

Abstract
Established tumors suppress anti-tumor immune responses and induce tolerance by incompletely characterized mechanisms, and this phenomenon is an important barrier to tumor immunotherapy. Single vaccination with tumor cells expressing gp96-Ig stimulates robust expansion of tumor-specific CTLs in tumor naïve mice and this expansion is inhibited by established tumors. Interestingly, frequent vaccinations restore anti-tumor immune responses in the presence of established tumors. Syngeneic EG7 tumor bearing mice have heterogeneous responses to frequent vaccination with EG7-gp96-Ig, with 32% complete responders and 68% partial responders. Comparison of responders to non-responders revealed an inverse correlation between tumor-specific CTL expansion in the peripheral blood and tumor size. To identify immune cells and molecules associated with effective anti-tumor immune responses, RT-PCR arrays were performed using cells isolated from the vaccination site. ELISAs, cellular phenotyping and tumor immunohistochemistry were also performed comparing vaccine responders to non-responders. These data demonstrate that upregulation of T-bet, RORγt, IFNγ, CCL8, CXCL9 and CXCL10 at the vaccination site are associated with vaccine-induced anti-tumor immunity. These data correlate with increased CTL expansion in the peripheral blood of responders, increased infiltration of responder tumors by CD8+ cells and IL-17+ cells and decreased infiltration of responder tumors by CD11b+Gr-1+ cells and FoxP3+ cells. Furthermore, serum ELISAs revealed a significant elevation of TGF-β in non-responders as compared to responders. Interestingly, CD8+ T cells isolated from responders and non-responders have equivalent cytotoxic activity in vitro. Taken together, our data suggest that established tumors may escape immunosurveillance by preventing clonal expansion of tumor specific CTL without inducing anergy.
Keywords: immunotherapy, immunosuppression, tolerance, cancer vaccine(s), immunosurveillance
Introduction
The hypothesis that cancer is controlled by the immune system to varying degrees during the life of the host has received substantial support over the last decade (1). These data have supported the concept that immunotherapy of cancer may provide valuable clinical benefits, but also led to the realization that the evolution of many tumors is accompanied by the acquisition of immunosuppressive properties. Recruitment of regulatory cells by tumors, tumor cell downregulation of immune costimulatory molecules or upregulation of immune negative regulatory molecules, preferential polarization toward Th2 responses and secretion of immunosuppressive cytokines by tumors and/or their surrounding stroma are all mechanisms that have been proposed to mediate tumor induced immunosuppression(2–8).
Effective cancer immunotherapy is widely believed to originate with appropriately activated CD8+ cytotoxic T cells (CTL), however these CTLs will not be capable of providing lasting clinical responses if they are circulating in the presence of multiple systemic and locally immunosuppressive cells and factors. There is a lack of studies which provide information regarding the context in which successful immune responses are generated as compared to failed responses for a given therapy, particularly in the therapeutic setting. Such information would provide important insight into which tumor-induced immunosuppressive mechanisms were most important to target.
We have previously demonstrated that replacing the KDEL ER retention sequence of the heat shock protein gp96 with the Fc portion of the IgG1 protein results in the expression of a gp96-Ig fusion protein capable of secreting tumor-specific antigens to the extracellular space and effectively stimulating antigen cross-presentation to CTL (9). Using this model we have demonstrated that immunization with a live tumor cell vaccine expressing gp96-Ig not only leads to CTL mediated rejection of the vaccine cells themselves, but is capable of rejecting established tumors (9, 10). Importantly, the ability of the vaccine to stimulate CTL expansion is significantly inhibited in the presence of an established tumor (10), however this study also demonstrated that more frequent vaccinations were sufficient to retard growth of the established tumors. The effect of frequent vaccinations on CTL expansion was not determined.
In the current study we demonstrate that the response of EG7 tumor bearing mice to frequent vaccinations with gp96-Ig is heterogeneous; with responses ranging from slightly delayed tumor growth to complete tumor rejection. The heterogeneity in anti-tumor responses allowed comparisons between productive and non-productive responses. Using a model system with the tumor specific neo-antigen, ovalbumin (ova), we show that there is an inverse correlation between tumor growth and tumor-specific CTL expansion. To elucidate the mechanisms inhibiting CTL expansion, a comparison of immune cells and products was performed at various sites in both responder and non-responder mice. CTL expansion and tumor rejection coincides with Th1 and Th17 polarizing conditions established at the site of immunization. Importantly, the Th1 polarizing conditions established at the site of immunization correlate with CTL expansion in the peripheral blood and infiltration into a regressing tumor, as well as reduced serum concentrations of CXCL10 and transforming growth factor beta (TGF-β).
Materials and Methods
Mice
Wild-type mice in the C57BL/6 (B6) background (CD45.2+) were obtained from Charles River Laboratories. CD45.1+ SJL mice were purchased from Jackson Laboratories. C57BL/6 OT-I mice were obtained from Dr. M. Bevan (University of Washington School of Medicine, Seattle, WA). GFP-transgenic mice were obtained by permission of the producers(11). All mice were used at 6–12 weeks of age.
Cell lines
EG7, the OVA-transfected EL4 lymphoma line, generously provided by Dr. M. Bevan, was further transfected with the vector pCMG-His containing gp96-Ig as described previously(9). NIH 3T3 cells were transfected with OVA in pAC-neo-OVA (generously provided by Dr. M. Bevan) and with pCMG-His containing gp96-Ig. Cell lines were maintained in IMDM containing 10% FBS, 1 µg/ml gentamycin and the appropriate antibiotics at the indicated concentrations: G418 (1 µg/ml), histidinol (2mM).
Antibodies
Fluorescent Abs were purchased from BD Pharmingen and eBioscience.
Tumor Inoculation
EG7 cells were isolated from log-phase cultures and washed in PBS. One million cells were then injected subcutaneously (s.c.) in the hind-flank of mice in 0.1 ml of PBS.
Purification and adoptive transfer of OT-I cells
GFP-marked OT-I cells (or CD45.2+ OT-I cells) were purified by positive selection with anti-CD8 using magnetic separation (≥95% pure, determined by flow cytometry; Miltenyi Biotec). Three days after tumor inoculation, 106 GFP-OT-I cells (or Ly5.2+ OT-I cells) were adoptively transferred through tail veins of C57BL/6 mice (or Ly5.1+ SJL mice) in 0.15 ml of HBSS.
Immunization
Two days after adoptive transfer of OT-I or GFP-OT-I (experimental day 5), 106 nonirradiated EG7-gp96-Ig cells or control EG7 cells were injected intraperitoneally (i.p.) in 0.1 ml of PBS. For some experiments mice were immunized i.p. with 3T3-OVA-gp96-Ig. Immunizations were repeated as described above on subsequent days as indicated.
Histology and Immunohistochemistry
EG7 tumors were excised between days 10–16 in pairs (progressors and regressors), submerged in OCT freezing compound and flash-frozen in liquid nitrogen. Sections were then cut at 5µm and mounted on glass slides. Sections were fixed and stained as previously described (12). Imaging was performed using a LSM-510 confocal microscope (Zeiss). Acquired images were analyzed using ImageJ (NIH, MacBioPhotonics) software.
RT-PCR
Total RNA was harvested from peritoneal exudate cells (PEC) between days 10–16 using the RNeasy mini-kit (Qiagen). RNA integrity was analyzed using an Agilent Bioanalyzer 2100; RNA with an integrity value ≤0.8 was discarded. cDNA was synthesized using the RT2 First Strand Kit (Superarray). A custom 96-well RT-PCR array was developed (supplemental figure 1), primer sets were synthesized (Sigma) and RT-PCR was performed using the RT2 SYBR green PCR master mix (Superarray) on an Applied Biosciences 7300 PCR platform.
ELISA
IL-1β, CXCL10 and TGF-β levels were determined using the Quantikine ELISA kits (R&D Systems) according to the manufacturer protocol. Antibody pairs for IFNγ (interferon gamma), IL-17 and IL-6 were obtained from BD Pharmingen and were analyzed in Sandwich ELISA assays by coating plates with capture antibody (1µg/ml), followed by loading serially diluted serum samples, biotin-conjugated detection antibodies (1µg/ml) and HRP-Streptavidin (0.5µg/ml, BD Biosciences).
In vitro cytotoxicity assays
Total splenocytes were harvested at the indicated time points and total CD8+ cells were isolated by magnetic bead separation as described above. EG7 target cells were maintained in cell culture according to standard protocols, labeled with PKH2 green fluorescent cell linker (Sigma) according to manufacturer protocols and plated in 96 well plates (104/well). Purified CD8+ cells were then added to the 96 well plates containing the labeled EG7 cells at the indicated ratios and incubated for 4 hours at 37°C. Following incubation, total cells were harvested and labeled with the 7-AAD vital stain (eBioscience) prior to analysis by flow cytometry.
Statistical Analysis
Paired comparisons were performed using the students t-test, multiple analysis was performed using a one-way ANOVA, p values are indicated as necessary.
Results
OT-I expansion correlates with tumor regression
To determine whether frequent vaccinations with gp96-Ig induce CTL expansion in the presence of an established tumor, EG7 tumor bearing mice were adoptively transferred with GFP+ OT-I cells and subsequently vaccinated with EG7-gp96-Ig or 3T3-ova-gp96-Ig cells (Figure 1a and supplementary figure 2). Peripheral blood was withdrawn from the tail at daily intervals and analyzed by flow cytometry for expression of GFP and CD8 (Figure 1b). Although the overall percentage of CD8+ cells out of the total lymphocyte population remains stable (data not shown), the percentage of OT-I cells out of total CD8+ cells increases following several vaccinations with EG7-gp96-Ig. The degree of OT-I expansion is variable between mice, with some animals maintaining OT-I levels in the peripheral blood similar to non-vaccinated control mice.
Figure 1. Tumor-specific CTL expansion correlates with tumor regression.
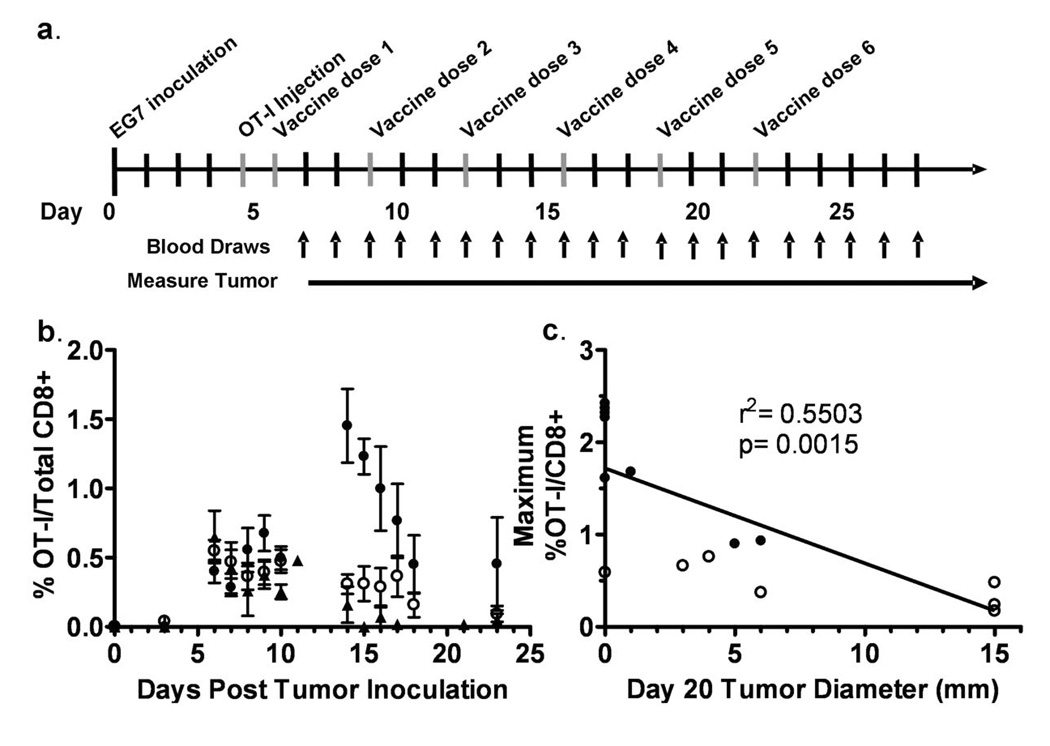
(a) The schedule of EG7 tumor inoculation, OT-I injection and immunizations is shown. (b) The percentage of OT-I cells out of total CD8+ cells in the lymphoid compartment was determined by flow cytometry. Open circles (○) and closed circles (●) indicate EG7-gp96-Ig immunized mice (7 representative mice per group, broken into two groups based on observed dichotomy in OT-I expansion), closed triangles (two representative mice shown) indicate non-vaccinated control mice (▲). (c) The maximum percentage of OT-I cells out of total CD8+ lymphoid cells between experimental days 10–20 is plotted on the y-axis and the tumor diameter at experimental day 20 is plotted on the x-axis. Open and closed circles correspond to the maximum OT-I expansion of mice displayed in (b). A linear regression analysis was performed and found to be statistically significant with p=0.0015 and with a correlation coefficient (r2) of 0.5503, with n=14.
Due to the heterogeneity in OT-I expansion between individual mice, we sought to determine whether there was a correlation between OT-I expansion and overall tumor size. The kinetics of OT-I expansion illustrated in figure 1b suggest that there is a gradual increase in the expansion of OT-I cells following the first three doses of gp96-Ig that peaks near the time of the fourth immunization and then rapidly drops back to baseline by the fifth vaccination. Therefore, to plot the correlation between tumor-specific CTL expansion and tumor size, we plotted the maximum percentage of OT-I cells out of total CD8+ cells achieved between days 10–20 on the y-axis and the tumor diameter at day 20 on the x-axis (Figure 1c). This analysis shows a significant inverse correlation between maximal OT-I expansion in the peripheral blood and tumor size.
The drop in peripheral blood OT-I cells during the third week of the experiment was unexpected. It was possible that a humoral or cellular anti-GFP response was generated which resulted in the rapid clearance of OT-I cells from the blood. To investigate this possibility, we performed experiments as shown in figure 1a using CD45.1+ mice as the tumor bearing mice and adoptively transferred CD45.2+ OT-I cells instead of GFP+ cells. Vaccinations were performed and peripheral blood OT-I cells were monitored by flow cytometry based on the dual expression of CD8 and CD45.2 (supplementary figure 2). GFP negative, CD45.2+ OT-I cells persist for at least 30 days in a CD45.1+ host in the peripheral blood.
Tumor regression is independent of extrinsic tumor-specific CTLs
An effective anti-EG7 tumor response in the absence of adoptively transferred OT-I cells requires that the host immune response prime and expand tumor specific T cells against ova or other uncharacterized EG7 tumor antigens. To address this requirement, experiments were performed as described in figure 1a, in the absence of adoptively transferred OT-I cells. Survival curves comparing vaccination in the absence or presence of OT-I cells are shown in figure 2a. To comply with IACUC regulations, the survival endpoint for these studies was tumor area ≥ 225 mm2. Overall survival was not significantly improved for mice receiving OT-I cells in addition to vaccination compared to mice receiving gp96-Ig vaccination alone (42% and 32%, respectively). These data indicate that immunization with gp96-Ig leads to expansion of endogenous tumor-specific T cell populations capable of mediating tumor rejection.
Figure 2. Extrinsic tumor-specific CTLs are not required for long-term tumor-free survival.
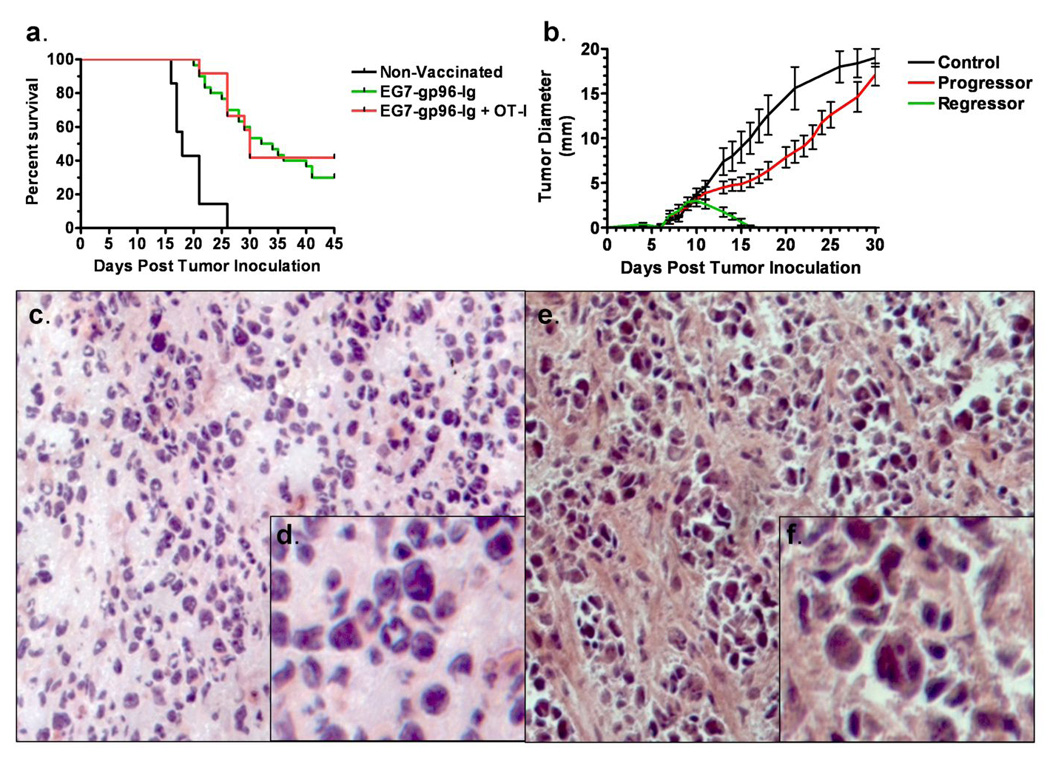
(a) WT mice were inoculated with EG7 tumor and then either adoptively transferred with OT-I cells and vaccinated with EG7-gp96-Ig (red line, n=12), vaccinated with EG7-gp96-Ig without an OT-I adoptive transfer (green line, n=30) or transferred with OT-I cells but not vaccinated (black line, n=7). Vaccinations were performed according to the schedule outlined in figure 1a. To comply with IACUC protocols, the survival endpoint for this study was tumor area ≥ 225 mm2. Median survival for non-vaccinated controls was 18 days, for EG7-gp96-Ig vaccinated was 33 days and for EG7-gp96-Ig vaccinated plus OT-I adoptive transfer was 30 days. Overall survival for the three groups was 0%, 32% and 42%, respectively. (b) WT mice bearing EG7 peripheral tumors were vaccinated as described in figure 1a in the absence of adoptively transferred OT-I cells. Following vaccination two outcomes were observed, with the first group having rapidly regressing tumors ‘regressor’ and the second with slightly delayed tumor growth as compared to controls, ‘progressor’. (c and close-up in d) ‘Progressor’ tumors were excised, sectioned and stained with hematoxylin and eosin. (e and close-up in f) ‘Regressor’ tumors were excised, sectioned and stained with hematoxylin and eosin. Tumors for c–f were excised at day 13 from a paired set of mice in the same experiment.
Responses to vaccination with gp96-Ig are heterogeneous
The observation that tumor specific CTL expansion, tumor size and survival following vaccination with gp96-Ig differs between individual mice suggests that vaccine induced anti-tumor responses are heterogeneous. After vaccinating over 40 mice, it became apparent that the anti-tumor responses to gp96-Ig divided into two groups; those that ‘progressed’ after a transient delay in tumor outgrowth and those that ‘regressed’. The determination between mice that ‘progressed’ or ‘regressed’ was tracked to approximately 10 days post tumor inoculation (figure 2b). After this time, mice bearing tumors that had decreased in diameter (regressors) would eventually become tumor-free and enjoy long-term disease-free survival. Mice bearing ‘progressor’ tumors displayed variable degrees of delayed tumor growth as compared to non-vaccinated controls, with responses ranging from transient tumor size stabilization to slightly delayed tumor growth. Due to this heterogeneity, the possibility arose to compare successful and failed gp96-Ig induced anti-tumor responses. Tumor masses were excised from mice bearing either ‘progressor’ and ‘regressor’ tumors at paired time-points between days 10–16 post tumor inoculation. Subsequent sectioning and staining of these sections revealed that ‘progressor’ tumor masses were relatively homogeneous and contained few small lymphocytes (figures 2c and d), whereas ‘regressor’ tumor masses consisted of focal accumulations of tumor cells and small lymphocytes between large tracts of fibrous extracellular matrix (figures 2e and f).
Tumor regression is associated with expression of Th1 and Th17 polarizing factors at the site of immunization
Because prior studies demonstrated that an established peripheral tumor (in the hind flank) suppressed gp96-Ig induced T cell expansion at the site of vaccination (peritoneal cavity), we compared the transcriptional profiles of peritoneal exudate cells (PECs) from tumor naïve to PECs from EG7 tumor bearing mice. RT-PCR was performed using total RNA extracted from PECs isolated from tumor naïve control mice or from mice bearing EG7 tumors at 10–16 days post tumor inoculation. This analysis revealed surprisingly few differences between the two groups, with only an increase in the expression of IL-1RA, CCR3, CXCL13 and RORγt (retinoid-related orphan receptor gamma) reaching statistical significance (p≤0.05, data not shown). Phenotypic analysis was also performed on PECs from TNM and TBM (Figure 3a), demonstrating that increased fractions of CD11c+ cells are present within the PEC from TBM as compared to TNM. The other cell populations examined appear relatively unchanged.
Figure 3. Tumor regression and CTL expansion occur following the expression of Th1 and Th17 polarizing factors at the site of vaccination.
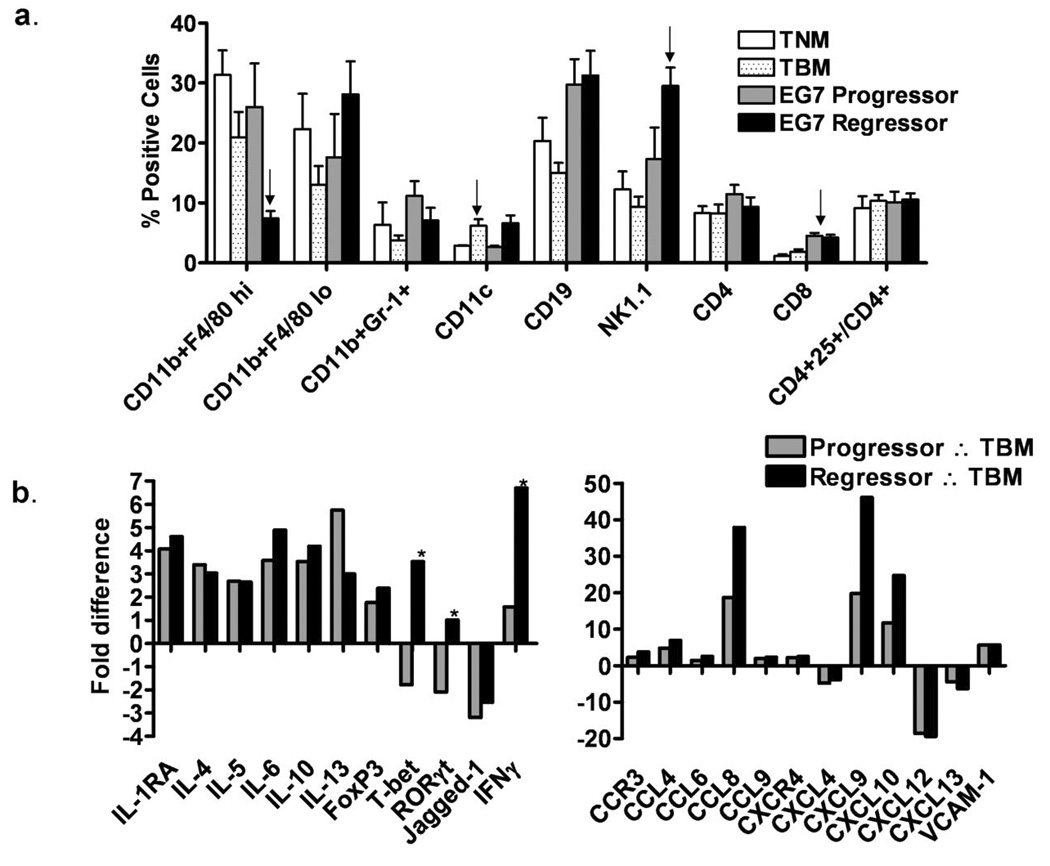
(a) PECs were collected from tumor naïve mice (TNM) or EG7 tumor bearing mice (EG7 TBM) and analyzed by flow cytometry for the indicated surface markers. There were no statistically significant differences between groups. (b) RT-PCR was performed using a custom 96-well array (supplementary figure 1) to examine differences between EG7 tumor ‘progressors’, tumor ‘regressors’ and TBM controls. As compared to TBM controls, each of the differences indicated for ‘progressors’ (gray bars) or ‘regressors’ (black bars) reached statistical significance (p<0.05). Statistically significant differences for the ‘regressors’ as compared to the ‘progressors’ are indicated above the blue bars with an asterisk (p<0.05). n≥5 arrays (with a single array per mouse) for each of the 4 conditions (TNM, TBM, progressor and regressor).
Furthermore, because heterogeneous responses to vaccination with gp96-Ig were observed, we compared the transcriptional profiles of PECs from mice that were rejecting the peripheral EG7 tumor (regressors) to those that were not (progressors) following vaccination with gp96-Ig. Because a clonally expanded population of transgenic OT-I cells may artificially alter the total RNA isolated from the PECs, and since they are not necessary for tumor rejection, these experiments were performed in the absence of adoptively transferred OT-I cells. These data (figure 3b) reveal many vaccination induced changes in the pattern of chemokines, cytokines and interleukins between both progressors and regressors to the control TBM. Importantly, regressors produce higher levels of the key Th1 and Th17 transcription factors, T-bet and RORγt, pro-inflammatory chemokines CCL8, CXCL9 and CXCL10, increased expression of IFNγ and reduced levels of the pro-Th2 interleukin-13. Regressors were also found to have elevated levels of CXCL10 in peritoneal fluid compared to progressors, 71.90 ± 30.73 and 36.72 ± 4.484 pg/ml, respectively (n=5 per group, peritoneal fluid diluted to 4 ml in PBS). Subsequent phenotypic analysis between progressors and regressors revealed increased peritoneal infiltration by CD11c+ cells (p=0.0128) and NK1.1+ cells (p=0.05), and decreased CD11b+F4/80-hi cells (p=0.0215) in regressors as compared to progressors (figure 3b). Conversely, there was a gradual increase in the percentage of peritoneal CD11b+F4/80dim cells in regressors. CD8+ T cells were increased in the peritoneal cavity in both groups of vaccinated mice as compared to non-vaccinated tumor bearing and tumor naïve control mice (p<0.0004).
Tumor regression is associated with increased tumor infiltration by CD8+ and IL-17+ cells and decreased CD11b+Gr-1+ and FoxP3+ cells
Previous data collected from the peritoneal cavity suggested that tumor regression following vaccination with gp96-Ig was associated with expansion of tumor specific CTLs in the peritoneal cavity (9, 13) as well as in the peripheral blood (figure 1). Furthermore, tumor regression is associated with increased expression of factors by cells at the site of vaccination with roles in the development of Th1 and Th17 polarized cells (T-bet and RORγt, respectively). To confirm that tumor regression was associated with the recruitment of CTLs within the tumor microenvironment, immunohistochemistry was performed on sections from both progressor and regressor tumors. Intratumoral staining illustrates that regressor tumors recruit significantly more CD8+ cells within the tumor microenvironment as compared to progressor tumors (Figure 4a–c). Interestingly, the number of IL-17+ cells within regressor tumors was greater than within progressor tumors (figure 4d and 4e, and data not shown), and the mean fluorescence intensity for IL-17 increased from 608.6±158.7 in progressors to 2,493±854.6 in regressors (p=0.0620). The relative differences between intratumoral CD8+ cells or IL-17+ cells and the associated transcription factors are similar (compare figure 3c to figure 4c and f). Progressor tumors were also found to have a greater fraction of infiltrating FoxP3+ cells to CD8+ cells as compared to regressor tumors (figure 4d–f).
Figure 4. Tumor regression is correlated with increased intratumoral effector cells and decreased immunosuppressive cells.
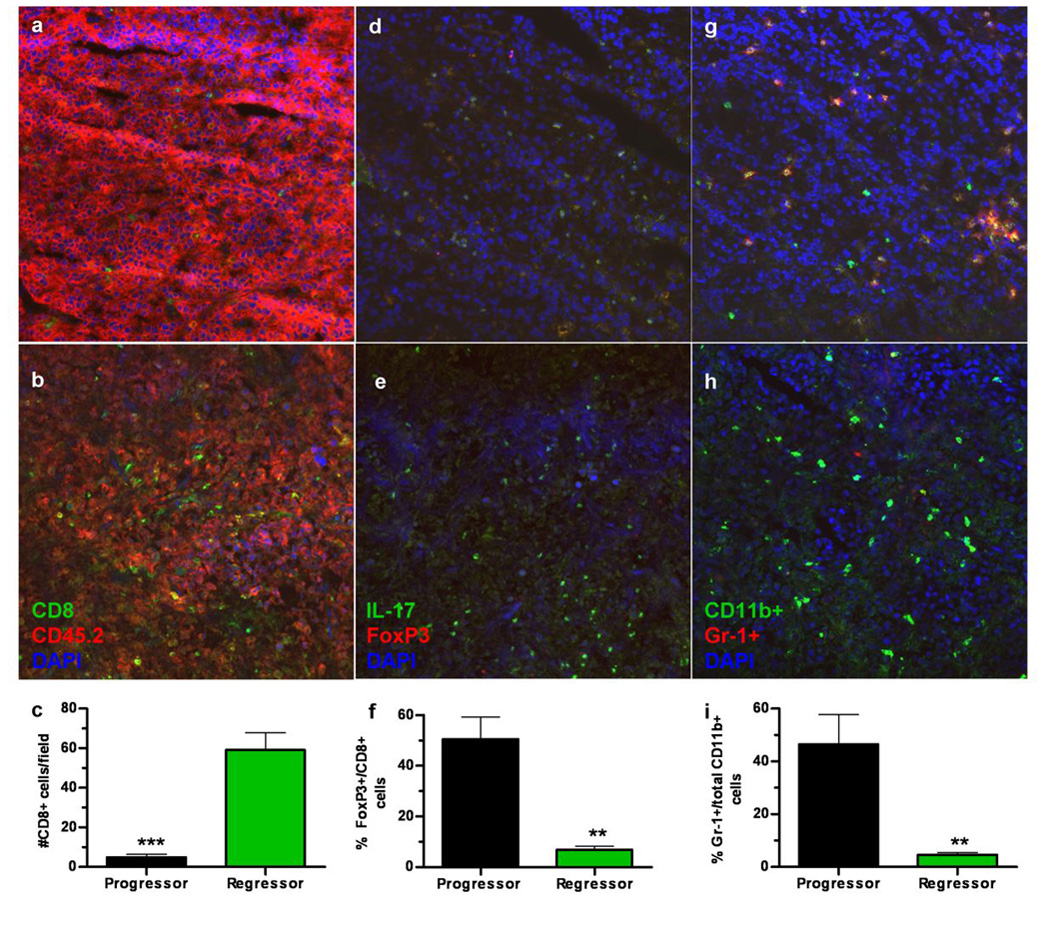
Flash frozen sections from EG7 tumors (CD45.2+) harvested from EG7-gp96-Ig vaccinated, CD45.1+ ‘progressor’ (a) mice or EG7-gp96-Ig vaccinated CD45.1+ ‘regressor’ (b) mice were stained with anti-CD8 and anti-CD45.2 (quantitated in c) or anti-IL-17 and FoxP3 (d ‘progressor’ & e ‘regressor’ and quantitated in f) or anti-CD11b and anti-Gr-1 (g ‘progressor’ & h ‘regressor’ and quantitated in i) and counterstained with DAPI (a–h). The number of positive cells was determined using ImageJ software and is plotted in panels c, f and i. ** indicates p<0.01, *** indicates p<0.001. Tumor sections were quantitated for ≥ 2 mice per group with a minimum of two sections/tumor for each staining condition with five fields per section used for counting.
CD11b+Gr-1+ immature myeloid cells (iMCs) are reported to play an important role in facilitating tumor growth and angiogenesis as well as in suppressing anti-tumor immunity, however this activity is controversial (4, 7, 14, 15). Furthermore, iMCs have been reported to be important for skewing anti-tumor immunity toward type-2 responses (16). Therefore, based upon the observation that type-1 responses are inhibited within the peritoneal cavity of tumor bearing control mice and progressor mice, we determined the co-expression of CD11b and Gr-1 within the tumor. Intratumoral staining for CD11b and Gr-1 demonstrates that progressor tumors recruit significantly more CD11b+Gr-1+ cells as compared to regressor tumors (figure 4g–i). Both progressor and regressor tumors recruited equivalent total numbers of CD11b+ cells (figure 4g and 4h, and data not shown).
Tumor progression is associated with elevated levels of serum TGF-β and CXCL10
A number of previous reports have suggested that high levels of TGF-β, produced by an unknown cell type, is associated with tumor progression (17). Furthermore, recent reports have demonstrated that neutralization of TGF-β facilitates tumor rejection (18–20). RT-PCR analysis of PECs from control, tumor bearing controls, ‘progressors’ and ‘regressors’ revealed no significant differences in the level of TGF-β produced within the peritoneal cavity (data not shown). TGF-β protein was also undetectable in peritoneal fluid (data not shown). The possibility remained, however, that TGF-β may be produced and secreted by the tumor or at another distant site, and exert systemic immunosuppressive effects. Therefore, we collected serum samples from ‘progressor’ and ‘regressor’ mice and compared the levels of TGF-β (Figure 5a). Mice bearing ‘progressor’ tumors were found to have 118.3 +/− 10.24 ng/ml TGF-β (n=8) whereas mice with ‘regressor’ tumors had 68.41 +/− 12.41 ng/ml TGF-β (n=6), p=0.0096. Serum TGF-β levels were 91.27 +/− 15.91 ng/ml (n=5) in non-tumor bearing control mice. Interestingly, whereas an increase in CXCL10 expression and protein within the peritoneal cavity was associated with tumor regression, elevated serum CXCL10 associates with tumor progressors. Serum CXCL10 was found to be 73.94 ± 6.149 (n=4), 224.3 ± 27.27 (n=9) and 79.08 ± 6.589 (n=6) pg/ml for tumor naïve mice, progressors and regressors, respectively (figure 5b). There is no difference between tumor naïve mice and regressors, however expression of CXCL10 by both groups is lower than in progressors (p=0.0002). No difference was detected in the level of serum IL-1β, IFNγ, IL-6 or IL-17 between progressors or regressors and tumor naïve control mice by ELISA (data not shown).
Figure 5. Tumor progression is correlated with increased serum TGF-β and CXCL10.
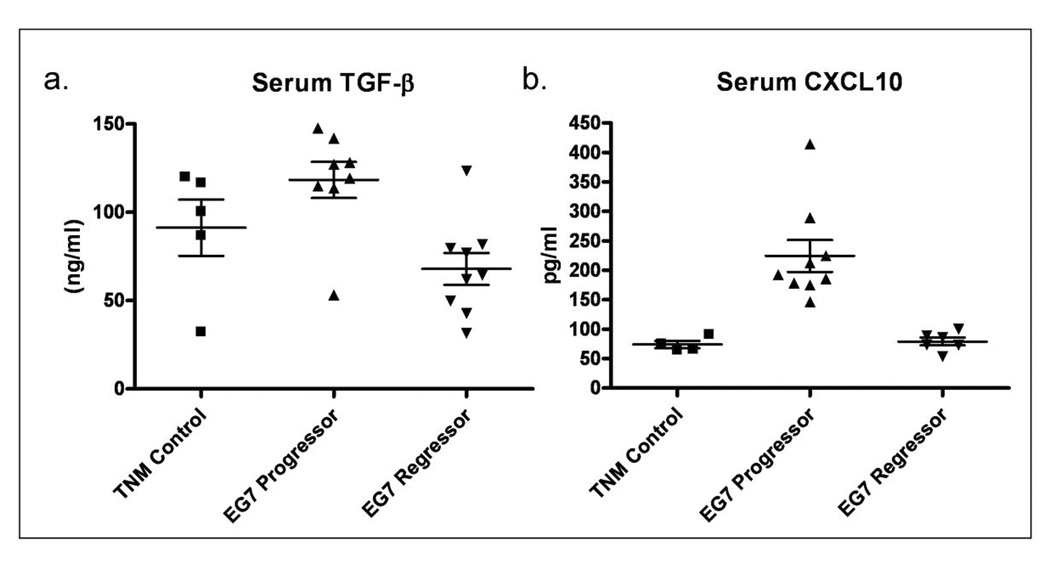
Blood samples were collected from ‘progressor’, ‘regressor’ or tumor naïve control mice (TNM) and serum was collected as described in materials and methods. (a) Serum levels of TGF-β in tumor naïve control mice were 91.27 ± 15.91 (N=5) progressor mice were 118.3 ± 10.24 (N=8) and 67.99 ± 9.001 ng/ml (N=9) for regressor mice, p= 0.0021. (b) Serum levels of CXCL10 in TNM were 73.94 ± 6.149 (N=4), 224.3 ± 27.27 (N=9) for progressors and 79.08 ± 6.589 (N=6) for regressors, with p<0.001 between progressors and either TNM or regressors.
CTLs from ‘progressor’ and ‘regressor’ mice have equivalent cytotoxic activity
Since expression of Th1-type cytokines and chemokines within the peritoneal cavity, peripheral expansion of tumor-antigen specific CTLs and tumor infiltration by CD8+ T cells all correlate with tumor regression, we hypothesized that CD8+ cells isolated from ‘regressor’ mice would have enhanced killing activity of EG7 target cells as compared to CD8+ cells isolated from ‘progressor’ mice. We performed 4-hr in vitro cytotoxicity assays with freshly isolated CD8+ splenocytes from ‘progressor’ and ‘regressor’ mice and found no difference in their ability to kill EG7 target cells (figure 6). Furthermore, no qualitative differences were found in the expression of IFNγ or granzyme-B between CD8+ cells from regressor and progressor mice (data not shown).
Figure 6. Equivalent in-vitro cytotoxicity between ‘progressor’ and ‘regressor’ CTLs.
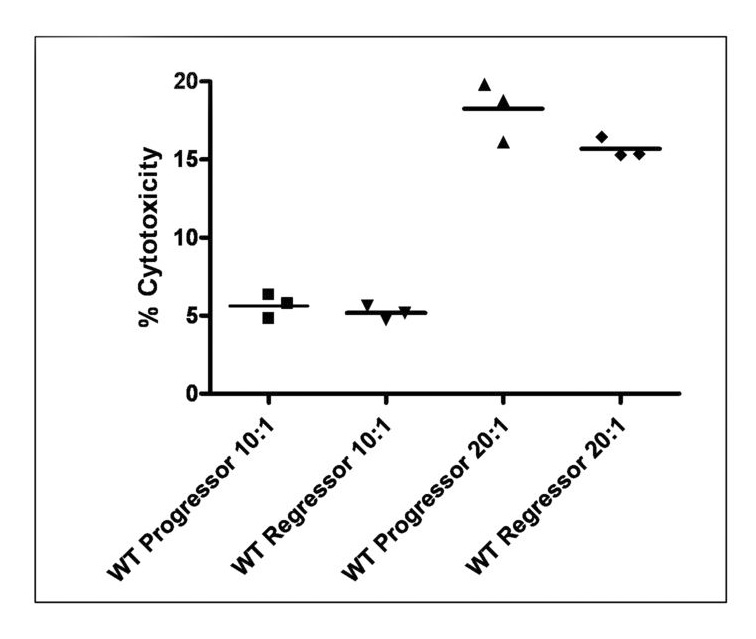
CD8+ splenocytes were isolated by magnetic bead separation as described and immediately incubated for 4 hours with EG7 target cells at the indicated effector:target ratios. No significant difference exists between the ability of ‘progressor’ or ‘regressor’ CTLs to kill EG7 targets in vitro (n=3 per group).
Discussion
The clinical utility of therapeutic anti-cancer vaccines is dependent upon the expression of tumor-specific or –associated antigens by individual patient tumors. It has been reported that malignant human tumors contain between 40–100 mutations that may result in the presentation of a tumor neo-antigen (21–23) in addition to the expression of developmental, unmutated antigens. CD8+ cytotoxic T lymphocytes reacting to these antigens are thought to play a primary role in immune-mediated control and rejection of established tumors (24). An obstacle to following the clonal expansion of a population of tumor-specific CTLs is that the dominant antigens to which those CTLs are reacting are rarely known. To better track the expansion of tumor-specific CTLs, we have utilized the ovalbumin-expressing EG7 tumor cell line as both the primary tumor bolus and the vaccine cells secreting gp96-Ig. This model system is artificial in that the tumor neo-antigen, ovalbumin, is not representative of a true tumor-specific or –associated antigen, but is nonetheless instructive for monitoring anti-tumor immune responses. In animals adoptively transferred with the TCR-transgenic ovalbumin-specific T-cell, OT-I, it was possible to monitor the proliferation of T cells responding to the tumor neo-antigen, ova.
Using this model system, we first advanced previous studies(10) by showing that more frequent vaccinations with gp96-Ig led to a dose-dependent increase in peripheral expansion of tumor-antigen specific CTLs. These studies also illustrated that not all mice respond equally well to the vaccine, with approximately 68% progressing slower than controls but without regression and 32% rejecting the established tumor and advancing to long term disease-free survival. The group of mice capable of rejecting the peripheral tumor was the same group that exhibited robust CTL expansion, providing significant evidence for a correlation between tumor-specific CTL expansion and tumor regression. Importantly, vaccination with gp96-Ig in the absence of adoptively transferred OT-I cells led to similar anti-tumor responses seen in the presence of OT-I cells, suggesting that expansion of tumor specific CTL with other specificities are responsible for tumor rejection. The experiments therefore validate the use of OT-I expansion as surrogate marker for CTL expansion upon vaccination without influencing the outcome of the therapeutic effect.
The reason why the response of syngeneic, age and sex-matched mice bearing similar size tumors to identical doses of the same batch of a vaccine is heterogeneous is unclear. One potential reason is likely to be a function of the tumor-specific antigen concentration within the cytosol of the administered vaccine cells. Since gp96 binds peptides nonspecifically it is likely to be a rare event that an appropriate tumor specific antigen encounters gp96 before it binds other cellular proteins or is secreted from the tumor cell (25, 26). This may also be one reason why cell-based gp96 vaccines, capable of continuously secreting gp96, are superior to a single bolus of purified gp96 or gp96-Ig (9, 13). Other potential reasons for the heterogeneity in vaccine responses may include the availability of CD91+ dendritic cells or activated NK cells at the appropriate time within the site of immunization. Recent clinical studies with purified autologous gp96 have also demonstrated similar heterogeneity in human patients, suggesting a conserved phenomena for gp96-induced anti-tumor immune responses(27, 28).
To better understand how a peripheral tumor growing subcutaneously in the flank conditions the host immune system such that it is unable to respond to a single dose of the gp96-Ig vaccine, we analyzed the phenotype and expression profile of PECs of tumor naïve and tumor bearing mice. The peritoneal cavity was selected because we have shown previously that peripheral tumors, regardless as to whether they contained ovalbumin, interfered with OT-I expansion upon intraperitoneal immunization with gp96-Ig(10). This finding suggested that peripheral tumors exerted long distance effects on peritoneal cells, which we decided to analyze. This analysis revealed significant elevations in the expression of IL-1RA, CCR3, CXCL13 and RORγt in peritoneal lavage cells. IL-1RA is the natural competitive inhibitor to IL-1β. IL-1β is known to be found at high concentrations within many tumors, and has a wide range of opposing effects on tumorigenesis involving tumor growth and invasiveness, but also enhancing tumor/immune interactions(29). CCR3 is one of the more promiscuous chemokine receptors, and is expressed by a wide range of leukocytes including eosinophils, monocytes, Th2 polarized T cells, neutrophils and dendritic cells. CXCL13 by comparison binds only to CXCR5 and is primarily associated with B cell homing to lymphoid tissue, and especially recruitment of auto-reactive B1 B cells (30). These findings may offer some evidence toward a Th1-adverse environment maintained by the peripheral tumor, however the majority of cytokines and chemokines analyzed showed no significant change between tumor bearing and control naïve mice. Further analysis is required to understand the antigen non-specific mechanism of tumor induced immunosuppression by peripheral tumors.
The results of the analysis of peritoneal cells of vaccinated, tumor bearing mice revealed interesting differences between mice with progressing as compared to regressing tumors. These studies were performed with the expectation that since CTL expansion is inhibited in ‘progressors’ despite receiving identical treatment with the vaccine, those factors critical in driving CTL expansion and overcoming tumor-induced immunosuppression would be found to be different between the two cohorts. Importantly, the expression of T-bet, RORγt and IFNγ was opposite between regressors and progressors, with regressors expressing significantly higher levels of each of these factors. Furthermore, regressors also produced more CCL8, CXCL9 and CXCL10. In addition to being induced by interferon-γ, CXCL9 and 10 are important NK and Th1-polarized cell chemoattractants; the increases in peritoneal NK1.1 positive cells correlate with the increases in these chemokines (figure 3a and b) and NK cells were previously shown to be critical for gp96-Ig mediated CTL expansion (13). Importantly, regressors were found to have increased levels of T-bet and IFNγ at the site of vaccination, increased frequencies of tumor specific CTLs in the peripheral blood and increased infiltration of CD8+ cells within a regressing tumor. These observations provide a logical continuum of events between the immunization with gp96-Ig and the rejection of the established tumor.
The observation that tumor bearing mice as well as tumor regressors produce more RORγt than tumor naïve mice or tumor progressors is incompletely understood. These findings are additionally supported by an increased frequency of IL-17+ cells, and increased IL-17 expression by those cells, within the tumor microenvironment in tumor regressors as compared to tumor progressors. The role of CD8+ CTLs in anti-tumor immunity has been long established, and a few recent reports have suggested that Th17 cells play an important role in anti-tumor immunity (31, 32), however there continues to be controversy in this developing area of research (9, 24, 33–37). Previous studies with gp96-Ig have clearly demonstrated that CTL expansion is independent of CD4+ cells (9, 13), so the contribution of gp96-Ig toward the development of IL-17 producing cells may or may not be a result of a direct interaction, and is under further investigation.
Recently, the role of TGF-β in suppressing anti-tumor immune responses has been more firmly established by experiments demonstrating that antibody blockade of TGF-β leads to tumor rejection (18, 20). The source of TGF-β in these studies is unknown, however it has been reported that TGF-β is produced by both tumor cells and their surrounding stroma (38). Therefore, although we saw no difference in TGF-β production by peritoneal cells (data not shown), we analyzed the levels of TGF-β present in the serum of both ‘progressor’ and ‘regressor’ mice and found reduced levels of TGF-β to be associated with tumor rejection (figure 5). In addition, tumor ‘progressors’ were found to have elevated levels of serum CXCL10, despite having reduced CXCL10 message and protein within the peritoneal cavity. The effect of this finding is under further investigation, however it is tempting to speculate that elevated levels of Th1-attracting chemokines within the serum may abolish a tissue-favored gradient of those chemokines and reduce the ability of Th1-polarized cells to extravasate to inflamed tissues by chemokine receptor desensitization (39). Other recent findings have suggested that defects in effector cell trafficking to tumors is a dominant mechanism of tumor induced immunosuppression(40).
Surprisingly, no difference was found between the ability of CD8+ splenocytes harvested from ‘progressor’ or ‘regressor’ mice to kill EG7 target cells in vitro. This observation suggests that tumor-specific CTLs are not necessarily tolerant of the ‘progressing’ tumor, but that the dominant mechanism of tumor-induced immunosuppression may be through increased serum levels of Th1-inhibitory chemokines and cytokines such as TGF-β and potentially by regulatory cells that are excluded from an in vitro killing assay. This view is supported by recent data demonstrating that a reagent capable of bringing CTLs into close proximity with tumor targets is sufficient to induce lysis of those tumor cells, indicating that no intrinsic cytotoxic defect exists in those CTLs (41).
Taken together, these data provide evidence that helps to explain the multiple mechanisms by which established tumors may dampen an effective anti-tumor immune response. Although the source remains unknown, elevated serum levels of suppressive cytokines such as TGF-β and Th1-chemoattractants such as CXCL10 may have the dual effect of suppressing T-effector function and recruitment to target tissues in an antigen non-specific fashion. Importantly, Th1-polarizing vaccine strategies, such as the one employed herein with gp96-Ig, may overcome both local and systemic tumor-induced immunosuppression to recapitulate an effective anti-tumor immune response leading to long-term disease free survival. Our data support the view that suppression of CTL expansion and subsequent tumor infiltration are major mechanisms of tumor immune evasion, without necessarily inducing T cell anergic tolerance. Future studies currently underway will seek to clarify the relative roles of CTLs, IL-17 producing cells and NK cells to tumor rejection, as well as identify secondary interventions that may facilitate the conversion of ‘progressors’ to ‘regressors’, including in vivo blockade of serum TGF-β.
Supplementary Material
Acknowledgments
Financial support provided by NCI-5P01CA109094-02 to Eckhard Podack
Footnotes
The authors declare no financial conflicts of interest.
References
- 1.Smyth MJ, Dunn GP, Schreiber RD. Cancer immunosurveillance and immunoediting: the roles of immunity in suppressing tumor development and shaping tumor immunogenicity. Adv Immunol. 2006;90:1–50. doi: 10.1016/S0065-2776(06)90001-7. [DOI] [PubMed] [Google Scholar]
- 2.Mapara MY, Sykes M. Tolerance and cancer: mechanisms of tumor evasion and strategies for breaking tolerance. J Clin Oncol. 2004;22:1136–1151. doi: 10.1200/JCO.2004.10.041. [DOI] [PubMed] [Google Scholar]
- 3.Rabinovich GA, Gabrilovich D, Sotomayor EM. Immunosuppressive strategies that are mediated by tumor cells. Ann Rev Immunol. 2007;25:267–296. doi: 10.1146/annurev.immunol.25.022106.141609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Willimsky G, Czeh M, Loddenkemper C, et al. Immunogenicity of premalignant lesions is the primary cause of general cytotoxic T lymphocyte unresponsiveness. J Exp Med. 2008;205:1687–1700. doi: 10.1084/jem.20072016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim R, Emi M, Tanabe K, Arihiro K. Tumor-driven evolution of immunosuppressive networks during malignant progression. Cancer Res. 2006;66:5527–5536. doi: 10.1158/0008-5472.CAN-05-4128. [DOI] [PubMed] [Google Scholar]
- 6.Leen AM, Rooney CM, Foster AE. Improving T cell therapy for cancer. Ann Rev Immunol. 2007;25:243–265. doi: 10.1146/annurev.immunol.25.022106.141527. [DOI] [PubMed] [Google Scholar]
- 7.Nagaraj S, Gabrilovich DI. Tumor escape mechanism governed by myeloid-derived suppressor cells. Cancer Res. 2008;68:2561–2563. doi: 10.1158/0008-5472.CAN-07-6229. [DOI] [PubMed] [Google Scholar]
- 8.Zou W, Chen L. Inhibitory B7-family molecules in the tumour microenvironment. Nat Rev Immunol. 2008;8:467–477. doi: 10.1038/nri2326. [DOI] [PubMed] [Google Scholar]
- 9.Yamazaki K, Nguyen T, Podack ER. Cutting edge: tumor secreted heat shock-fusion protein elicits CD8 cells for rejection. J Immunol. 1999;163:5178–5182. [PubMed] [Google Scholar]
- 10.Oizumi S, Deyev V, Yamazaki K, et al. Surmounting tumor-induced immune suppression by frequent vaccination or immunization in the absence of B cells. J Immunother. 2008;31:394–401. doi: 10.1097/CJI.0b013e31816bc74d. [DOI] [PubMed] [Google Scholar]
- 11.Ikawa M, Yamada S, Nakanishi T, Okabe M. 'Green mice' and their potential usage in biological research. FEBS Lett. 1998;430:83–87. doi: 10.1016/s0014-5793(98)00593-6. [DOI] [PubMed] [Google Scholar]
- 12.Hofman F, et al. Chapter 21: Unit 21 4. In: Coligan John E., editor. Immunohistochemistry. Current protocols in immunology. 2002. [DOI] [PubMed] [Google Scholar]
- 13.Oizumi S, Strbo N, Pahwa S, Deyev V, Podack ER. Molecular and cellular requirements for enhanced antigen cross-presentation to CD8 cytotoxic T lymphocytes. J Immunol. 2007;179:2310–2317. doi: 10.4049/jimmunol.179.4.2310. [DOI] [PubMed] [Google Scholar]
- 14.Shojaei F, Ferrara N. Refractoriness to antivascular endothelial growth factor treatment: role of myeloid cells. Cancer Res. 2008;68:5501–5504. doi: 10.1158/0008-5472.CAN-08-0925. [DOI] [PubMed] [Google Scholar]
- 15.Shojaei F, Zhong C, Wu X, Yu L, Ferrara N. Role of myeloid cells in tumor angiogenesis and growth. Trends Cell Biol. 2008 doi: 10.1016/j.tcb.2008.06.003. [DOI] [PubMed] [Google Scholar]
- 16.Sinha P, Clements VK, Bunt SK, Albelda SM, Ostrand-Rosenberg S. Cross-talk between myeloid-derived suppressor cells and macrophages subverts tumor immunity toward a type 2 response. J Immunol. 2007;179:977–983. doi: 10.4049/jimmunol.179.2.977. [DOI] [PubMed] [Google Scholar]
- 17.Bierie B, Moses HL. Tumour microenvironment: TGFbeta: the molecular Jekyll and Hyde of cancer. Nature Rev. 2006;6:506–520. doi: 10.1038/nrc1926. [DOI] [PubMed] [Google Scholar]
- 18.Nam JS, Terabe M, Kang MJ, et al. Transforming growth factor beta subverts the immune system into directly promoting tumor growth through interleukin-17. Cancer Res. 2008;68:3915–3923. doi: 10.1158/0008-5472.CAN-08-0206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nam JS, Terabe M, Mamura M, et al. An anti-transforming growth factor beta antibody suppresses metastasis via cooperative effects on multiple cell compartments. Cancer Res. 2008;68:3835–3843. doi: 10.1158/0008-5472.CAN-08-0215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wei S, Shreiner AB, Takeshita N, Chen L, Zou W, Chang AE. Tumor-induced immune suppression of in vivo effector T-cell priming is mediated by the B7-H1/PD-1 axis and transforming growth factor beta. Cancer Res. 2008;68:5432–5438. doi: 10.1158/0008-5472.CAN-07-6598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wood LD, Parsons DW, Jones S, et al. The genomic landscapes of human breast and colorectal cancers. Science. 2007;318:1108–1113. doi: 10.1126/science.1145720. [DOI] [PubMed] [Google Scholar]
- 22.Jones S, Zhang X, Parsons DW, et al. Core Signaling Pathways in Human Pancreatic Cancers Revealed by Global Genomic Analyses. Science. 2008;321(5897):1801–1806. doi: 10.1126/science.1164368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Parsons DW, Jones S, Zhang X, et al. An Integrated Genomic Analysis of Human Glioblastoma Multiforme. Science. 2008;321(5897):1807–1812. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Koebel CM, Vermi W, Swann JB, et al. Adaptive immunity maintains occult cancer in an equilibrium state. Nature. 2007;450:903–907. doi: 10.1038/nature06309. [DOI] [PubMed] [Google Scholar]
- 25.Demine R, Walden P. Testing the role of gp96 as peptide chaperone in antigen processing. J Biol Chem. 2005;280:17573–17578. doi: 10.1074/jbc.M501233200. [DOI] [PubMed] [Google Scholar]
- 26.Srivastava P. Interaction of heat shock proteins with peptides and antigen presenting cells: chaperoning of the innate and adaptive immune responses. Ann Rev Immunol. 2002;20:395–425. doi: 10.1146/annurev.immunol.20.100301.064801. [DOI] [PubMed] [Google Scholar]
- 27.Testori A, Richards J, Whitman E, et al. Phase III comparison of vitespen, an autologous tumor-derived heat shock protein gp96 peptide complex vaccine, with physician's choice of treatment for stage IV melanoma: the C-100-21 Study Group. J Clin Oncol. 2008;26:955–962. doi: 10.1200/JCO.2007.11.9941. [DOI] [PubMed] [Google Scholar]
- 28.Wood C, Srivastava P, Bukowski R, et al. An adjuvant autologous therapeutic vaccine (HSPPC-96; vitespen) versus observation alone for patients at high risk of recurrence after nephrectomy for renal cell carcinoma: a multicentre, open-label, randomised phase III trial. Lancet. 2008;372:145–154. doi: 10.1016/S0140-6736(08)60697-2. [DOI] [PubMed] [Google Scholar]
- 29.Apte RN, Voronov E. Is interleukin-1 a good or bad 'guy' in tumor immunobiology and immunotherapy? Immunol Rev. 2008;222:222–241. doi: 10.1111/j.1600-065X.2008.00615.x. [DOI] [PubMed] [Google Scholar]
- 30.Ishikawa S, Sato T, Abe M, et al. Aberrant high expression of B lymphocyte chemokine (BLC/CXCL13) by C11b+CD11c+ dendritic cells in murine lupus and preferential chemotaxis of B1 cells towards BLC. J Exp Med. 2001;193:1393–1402. doi: 10.1084/jem.193.12.1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kottke T, Sanchez-Perez L, Diaz RM, et al. Induction of hsp70-mediated Th17 autoimmunity can be exploited as immunotherapy for metastatic prostate cancer. Cancer Res. 2007;67:11970–11979. doi: 10.1158/0008-5472.CAN-07-2259. [DOI] [PubMed] [Google Scholar]
- 32.Muranski P, Boni A, Antony PA, et al. Tumor-specific Th17-polarized cells eradicate large established melanoma. Blood. 2008;112:362–373. doi: 10.1182/blood-2007-11-120998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bronte V. Th17 and cancer: friends or foes? Blood. 2008;112:214. doi: 10.1182/blood-2008-04-149260. [DOI] [PubMed] [Google Scholar]
- 34.Dunn GP, Old LJ, Schreiber RD. The three Es of cancer immunoediting. Ann Rev Immunol. 2004;22:329–360. doi: 10.1146/annurev.immunol.22.012703.104803. [DOI] [PubMed] [Google Scholar]
- 35.Shankaran V, Ikeda H, Bruce AT, et al. IFNgamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature. 2001;410:1107–1111. doi: 10.1038/35074122. [DOI] [PubMed] [Google Scholar]
- 36.Smyth MJ, Thia KY, Cretney E, et al. Perforin is a major contributor to NK cell control of tumor metastasis. J Immunol. 1999;162:6658–6662. [PubMed] [Google Scholar]
- 37.Strbo N, Oizumi S, Sotosek-Tokmadzic V, Podack ER. Perforin is required for innate and adaptive immunity induced by heat shock protein gp96. Immunity. 2003;18:381–390. doi: 10.1016/s1074-7613(03)00056-6. [DOI] [PubMed] [Google Scholar]
- 38.Massague J. TGFbeta in Cancer. Cell. 2008;134:215–230. doi: 10.1016/j.cell.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kurt RA, Baher A, Wisner KP, Tackitt S, Urba WJ. Chemokine receptor desensitization in tumor-bearing mice. Cell Immunol. 2001;207:81–88. doi: 10.1006/cimm.2000.1754. [DOI] [PubMed] [Google Scholar]
- 40.Quezada SA, Peggs KS, Simpson TR, Shen Y, Littman DR, Allison JP. Limited tumor infiltration by activated T effector cells restricts the therapeutic activity of regulatory T cell depletion against established melanoma. J Exp Med. 2008;205:2125–2138. doi: 10.1084/jem.20080099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bargou R, Leo E, Zugmaier G, et al. Tumor regression in cancer patients by very low doses of a T cell-engaging antibody. Science. 2008;321:974–977. doi: 10.1126/science.1158545. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.