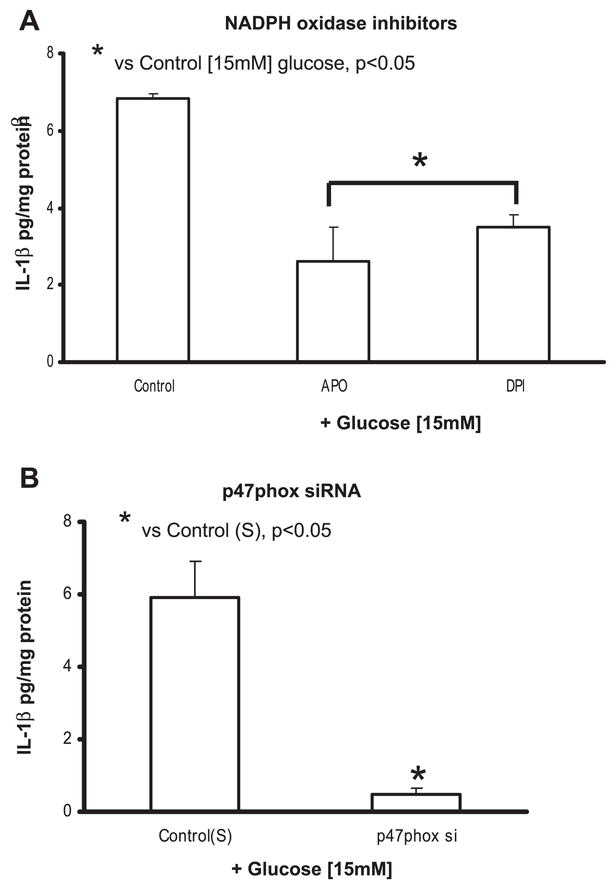
A: effect of NADPH oxidase inhibitors on IL-1β release from THP-1 cells under high glucose. Cells were cultured as described in Fig. 1 except for exposure to apocyanin (Apo; 30 μmol/l) and diphenyleneiodonium chloride (DPI; 10 μmol/l) for 2 h followed by 15 mmol/l glucose challenge for 48 h. IL-1β release was then measured as described in MATERIALS AND METHODS (n = 5). *P < 0.05 vs. control (15 mmol/l). B: effect of siRNA to p47phox on IL-1β release from THP-1 cells under high glucose. Cells were transfected with p47phox and scrambled siRNAs. After 48 h, cells were challenged with 15 mmol/l glucose. IL-1β release was then measured as described in MATERIALS AND METHODS (n = 5). *P < 0.05 vs. control (scrambled siRNA + 15 mmol/l glucose).
PKC-α signaling is proximal under HG conditions in THP-1 cells
After the identification of signaling mediators under HG conditions, we sought to determine the proximal signaling mediator via siRNA technology.
First, because PKC is upstream to p38 and ERK1/2, we examined the effect of PKC inhibition on p38 and ERK1/2 phosphorylation. THP-1 cells were transfected with PKC-α/β siRNAs and exposed to HG (15 mM). As assessed by Western blots, p38 and ERK1/2 phosphorylation were significantly decreased (Fig. 9A), indicating that PKC-α is proximal to p38 and ERK1/2.
Fig. 9.
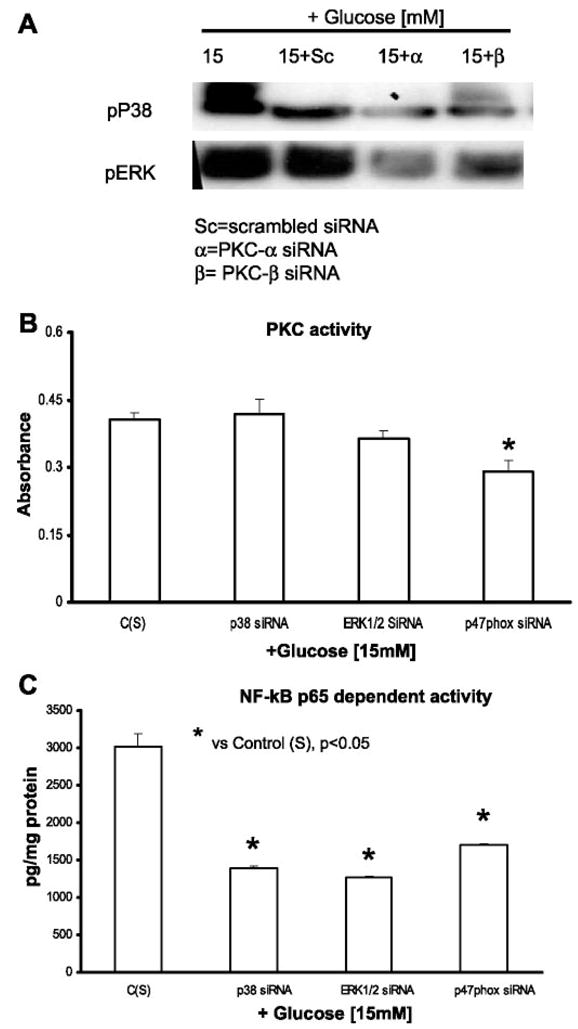
PKC-α is proximal mediator in IL-1β release from THP-1 cells under high glucose. A: cells were transfected with PKC-α, PKC-β, and scrambled siRNAs. After 48 h, cells were challenged with 15 mmol/l glucose. Cells were then lysed, and lysates were blotted for phosphorylated p38 and ERK (n = 3). B: cells were transfected with p38, ERK, p47phox, and scrambled siRNAs. After 48 h, cells were challenged with 15 mmol/l glucose. Cell lysates were then isolated and used for measuring PKC activity as described in MATERIALS AND METHODS (n = 5). *P < 0.05 vs. control (scrambled siRNA + 15 mmol/l glucose). C: cells were transfected with p38, ERK, p47phox, and scrambled siRNAs. After 48 h, cells were challenged with 15 mmol/l glucose. Nuclear extracts were then isolated and used for measuring NF-κB p65-dependent activity, as described in MATERIALS AND METHODS (n = 5). *P < 0.05 vs. control (scrambled siRNA + 15 mmol/l glucose).
Second, we transfected THP-1 cells with siRNA smart pools for p38, ERK1/2, and p47phox and challenged these with HG. PKC activity in cell lysates did not change with p38 or ERK1/2 inhibition (Fig. 9B) but were significantly altered with p47phox inhibition, suggesting that p38 and ERK1/2 are downstream of PKC and that NADPH oxidase-mediated IL-1β release is independent of PKC.
Third, knockdown of p38, ERK1/2, and p47phox resulted in significant inhibition in NF-κB p65 activity (Fig. 9C) in THP-1 cell nuclear extracts, indicating that both MAPK and NADPH oxidase led to a p65-dependent NF-κB pathway activation resulting in IL-1β release.
Thus IL-1β release from human monocytes under HG appears to be mediated by PKC-α, via phosphorylation of p38 MAPK and ERK1/2 leading to NF-κB activation, resulting in increased mRNA and protein for IL-1β. At the same time, it appears that NADPH oxidase activates NF-κB, resulting in increased IL-1β secretion (Fig. 10). Simultaneously, we corroborated the in vitro mechanism of IL-1β release under HG conditions in human monocytes (n = 4 healthy volunteers) using identical assays, including the pharmacological inhibitors, except siRNAs (data not shown).
Fig. 10.
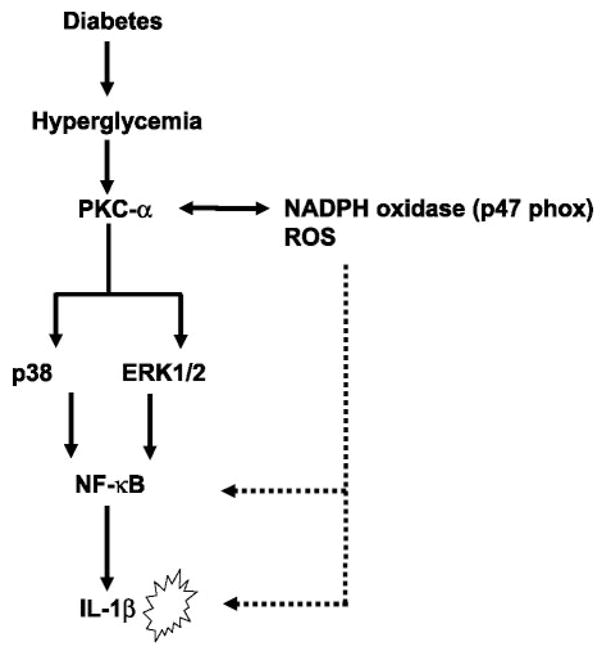
Schema illustrating the mechanism of IL-1β release under high-glucose conditions in THP-1 cells. Inflammatory effects of high glucose are mediated through NADPH oxidase and activation of PKC-α. Activation of PKC-α results in the phosphorylation of p38 MAPK and ERK1/2 and culmination in NF-κB activation, leading to increased IL-1β secretion in monocytes. ROS, reactive oxygen species.
DISCUSSION
In this study, we make the novel observation that levels of inflammatory cytokine IL-1β in human monocytes under HG conditions are increased and also delineated the corresponding signaling events. Our results show that HG treatment (15 and 25 mM) time dependently induces a marked increase in IL-1β and IL-1ra expression and IL-1β-to-IL-1ra molar ratio, and these effects are specific because mannitol had no effect. In this study, we used THP-1 cells (a valid human monocytic cell line), thus confirming our findings in human monocytes. We have presented data showing that the levels of IL-1β, as measured by ELISA, Western blot, and mRNA expression, are increased in the monocytes under HG. These HG-induced changes are similar to those observed in bovine retinal endothelial cells (26), LPS-activated human monocytes (38), human macrophages (44), human pancreatic islets (33), and human aortic endothelial cells (4). All of these studies addressed the role of IL-1β and its downstream biological events with minimal information on upstream events that lead to its release under HG conditions. In this context, we have provided sequential data that human monocytes under HG conditions via NADPH oxidase and PKC-α activation phosphorylate p38 MAPK and ERK1/2, culminating in NF-κB transactivation leading to significant IL-1β secretion, thus elaborating the steps that potentiate increased production of IL-1β seen in diabetes.
IL-1β is produced by a variety of inflammatory cells that then release a cascade of inflammatory signals (12). Several lines of evidence suggest that IL-1β is proatherogenic (6, 8, 14, 30, 34, 40). IL-1β appears to have procoagulant activity, stimulates monocytes-endothelial cell adhesion and cholesterol esterification in macrophages, and stimulates smooth muscle cell proliferation by platelet-derived growth factor (6, 8, 14, 30, 34, 40). IL-1β protein has been demonstrated in coronary arteries of patients with ischemic heart disease vs. that shown with nonischemic cardiomyopathy and has been shown to correlate with the severity of atherosclerosis (15). Kirii et al. (24) showed that the lack of IL-1β decreased the severity of atherosclerosis. Ghanim et al. (16) showed that, in obesity, an insulin-resistant prediabetic state, mononuclear cell NF-κB and proinflammatory cytokine levels were increased (16). Because type 2 diabetes mellitus manifests obesity and hyperglycemia, it is conceived that the proinflammatory state is accentuated. Monocytes are classic phagocytes that are crucial and the most readily accessible cell in the artery wall, contributing substantially to diabetes and atherosclerosis (18). Here, we provide evidence that IL-1β-to-IL-1β/IL-1ra ratios are increased in the monocytes under HG conditions. Furthermore, recent data from our group (11) have shown that monocyte IL-1β in diabetic subjects is increased compared with that shown in control subjects.
Diabetes and cells cultured under HG conditions are associated with increases in PKC activity (10, 47). Our group (47) has previously shown that superoxide release from monocytes under HG is mediated via activation of PKC-α. In contrast to our previous data on reactive oxygen species inhibitors failing to affect IL-6 release from monocytes under HG (10), in the present study, DPI and apocyanin significantly decreased IL-1β secretion. These findings suggest that there are different mechanisms mediating proinflammatory cytokine release by excess production under HG conditions. We then examined the effect of PKC inhibitors on IL-1β release from THP-1 cells under HG. The PKC inhibitor calphostin C significantly decreased IL-1β release from monocytes under HG conditions. Because different PKC isoforms are activated in different tissues, we tested whether PKC-α/β isoforms are involved in monocyte IL-1β release under HG conditions using inhibitors for PKC-α/β isoforms (HBDDE) and for PKC-β2 (LY-379196). HBDDE decreased HG-induced IL-1β release, whereas LY-379196 had no effect; different from this observation is one made by Koya and King (27) that PKC-β2 isoform mediates glucose-induced vascular dysfunction in streptozotocin-induced diabetic rats. siRNA technology was used to further confirm our inhibitor data. PKC-α siRNA significantly decreased HG-induced IL-1β release, whereas PKC-β siRNA had minimal effect, corroborating inhibitor studies. This suggests that monocytic IL-1β secretion is mediated via PKC-α and not via PKC-β under HG, since PKC-α siRNA transfection effectively ameliorated IL-1β secretion, whereas PKC-β siRNA had no effect.
HG has been shown to activate p38 MAPK in vascular smooth muscle cells (21). In addition, production of inflammatory cytokines such as TNF and IL-6 by activated rat smooth muscle cells was regulated by the p38 MAPK pathway (50). Also, activation of PKC could directly activate the MAPK pathway leading to altered gene expression. Igarashi et al. (21) showed that p38 MAPK and not JNK was activated by HG in smooth muscle cells. In this study, specific inhibitors of p38 MAPK and ERK significantly decreased IL-1β release from THP-1 cells under HG. The JNK inhibitor did not influence IL-1β release. Western blot analysis of PKC-α and -β siRNA-transfected cell lysates revealed that PKC-mediated signaling is proximal to p38 and ERK1/2, which is indicated by reduction in their phosphorylation with PKC-α knockdown only. Inhibition of p38 MAPK and ERK1/2 significantly decreased IL-1β release from human monocytes under HG, suggesting that PKC-α-mediated signaling is proximal to p38 and ERK1/2 activation.
HG levels upregulate expression of transcription factors like NF-κB and activating protein-1. NF-κB mediates the expression of many genes (e.g., IL-1β, IL-6, TNF-α) whose promoters are regulated through complex interactions (5, 29). We show using specific inhibitors of NF-κB that IL-1β release from monocytes in HG can be ameliorated. Data provided here suggest that NF-κB mediates IL-1β secretion in monocytes and is accentuated under HG conditions. Our data are further supported by previously reported work by Aljada et al. (1) and Hofmann et al. (1, 20), who separately showed that hyperglycemia induces activation of NF-κB in ex vivo-isolated peripheral blood mononuclear cells after HG ingestion by healthy human volunteers and patients with type 1 diabetes, respectively.
Increased oxidative stress in diabetes may be mediated via the mitochondrial electron transport chain, activation of phagagocyte NADPH oxidase, or glycoxidation via interactions with advanced glycation end-product receptors. Several laboratories have shown that PKC-dependent signaling is involved in the activation of NADPH oxidase and production in neutrophils (23, 37). Our group and others have previously shown that HG activates monocytes and endothelial cells to produce reactive oxygen species (11). Our group (10) recently showed that IL-6 levels are increased in hyperglycemic conditions and that this increase was mediated through increased reactive oxygen species and activation of PKC-α/β, leading to increased IL-6 secretion via NF-κB (10). Our group and others have also shown that p47phox, an essential subunit of NADPH oxidase (translocation to membranes), is involved in monocytic release under HG conditions (2, 10, 36, 40, 44, 47). Hence, we studied NADPH oxidase and p47phox inhibition on HG-induced IL-1β release using specific inhibitors and siRNA technology. Our results confirmed that inhibition of NADPH oxidase in monocytes reduced IL-1β secretion under HG conditions.
Increased oxidative stress under HG activates p38 MAPK in endothelial and U937 cells, leading to IL-8 and TNF-α production via NF-κB (22, 45). Present findings further emphasize the fact that many biochemical pathways are involved in hyperglycemia that contribute to increased proinflammatory cytokine release principle to cellular damage associated with diabetes and also dependent on the cells exposed (41, 49). We definitively show in the present study the involvement of PKC-α/p38 and NF-κB pathway leading to increased IL-1β release by monocytes under HG conditions. Our data add to earlier findings that the IL-1β and NF-κB pathways are detrimental in the pathogenesis of glucotoxicity in diabetic human pancreatic islets (33). Furthermore, these data support the findings of Kowluru et al. (26), implicating IL-1β in diabetic retinopathy via inducing apoptosis and NF-κB activation in hyperglycemia.
The important mechanistic finding of our studies is that inflammatory effects of HG are mediated through NADPH oxidase and activation of PKC-α. Activation of PKC-α results in the phosphorylation of p38 MAPK and ERK1/2, culminating in NF-κB activation and leading to increased IL-1β secretion in monocytes (as shown in Fig. 10). The elucidation of mechanisms of IL-1β release under HG conditions is significant in terms of its relationship to diabetic retinopathies and β-cell toxicity. Targeting these IL-1β-mediated pathways could result in the amelioration of inflammation and possibly diabetic vasculopathies.
Acknowledgments
GRANTS
Grant support from the National Institutes of Health (K24 AT-00596 and R21 DK-69801) and the American Diabetes Association is gratefully acknowledged.
References
- 1.Aljada A, Friedman J, Ghanim H, Mohanty P, Hofmeyer D, Chaudhuri A, Dandona P. Glucose ingestion induces an increase in intranuclear NF-κB, a fall in cellular inhibitor κB, and an increase in TNF-alpha mRNA by mononuclear cells in healthy human subjects. Metabolism. 2006;55:1177–1185. doi: 10.1016/j.metabol.2006.04.016. [DOI] [PubMed] [Google Scholar]
- 2.Aljada A, Ghanim H, Dandona P. Translocation of p47phox and activation of NADPH oxidase in mononuclear cells. Methods Mol Biol. 2002;196:99–103. doi: 10.1385/1-59259-274-0:99. [DOI] [PubMed] [Google Scholar]
- 3.Arondel J, Singer M, Matsukawa A, Zychlinsky A, Sansonetti PJ. Increased interleukin-1 (IL-1) and imbalance between IL-1 and IL-1 receptor antagonist during acute inflammation in experimental Shigellosis. Infect Immun. 1999;67:6056–6066. doi: 10.1128/iai.67.11.6056-6066.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Asakawa H, Miyagawa J, Hanafusa T, Kuwajima M, Matsuzawa Y. High glucose and hyperosmolarity increase secretion of interleukin-1β incultured human aortic endothelial cells. J Diabetes Complications. 1997;11:176–179. doi: 10.1016/s1056-8727(97)00004-4. [DOI] [PubMed] [Google Scholar]
- 5.Baeuerle PA. The inducible transcription activator NF-κB: regulation by distinct protein subunits. Biochim Biophys Acta. 1991;1072:63–80. doi: 10.1016/0304-419x(91)90007-8. [DOI] [PubMed] [Google Scholar]
- 6.Bevilacqua MP, Pober JS, Majeau GR, Cotran RS, Gimbrone MA. Interleukin 1 (IL-1) induces biosynthesis and cell surface expression of procoagulant activity in human vascular endothelial cells. J Exp Med. 1984;163:1595–1600. doi: 10.1084/jem.160.2.618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bevilacqua M, Gimbrone M. Inducible cell functions in inflammation and coagulation. Semin Thromb Hemost. 1987;138:425–433. doi: 10.1055/s-2007-1003519. [DOI] [PubMed] [Google Scholar]
- 8.Bevilacqua MP, Pober JS, Wheeler ME, Cotran RS, Gimbrone MA. Interleukin 1 acts on cultured human vascular endothelium to increase the adhesion of polymorphonuclear leukocytes, monocytes, and related leukocyte cell lines. J Clin Invest. 1985;76:2003–2008. doi: 10.1172/JCI112200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brownlee M, Lilly Lecture. Glycation and diabetic complications. Diabetes. 1993;43:826–841. doi: 10.2337/diab.43.6.836. 1994. [DOI] [PubMed] [Google Scholar]
- 10.Devaraj S, Venugopal SK, Singh U, Jialal I. Hyperglycemia induces monocytic release of interleukin-6 via induction of protein kinase C-α and -β. Diabetes. 2005;54:85–91. doi: 10.2337/diabetes.54.1.85. [DOI] [PubMed] [Google Scholar]
- 11.Devaraj S, Glaser N, Griffen S, Wang-Polagruto J, Miguelino E, Jialal I. Increased monocytic activity and biomarkers of inflammation in patients with type 1 diabetes. Diabetes. 2006;55:774–779. doi: 10.2337/diabetes.55.03.06.db05-1417. [DOI] [PubMed] [Google Scholar]
- 12.Dinarello CA. Interleukin-1β. Crit Care Med. 2005;33:S460–S462. doi: 10.1097/01.ccm.0000185500.11080.91. [DOI] [PubMed] [Google Scholar]
- 13.Donahue R, Abbott R, Reed D, Yano K. Postchallenge glucose concentration and coronary heart disease in men of Japanese ancestry. Diabetes. 1987;36:689–692. doi: 10.2337/diab.36.6.689. [DOI] [PubMed] [Google Scholar]
- 14.Eizirik DL, Mandrup-Poulsen T. A choice of death-the signal-transduction of immune-mediated β-cell apoptosis. Diabetologia. 2001;44:2115–2133. doi: 10.1007/s001250100021. [DOI] [PubMed] [Google Scholar]
- 15.Galea J, Armstrong J, Gadsdon P, Holden H, Francis SE, Holt CM. IL-1β in coronary arteries patients with ishcemic heart disease. Arterioscler Thromb Vasc Biol. 1996;16:1000–1006. doi: 10.1161/01.atv.16.8.1000. [DOI] [PubMed] [Google Scholar]
- 16.Ghanim H, Aljada A, Hofmeyer D, Syed T, Mohanty P, Dandona P. Circulating mononuclear cells in the obese are in a proinflammatory state. Circulation. 2004;110:1564–1571. doi: 10.1161/01.CIR.0000142055.53122.FA. [DOI] [PubMed] [Google Scholar]
- 17.Hansson GK. Inflammation, atherosclerosis, and coronary disease. N Engl J Med. 2005;352:1685–1695. doi: 10.1056/NEJMra043430. [DOI] [PubMed] [Google Scholar]
- 18.Hill H, Hogan N, Rallison M, Santo JI, Charette RP, Kitahara M. Functional and metabolic abnormalities of diabetic monocytes. Adv Exp Med Biol. 1980;69:621–628. doi: 10.1007/978-1-4684-8088-7_61. [DOI] [PubMed] [Google Scholar]
- 19.Hiramatsu K, Rosen H, Heinecke J, Wolfbauer G, Chait A. Superoxide initiates oxidation of low density lipoprotein by human monocytes. Arteriosclerosis. 1987;7:55–60. doi: 10.1161/01.atv.7.1.55. [DOI] [PubMed] [Google Scholar]
- 20.Hofmann MA, Schiekofer S, Kanitz M, Klevesath MS, Joswig M, Lee V, Morcos M, Tritschler H, Ziegler R, Wahl P, Bierhaus A, Nawroth PP. Insufficient glycemic control increases NF-κB binding activity in peripheral blood mononuclear cells isolated from patients with type 1 diabetes. Diabetes Care. 1998;21:1310–1316. doi: 10.2337/diacare.21.8.1310. [DOI] [PubMed] [Google Scholar]
- 21.Igarashi M, Wakasaki H, Takahara N, Ishii H, Jiang ZY, Yamauchi T, Kuboki K, Meier M, Rhodes CJ, King GL. Glucose or diabetes activates p38 mitogen-activated protein kinase via different pathways. J Clin Invest. 1999;103:185–195. doi: 10.1172/JCI3326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jain SK, Kannan K, Lim G, McVie R, Bocchini JA., Jr Hyperketonemia increases tumor necrosis factor-α secretion in cultured U937 monocytes and Type 1 diabetic patients and is apparently mediated by oxidative stress and cAMP deficiency. Diabetes. 2002;51:2287–2293. doi: 10.2337/diabetes.51.7.2287. [DOI] [PubMed] [Google Scholar]
- 23.Kadri-Hassani N, Leger CL, Descomps B. The fatty acid bimodal action on superoxide production by human adherent monocytes under phorbol 12-myristate 13-acetate or diacylglycerol activation can be explained by the modulation of protein kinase C and p47phox translocation. J Biol Chem. 1995;270:15111–15118. doi: 10.1074/jbc.270.25.15111. [DOI] [PubMed] [Google Scholar]
- 24.Kirii H, Niwa T, Yamada Y, Wada H, Saito K, Iwakura Y, Asano M, Moriwaki H, Seishima M. Lack of interleukin-1β decreases the severity of atherosclerosis in ApoE-deficient mice. Arterioscler Thromb Vasc Biol. 2003;23:656–660. doi: 10.1161/01.ATV.0000064374.15232.C3. [DOI] [PubMed] [Google Scholar]
- 25.Kitahara M, Eyre H, Lynch R, Rallison ML, Hill HR. Metabolic activity of diabetic monocytes. Diabetes. 1980;29:251–256. doi: 10.2337/diab.29.4.251. [DOI] [PubMed] [Google Scholar]
- 26.Kowluru RA, Odenbach S. Role of interleukin-1β in the pathogenesis of diabetic retinopathy. Br J Ophthalmol. 2004;88:1343–1347. doi: 10.1136/bjo.2003.038133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Koya D, King GL. Protein kinase C activation and the development of diabetic complications. Diabetes. 1998;47:859–866. doi: 10.2337/diabetes.47.6.859. [DOI] [PubMed] [Google Scholar]
- 28.Kuusisto J, Mykkanen L, Pyorala K, Laakso M. NIDDM and its metabolic control predict coronary heart disease in elderly subjects. Diabetes. 1994;43:960–967. doi: 10.2337/diab.43.8.960. [DOI] [PubMed] [Google Scholar]
- 29.Lenardo MJ, Baltimore D. NF-κB: a pleiotropic mediator of inducible and tissue-specific gene control. Cell. 1989;58:227–229. doi: 10.1016/0092-8674(89)90833-7. [DOI] [PubMed] [Google Scholar]
- 30.Libby P, Hansson GK. Involvement of the immune system in human atherogenesis: current knowledge and unanswered questions. Lab Invest. 1991;64:5–15. [PubMed] [Google Scholar]
- 31.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 32.Lyons T. Glycation and oxidation: a role in the pathogenesis of atherosclerosis. Am J Cardiol. 1993;71:26B–31B. doi: 10.1016/0002-9149(93)90142-y. [DOI] [PubMed] [Google Scholar]
- 33.Maedler K, Sergeev P, Ris F, Oberholzer J, Joller-Jemelka HI, Spinas GA, Kaiser N, Halban PA, Donath MY. Glucose-induced beta cell production of IL-1β contributes to glucotoxicity in human pancreatic islets. J Clin Invest. 2002;110:851–860. doi: 10.1172/JCI15318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Maziere C, Barbu V, Auclair M, Maziere JC. Interleukin 1 stimulates cholesterol esterification and cholesterol deposition in J774 monocytes-macrophages. Biochim Biophys Acta. 1996;1300:30–34. doi: 10.1016/0005-2760(95)00232-4. [DOI] [PubMed] [Google Scholar]
- 35.McGill H, Jr, McMahan A, Malcom G, Oalmann MC, Strong JP. Relation of glycohemoglobin and adiposity to atherosclerosis in youth. Pathobiological Determinants of Atherosclerosis in Youths (PDAY) Research Group. Arterioscler Thromb Vasc Biol. 1995;15:431–440. doi: 10.1161/01.atv.15.4.431. [DOI] [PubMed] [Google Scholar]
- 36.Mohanty P, Hamouda W, Garg R, Aljada A, Ghanim H, Dandona P. Glucose challenge stimulates reactive oxygen species (ROS) by leucocytes. J Clin Endocrinol Metab. 2000;85:2970–2973. doi: 10.1210/jcem.85.8.6854. [DOI] [PubMed] [Google Scholar]
- 37.Nishikawa T, Edelstein D, Du XL, Yamagishi S, Matsumura T, Kaneda Y, Yorek MA, Beebe D, Oates PJ, Hammes HP, Giardino I, Brownlee M. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycemic damage. Nature. 2000;404:787–790. doi: 10.1038/35008121. [DOI] [PubMed] [Google Scholar]
- 38.Orlinska U, Newton RC. Role of glucose in IL-1β production by lipopolysaccharide activated human monocytes. J Cell Physiol. 1993;157:201–209. doi: 10.1002/jcp.1041570126. [DOI] [PubMed] [Google Scholar]
- 39.Perrier S, Darakhshan F, Hajduch E. IL-1 receptor antagonist in metabolic diseases: Dr. Jekyll or Mr. Hyde? FEBS Lett. 2006;580:6289–6294. doi: 10.1016/j.febslet.2006.10.061. [DOI] [PubMed] [Google Scholar]
- 40.Raines EW, Dower SK, Ross R. Interleukin-1 mitogenic activity for fibroblasts and smooth muscle cells is due to PDGF-AA. Science. 1989;243:393–395. doi: 10.1126/science.2783498. [DOI] [PubMed] [Google Scholar]
- 41.Ruderman NB, Williamson JR, Brownlee M. Glucose and diabetic vascular disease. FASEB J. 1992;6:2905–2914. doi: 10.1096/fasebj.6.11.1644256. [DOI] [PubMed] [Google Scholar]
- 42.Schreiber E, Matthias P, Müller MM, Schaffner W. Rapid detection of octamer binding proteins with “mini-extracts,” prepared from a small number of cells. Nucleic Acids Res. 1989;17:6419. doi: 10.1093/nar/17.15.6419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shanmugam N, Reddy MA, Guha M, Natarajan R. High glucose-induced expression of proinflammatory cytokine and chemokine genes in monocytic cells. Diabetes. 2003;52:1256–1264. doi: 10.2337/diabetes.52.5.1256. [DOI] [PubMed] [Google Scholar]
- 44.Shashkin PN, Jain N, Miller YI, Rissing BA, Huo Y, Keller SR, Vandenhoff GE, Nadler JL, McIntyre TM. Insulin and glucose play a role in foam cell formation and function. Cardiovasc Diabetol. 2006;20:5–13. doi: 10.1186/1475-2840-5-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Srinivasan S, Bolick DT, Hatley ME, Natarajan R, Reilly KB, Yeh M, Chrestensen C, Sturgill TW, Hedrick CC. Glucose regulates IL-8 production in aortic endothelial cells through activation of the p38 mitogen-activated protein kinase pathway in diabetes. J Biol Chem. 2004;279:31930–31936. doi: 10.1074/jbc.M400753200. [DOI] [PubMed] [Google Scholar]
- 46.Turner R, Millns H, Neil H, Stratton IM, Manley SE, Matthews DR, Holman RR. Risk factors for coronary artery disease in non-insulin dependent diabetes mellitus: UK Prospective Diabetes Study. Br Med J. 1998;316:823–828. doi: 10.1136/bmj.316.7134.823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Venugopal SK, Devaraj S, Yang T, Jialal I. α-Tocopherol decreases superoxide anion release in human monocytes under hyperglycemic conditions via inhibition of protein kinase C-α. Diabetes. 2002;51:3049–3054. doi: 10.2337/diabetes.51.10.3049. [DOI] [PubMed] [Google Scholar]
- 48.Vlassara H, Bucala R, Sturcker L. Pathogenic effects of advanced glycosylation: biochemical, biologic, and clinical implications for diabetes and aging. Lab Invest. 1994;70:138–151. [PubMed] [Google Scholar]
- 49.Wolff S, Jiang ZY, Hunt J. Protein glycation and oxidative stress in diabetes mellitus and aging. Free Radic Biol Med. 1991;10:339–352. doi: 10.1016/0891-5849(91)90040-a. [DOI] [PubMed] [Google Scholar]
- 50.Yamakawa T, Eguchi S, Matsumoto T, Yamakawa Y, Numaguchi K, Miyata I, Reynolds CM, Motley ED, Inagami T. Intracellular signaling in rat cultured vascular smooth muscle cells: roles of nuclear factor-κB and p38 MAP kinase on TNF-α production. Endocrinology. 1999;140:3562–3572. doi: 10.1210/endo.140.8.6914. [DOI] [PubMed] [Google Scholar]