

SUMMARY
Targeted therapy for metastatic diseases relies on the identification of functionally important metastasis genes from a large number of random genetic alterations. Here we use a computational algorithm to map minimal recurrent genomic alterations associated with poor-prognosis breast cancer. 8q22 genomic gain was identified by this approach and validated in an extensive collection of breast tumor samples. Regional gain of 8q22 elevates the expression of metastasis gene Metadherin (MTDH), which is overexpressed in more than 40% of breast cancers and is associated with poor clinical outcomes. Functional characterization of MTDH revealed its dual role in promoting metastatic seeding and enhancing chemoresistance. These findings establish MTDH as an important therapeutic target for simultaneously enhancing chemotherapy efficacy and reducing metastasis risk.
SIGNIFICANCE
Genomic profiling of breast cancer has established several clinically applicable poor-prognosis gene signatures. However, the lack of overlap between independent signatures prevents the identification of functionally important genes in the signatures. Here we report an integrative strategy to identify recurrent genomic alterations that are both clinically relevant and functionally important for breast cancer progression. Successful application of this approach lead to the identification of MTDH at the recurrent 8q22 poor-prognosis genomic gain with important functions in both metastasis and chemoresistance. The dual-functionality of MTDH further provides an explanation for the long standing conceptual dilemma regarding the selection of metastasis genes in the primary tumor. Overall, our data illustrate the synergistic value of integrating bioinformatics with clinical and experimental metastasis research.
INTRODUCTION
The progression of cancer from an abnormal outgrowth to a life-threatening metastatic tumor is accompanied by a myriad of genetic and epigenetic alterations accumulated along the way (Chin and Gray, 2008; Fidler, 2003; Gupta and Massague, 2006; Hanahan and Weinberg, 2000; Steeg, 2006). The challenge of distinguishing crucial drivers of metastasis from thousands of by-stander alterations remains a major bottleneck in metastasis research. The turn of the century has witnessed the advent of two parallel, but individually incomplete, genomic approaches to unravel the genetic mystery of cancer metastasis (Kang, 2005). Comparative expression profiling analyses of cancer cell line variants with different metastasis potentials, often obtained by in vivo selection in animal models, have led to the identification of several metastasis genes (Clark et al., 2000; Kang et al., 2003; Minn et al., 2005; Yang et al., 2004). However, much work remains to be done to validate the clinical relevance of metastasis genes identified in animal model studies. As a second approach, gene expression profiling of human tumor specimens has enabled the identification of several poor-prognosis signatures that are predictive of recurrence and metastasis risk in human cancers (Ramaswamy et al., 2003; van ’t Veer et al., 2002; van de Vijver et al., 2002; Wang et al., 2005). Although different poor-prognosis signatures have proven to be operationally interchangeable for class prediction purposes in the clinic (Fan et al., 2006), the lack of overlap between different poor-prognosis signatures has posed a major challenge for understanding the biological underpinnings of cancer progression and metastasis, thereby hindering the development of targeted therapeutics. Identifying functionally important and clinically relevant metastasis genes requires innovative strategies to synergize advances in both clinical and experimental metastasis studies.
Recurrent DNA copy number alterations (CNAs) have been observed in a wide range of human cancers and such genetic events often indicate the presence of key mediators of malignancy in the affected genomic loci. For example, elevated expression of oncogenes, such as c-Myc, CCND1, Her2 and EGFR1, is often a result of amplification of their corresponding genomic segments (Chin and Gray, 2008). However, CNAs responsible for cancer metastasis are poorly characterized. Various techniques have been developed to detect genomic alterations, including fluorescence in situ hybridization (FISH), comparative genomic hybridization (CGH) and high density single nucleotide polymorphism (SNP) genotyping. Detection of CNAs by expression profiling analysis is theoretically possible since a strong correlation between genomic alterations and aberrant expression of genes in affected loci has been observed (Pollack et al., 2002). Accurate detection of CNAs using expression analysis, however, is technically difficult because gene expression data reflect multiple layers of gene regulation beyond genomic alterations. Such analysis is particularly challenging with clinical tumor samples due to the inherent heterogeneity of clinical specimens and the rampant genomic instability of late stage tumors. Here we used a computational algorithm to identify a recurrent 8q22 genomic gain in poor-prognosis human breast cancers, which harbors the metastasis gene Metadherin (MTDH; also called Lyric, AEG1 (Britt et al., 2004; Brown and Ruoslahti, 2004; Kang et al., 2005). Functional characterization of MTDH revealed its dual functions in promoting metastasis and chemoresistance of breast cancers.
RESULTS
Recurrent poor-prognosis genomic alterations
To sensitively detect CNAs that affect regional gene expression, we developed a bioinformatic strategy called ACE (Analysis of CNAs by Expression data, Figure 1A). ACE first calculates the expression scores of all genes according to their expression differences between comparison groups, and orders them by genomic positions. To measure the regional expression pattern, a neighborhood score (NS) is calculated for each genomic locus using a geometry-weighted sum of expression scores of all the genes on the chromosome. The expression scores of the genes in proximity to the locus in consideration are assigned greater weights than those farther away, because the locus linkage strength decays with distance. The significance of the NS is estimated by permutation and regions with a stretch (≥20) of aberrant NS are declared as potential CNA regions.
Figure 1. ACE analysis identifies a recurrent genomic gain at 8q22 in poor-prognosis breast cancer.
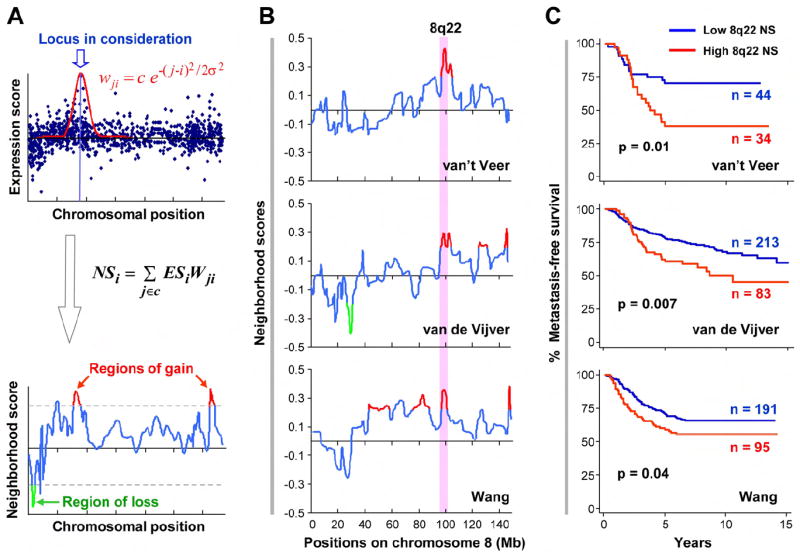
A, In the ACE approach, the expression score (ES) of each gene is calculated by comparing samples of different phenotypes, and then a neighborhood score (NS), indicative of the DNA copy number status, is computed for each locus as the geometry-weighted ES sum of all the genes on the chromosome. Regions of gain (red, bottom panel) and loss (green) were defined by applying NS cutoffs (dotted lines) obtained from permutations. i, j, gene index when they are ordered on the chromosome by genomic positions; c, normalizing constant; wji, weight of gene j when locus i is in consideration (see text and Experimental Procedures for details). B, Poor-prognosis genomic gain at 8q22 was detected in all three datasets (van ’t Veer et al., 2002; van de Vijver et al., 2002; Wang et al., 2005). The traces are the NS scores on chromosome 8 produced by ACE. The shaded area highlights the consensus region of gain at 8q22. Red and green peaks represent statistically significant regions of gains or loss, respectively. C, Kaplan-Meier metastasis-free survival curves of patients with high or low 8q22 NS.
After validating the efficacy of the ACE method using a number of existing gene expression profiling datasets that have corresponding genomic alteration information (See Figure S1 and Supplemental Experimental Procedures), we applied it to the study of genomic alterations associated with poor prognosis of human breast cancer. Three separate studies have previously identified two poor-prognosis gene sets, of 70 and 76 genes, respectively, that can be used to robustly predict the clinical outcome of human breast cancers. However, only a single gene (CCNE2) is present in both signatures. When the ACE method was applied to analyze these three datasets, five common genomic gains were observed in at least two datasets (Table 1) and 15 other genomic gains were observed in one of the three datasets (Table S1). The smallest regions of overlap (SRO) of common CNA events, namely, gains at 3q26-27, 8q22, 8q24.3, 17q23-25 and 20q13.3, are among a large number of genomic alterations previously observed in high frequencies in breast cancer, although their links to poor prognosis and tumor progression have not been established (Bergamaschi et al., 2006). We did not detect any genomic losses associated with more than one dataset, which is consistent with previous observations that genomic gains are more prevalent than genomic losses in breast cancer, especially in patients with poor outcomes (Naylor et al., 2005). Out of the five prevalent genomic events, the 8q22 gain was consistently observed in all three datasets (Figure 1B). We calculated the NS of this region for each sample in the three datasets, and used it to classify tumor samples into two groups with high and low NS. As shown in Figure 1C and Table S2, the probability of metastasis-free survival of patients with a high 8q22 NS was significantly lower than the control group in all three datasets. These analyses suggested that the genomic gain of 8q22 is a strong predictor of breast cancer poor prognosis.
Table 1.
Recurrent regions of gain associated with poor-prognosis breast cancer as detected by ACE
| # | Location | Genomic Position (Mb) | Size (Mb) | Gene No. | Predicted in datasets |
|---|---|---|---|---|---|
| 1 | 8q22 | 96.3-99.2 | 2.9 | 20 | All |
| 2 | 3q26.33-q27.1 | 180.2-184.4 | 4.2 | 27 | Wang, van’t Veer |
| 3 | 8q24.3 | 144.4-145.7 | 1.3 | 82 | Wang, van de Vijver |
| 4 | 17q23.3-q25 | 54.8-58.2 | 3.4 | 42 | Wang, van de Vijver |
| 5 | 20q13.3 | 60.7-62.0 | 1.3 | 60 | Wang, van de Vijver |
During tumor progression cancer cells usually acquire multiple genomic alterations. These genetic events may contribute to tumor aggressiveness independently or synergistically. To look at whether the other four regions interact with 8q22 gain in poor prognosis, we clustered the tumor samples in the three datasets according to the NS of the five regions (Figure S2). A significant fraction of samples with 8q22 gain also show increased copy numbers at 8q24.3 (p<0.01, χ2 test) in all the three datasets, but no obvious link was found between 8q22 and the other 3 regions. The concurrent copy number gains of the two 8q subregions is consistent with previous observation of frequent genomic gain of the whole 8q arm in breast cancer tumors (Ried et al., 1995). However, survival analysis revealed that there is no significant contribution of 8q24.3 to the prognostic power of 8q22, as the patients with genomic gains at both regions display similar, or even better, survival records than the patients with 8q22 gain only (Figure S2), suggesting that 8q22 gain functions independently in poor prognosis.
Validation of 8q22 genomic gain in breast tumors
We used FlSH (Fluorescence in situ hybridization) and genomic DNA real-time PCR (qPCR) to confirm 8q22 genomic gain in breast tumor samples. First, we analyzed a panel of microdissected tumor samples from fresh frozen breast cancer specimens by qPCR using four pairs of primers that amplify DNA sequences at chromosome 8q21, q22 and q23 (Figure 2A, B). Ten out of 36 tumors (27.8%) were found to have aberrantly higher copy numbers (>3.6) at 8q22 than the control human DNA sample (Table S3). As shown in Figure 2B, the 10 genomic gain events spanned from 8q21 to 8q23 with a consensus region at 8q22, which is consistent with our computational prediction. DNA copy numbers detected by genomic qPCR analysis are consistent with FISH analysis of the same tumor specimens (Figure S3). To confirm the link between 8q22 genomic gain and elevated expression of genes located in this region, we used qRT-PCR to investigate the expression patterns of three genes at 8q22 (PTDSS1, MTDH and LAPTM4β) in these tumors. A strong positive correlation was found between the expression of these genes and the 8q22 copy numbers (Figure 2B). Similar results were obtained when we analyzed a separate panel of 18 paraffin-embedded breast tumors (Table S3). Analysis of a panel of breast cancer cell lines also found a good correlation of 8q22 gain with a higher level of MTDH expression (Figure S3).
Figure 2. Validation of 8q22 genomic gain in human breast tumors.

A, Expression of the genes at the 8q22 region in poor-prognosis vs. good-prognosis samples of the three datasets. Red indicates overexpression, while green denotes underexpression. B, To validate the 8q22 genomic gain, a panel of breast tumor were analyzed for 8q22 genomic alterations and gene expression using qPCR. Shown are the DNA copy numbers of 4 genomic loci at 8q21-23 (filled circles) and the expression levels of 3 genes at 8q22 (diamonds). Student’s t-test p-values of expression comparison in samples with and without 8q22 gain are shown in parenthesis after each gene. C, Breast cancer tissue microarray FISH analysis with the green SpectrumGreen and red SpectrumOrange probes detecting chromosome 8 centromere and the 8q22 region, respectively. A case of 8q22 gain (left) and a diploid case (right) are shown. D, Kaplan–Meier survival analysis in breast cancer patients with or without 8q22 gain.
We further analyzed a breast cancer tissue microarray with detailed clinicopathological records by FISH using a bacterial artificial chromosome (BAC) probe located at the 8q22 region, and found that 22 (26.8%) of the 82 hybridized primary tumor samples had an average 8q22 copy number larger than 3 (Figure 2C, Table S4). Notably, 8q22 gain was associated with a higher propensity of metastatic recurrence (Figure 2D). Together with the qPCR analysis described above, these data confirmed the ACE prediction that recurrent genomic gain at 8q22 leads to regional gene activation. More importantly, these results established 8q22 gain as a breast cancer poor-prognosis marker event.
MTDH promotes breast cancer metastasis
There are 13 known genes and 7 hypothetical genes in the 8q22 poor-prognosis genomic gain region (Figure 2A). Overexpression of several genes in this region was independently linked to poor-prognosis in one or several datasets (Table S5). To determine the functional targets of 8q22 gain, we tested six known genes most likely to promote cancer progression based on statistical analysis and their known biological functions: UQCRB, PTDSS1, TSPYL5, MTDH and LAPTM4β, which were significantly overexpressed in metastatic diseases in at least two of these datasets (student’s t-test, p<0.05), and SDC2 which was reported to mediate cell adhesion and proliferation in colon cancer (Park et al., 2002) (Figure S4). To test the role of these genes in metastasis, we stably overexpressed them in the SCP28 cell line, a subline of the human breast cell line MDA-MB-231 that is mildly metastatic to lung and bone when injected into mice (Kang et al., 2003). The cell line was labeled with a retroviral construct expressing a GFP/luciferase fusion protein (Minn et al., 2005), and its in vivo metastasis capability was monitored by non-invasive bioluminescent imaging (BLI) after intravenous injection. Our data showed that MTDH overexpression significantly accelerated the development of lung metastasis and shortened the survival of mice that received tumor cell xenografts (Figure 3A-D and Figure S4). Animal metastasis burden caused by MTDH overexpression was nearly 7-fold higher than the controls six weeks after cancer cell injection. In contrast, overexpression of the other five genes, either individually or in combination, failed to enhance the metastasis ability of SCP28 (Figure S4). Furthermore, overexpression of these genes together with MTDH did not further enhance lung metastasis beyond MTDH overexpression alone (Figure S4). Therefore, MTDH is likely to be the most significant functional mediator of this poor-prognosis genomic gain, although possible contributions from untested genes in the 8q22 region could not be completely ruled out. MTDH is located at the center of the minimal common region of the 8q22 genomic gain as indicated by the ACE computational analyses of three microarray datasets and has been shown to encode a cell surface protein responsible for promoting mouse mammary tumor cell adhesion to lung endothelial cells (Brown and Ruoslahti, 2004). However, the functional role of MTDH in human breast cancer and the mechanism of its deregulation have not been previously investigated.
Figure 3. MTDH mediates lung metastasis of human breast cancer.

A, MTDH is constitutively overexpressed in the mildly metastatic cells SCP28, and stably knocked down in the highly lung-metastatic cells LM2 with two independent hairpin constructs. B, In vivo metastasis assays of SCP28 cells with or without MTDH overexpression. Lung metastasis burden of xenografted animals was monitored weekly using BLI. Shown are BLI images of representative mice at the sixth week after injection. The color scale depicts the photon flux (photon per second) emitted from the metastasis cells. C, BLI quantification of lung metastasis of SCP28 cells. D, Kaplan-Meier survival curves of mice inoculated with SCP28 cells. E, Representative BLI images and lung sections of the inoculated mice at the sixth week after injection of LM2 with or without MTDH knockdown. Arrows point to the sporadic lesions by MTDH knockdown cells as compared to much more prevalent tumor lesions by control cells. Scale bars, 0.5 mm. F, BLI quantification of lung metastasis by LM2 cells. G. Kaplan-Meier survival analysis of the mice injected with LM2 cells. C, F, Data represent averages ± SEM of 10 mice. *p<0.05; **p<0.01 based on a two-sided Wilcoxon rank test. H, Genetically modified SCP28 or LM2 cells were seeded on top of a monolayer of endothelial cells from lung (HMVEC-L), umbilical vein (HUVEL), bone marrow (HBMEC60) and control fibroblast cells (WI38). Cancer cells were seeded on top of the endothelial or fibroblast monolayer and the attached cells were quantified 3 hours later. Data represent averages ± SEM.
To further validate the role of MTDH in metastasis, we used two different short-hairpin RNA (shRNA) constructs to knock down the expression of MTDH in the LM2 cell line, a MDA-MB-231 subline selected in vivo for its high lung metastasis propensity (Minn et al., 2005). MTDH knockdown significantly reduced the lung metastasis burden of LM2 by 3-5 folds and extended the survival of the mice by 1-2 weeks (Figure 3A, E-G and Figure S4). Histological analysis of primary tumors and lung metastasis revealed poorly differentiated adenocarcinoma at both sites with very similar histological features (Figure S5). We further quantified the mRNA level of MTDH in the cancer cells isolated from lung metastasis (Figure S5). Cells isolated from lung lesions produced by MTDH-knockdown cells continue to have a low level of MTDH expression but is modestly higher than the MTDH level in the cells grown in culture, suggesting that in vivo lung metastasis assay may select for “escapers” that have regained a higher level of MTDH expression.
We also examined the effect of altered MTDH expression on bone and brain metastasis by injecting the genetically modified breast cancer cell lines into the left cardiac ventricle of recipient nude mice. MTDH knockdown in LM2 resulted in a modest but significant improvement of post-injection survival, although bioluminescent quantification of the decrease of bone and brain metastasis burden did not reach statistical significance. Conversely, overexpression of MTDH in SCP28 cells led to a modest but significant increase of bone and brain metastasis (Figure S6). These results suggested that MTDH preferentially promotes metastasis to lung, while having a modest effect on metastasis to other organs.
We further investigated the functional role of MTDH in the multistep process of metastasis. MTDH knockdown or overexpression did not affect the growth, migration or invasiveness of various breast cancer cell lines, including MDA-MB-231 sublines (Figure S7), MCF7 and T47D (Figure S8 and data not shown). However, MTDH knockdown significantly reduced the adhesion of the cancer cells to lung microvascular endothelial cells (HMVEC-L), as well as to endothelial cells of the bone marrow (HBMEC60) and the umbilical vein (HUVEC), albeit to a lesser extent. A reciprocal change was observed when MTDH was overexpressed (Figure 3H). In contrast, the adhesion of cancer cells to the WI-38 lung fibroblast cell line was not affected. MTDH-mediated enhancement of tumor cell adhesion to endothelial cells was also observed in other breast cancer cell lines, including MCF7 and T47D (Figure S8). MTDH did not promote intravasation or extravasation through endothelial layers based on both in vitro transendothelial assays and in vivo metastasis assays using an orthotopic xenograft method (data not shown). Instead, MTDH appeared to specifically enhance the seeding of tumor cells to the target organ endothelium.
MTDH promotes chemoresistance
Poor prognosis of breast cancer at the time of diagnosis or surgery indicates a higher probability of death as the result of recurrent tumors and development of metastases in vital organs. Emergence of metastasis reflects not only the ability of cancer cells to overcome hurdles during the multi-step process of metastasis, but also the capability to survive standard adjuvant therapy and other physiological stresses. Therefore, the driver gene of a poor-prognosis genetic alteration might function to promote chemoresistance in addition to enabling the metastasis process. A bioinformatic analysis of the available NCI60 pharmacogenomic data (Garraway et al., 2005) indicated a potential contribution of the genes at 8q22 to chemoresistance. The NCI60 data include the cytogenetic and expression profiles of 58 cancer cell lines as well as their sensitivity profiles to 24,000 small molecule compounds. Analysis of such data revealed that genomic gain at 8q22 strongly correlates with a higher overall gene expression of this region (Pearson’s r = 0.578, Figure S9); intriguingly, this higher NS is in turn associated with a significantly higher mean GI50 (the drug concentration for 50% growth inhibition) for 1,123 compounds, as compared to 211±178 compounds expected by random permutation (p= 0.019, Figure 4A). In contrast, copy number of the other 8q poor-prognosis region, 8q24.3, was not associated with any significant changes in GI50 values (Figure S10). Similar analysis with the mRNA expression data of the genes at 8q22 revealed that MTDH is the only gene that has a significant correlation with higher chemoresistance (Table S6).
Figure 4. MTDH enhances chemoresistance of the breast cancer cells.

A, Genomic gain of 8q22 is associated with higher resistance to chemical compounds in the 58 human cancer cell lines. logGI50 (drug concentration for 50% growth inhibition) of each of the 24,642 compounds in cell lines with 8q22 gain was compared to those in cells without 8q22 gain. The numbers of compounds with significantly increased logGI50 associated with 8q22 gain, counted by applying various significance thresholds of the logGI50 differences (p<0.05, 0.01 and 0.001), was compared to a null distribution obtained by permuting the 8q22 copy numbers of the cell lines. Median values from permutations are shown with mean absolute deviation (MAD) as the error bar. B, Chemoresistance of LM2 cells was analyzed by clonogenic assays after treatment with various apoptosis-inducing agents with or without HMVEC-L co-culture. Shown are the relative clonogenic abilities as percentages of the non-treatment control. C, Representative images of the clonogenic assays of LM2 cells. D, Clonogenic assays of SCP28 cells with overexpression of MTDH or other genes in the 8q22 region. Shown are the data with HMVEC-L co-culture. E, In vivo chemoresistance assay of LM2 cells. Shown are the xenograft tumor sizes when mice were treated with paclitaxel or drug vehicle. 6 mice per group were used. F, Representative tumors isolated from the mice 25 days after injection in the in vivo chemoresistance assay. B,D,E, results represent average values ± SEM. *p<0.05; **p<0.01; ***p<0.001 with a two-sided student’s t-test.
To investigate the chemoresistance function of MTDH and other genes in 8q22, genetically modified LM2 breast cancer cell lines used for in vivo metastasis assays were treated with chemotherapeutic or other stress agents including paclitaxel, doxorubicin, cisplatin, and hydrogen peroxide with or without co-culture with the HMVEC-L endothelial cell line. Long-term survival of the cells was then quantified by clonogenic assays. Inhibition of MTDH expression sensitized the LM2 cell line to chemotherapeutic and stress agents, while overexpression of MTDH rendered SCP28 cells more resistant to these treatments (Figure 4B-D). In contrast, overexpression of up to 4 other genes in the 8q22 locus did not significantly alter the chemosensitivity of cancer cells (Figure 4D). MTDH-dependent chemoresistance was further enhanced when cancer cells were co-cultured with HMVEC-L lung endothelial cells (Figure 4B, C). MTDH-induced chemoresistance was not limited to the MDA-MB-231 cells, as MTDH knockdown in several additional breast cancer cell lines, including MCF7 and T47D, also significantly sensitized cells to chemotherapeutic challenges (Figure S8).
Next, we studied the chemoresistance function of MTDH in vivo using xenograft models. LM2 cells with or without MTDH knockdown were injected into nude mice subcutaneously. Twice-weekly treatment of tumors with paclitaxel or the drug vehicle was initiated at one week after injection. Subcutaneous tumor volumes were monitored by direct caliper measurement. When the mice were treated with the drug vehicle, the LM2 tumors grew rapidly, reaching five times the initial volume in 18 days after treatment (Figure 4E). Tumors from the MTDH knockdown cells grew at an equal rate, an observation consistent with the finding that MTDH does not affect primary tumor growth (Figure S7). Paclitaxel treatment significantly hampered tumor growth in mice injected with the control LM2 cells. However, the tumors still grew to 140% in volume 18 days after treatment, indicating a considerable degree of chemoresistance of these cancer cells. MTDH knockdown significantly sensitized the cells to paclitaxel treatment as tumor regression was observed immediately after the first treatment. The tumors eventually shrank to about 30% of the pre-treatment sizes 18 days after the initiation of treatment (Figure 4E, F). Similar results were obtained with another commonly used chemotherapeutic agent, doxorubicin (Figure S11).
ALDH3A1 and MET contribute to MTDH-induced chemoresistance
We performed drug uptake and retention assays for paclitaxel and doxorubicin in cancer cells with modified MTDH expression and found that MTDH does not decrease drug uptake or retention in these cells (Figure S12). Without a direct function in altering drug accumulation, MTDH may instead increase chemoresistance by promoting cellular survival against anti-neoplastic stresses. To further elucidate the molecular mechanism of MTDH-dependent chemresistance, we compared gene expression profiles of two different MTDH-knockdown LM2 cell lines with control cells. A similar comparison was also performed with LM2 cells co-cultured with HMVEC-L cells (Figure 5A and Table S7). In the latter analysis, LM2 and HMVEC-L cells were labeled with GFP and the SNARF dye, respectively, to allow FACS-sorting of the two cell populations before RNA extraction (Figure 5B). Since MTDH induces significant chemoresistance with or without HMVEC-L co-culture, we focus our attention on the significant genes (>2.5-fold change in expression and student’s t-test p<0.05) that are consistently present in both conditions. Twenty-three genes (including MTDH) were found to be under-expressed in MTDH-knockdown cells while 10 genes were overexpressed. Among the MTDH downregulated genes (i.e. genes upregulated following MTDH knockdown) are two cell death inducing genes TRAIL and BINP3. TRAIL encodes a TNF family cytokine that induces apoptosis in tumor cells. Combining TRAIL with conventional anticancer drugs has been showed to improve therapeutic efficacy of chemotherapies (Ballestrero et al., 2004). BNIP3 is a pro-apoptotic Bcl-2 family gene that has been shown to be involved in apoptotic, necrotic, and autophagic cell death (Mellor and Harris, 2007). Among the MTDH upregulated genes are several genes previously implicated in chemoresistance of cancer cells, including ALDH3A1, MET, HSP90AB1 (Bertram et al., 1996), HSP90AB3P, and HMOX1 (Tanaka et al., 2003). The expression pattern of these genes in MTDH knockdown cells was confirmed by qPCR analysis using samples from both cell cultures and xenograft tumors of LM2 (Figure 5C). The regulation of these genes by MTDH was additionally validated in three other breast cancer cell lines (MCF7, T47D and BT474) using MTDH knockdown (Figure S8). Furthermore, a significant correlation of the expression of MTDH and its downstream genes was found in the NCI60 panel of human cancer cell lines as well as in primary breast tumor samples by computational analysis (Figure S13).
Figure 5. ALDH3A1 and MET contribute to MTDH-mediated chemoresistance.

A, Expression pattern of the genes regulated in MTDH knockdown cells with or without HMVEC-L co-culture. Some genes previously implicated to promote (red) or suppress (green) cellular chemoresistance are highlighted. B, In the co-culture microarray experiment, HMVEC-L were pre-labeled with the SNARF dye and separated from GFP+ LM2 cells by FACS before microarray profiling. C, qPCR analysis of the expression patterns of genes down- (red) or upregulated (green) in MTDH knockdown cells identified by microarray analysis. D, ALDH3A1 expression levels (top) and the clonogenic ability (bottom) of the cells engineered with ALDH3A1 inducible knockdown. E. Gene expression (top) and clonogenic assays (bottom) of MET knockdown and MET/ALDH3A1 double knockdown. F, Gene expression (top) and clonogenic assays (bottom) of ALDH3A1 or MET overexpression rescue in LM2 cells with MTDH knockdown. D-F, data represent average ± SEM of three replicates. *p<0.05; **p<0.01 with a two-sided student’s t-test.
Among these candidate MTDH-downstream genes, ALDH3A1 (aldehyde dehydrogenase 3 family, member A1) and MET (hepatocyte growth factor receptor) are attractive targets because of their physiological functions and their expression pattern. Anti-neoplastic agents have been shown to produce oxidative stress in tumors during cancer chemotherapy. The effects are mediated, in part, by the generation of aldehydes that result from oxidative stress-induced lipid peroxidation. ALDH3A1 encodes an anti-oxidant enzyme with several postulated protective roles that include detoxification of peroxidic aldehydes and scavenging of free radicals. Its expression has been implicated in clinical resistance to cyclophosphomide (Sreerama and Sladek, 2001), which is a mainstay of chemotherapeutic regimens used to treat breast cancers. Aldehyde dehydrogenase activity has also been shown to be a marker of cancer stem cells and may contribute to their increased chemoresistance (Croker et al., 2008; Ginestier et al., 2007). Interestingly, as revealed by microarray analysis (Figure 5A) and further confirmed by qRT-PCR (data not shown), ALDH3A1 expression is 2 to 3-fold higher in the HMVEC-L co-culture as compared to the non-co-culture condition, while MTDH knockdown effectively represses ALDH3A1 expression in both conditions. Such an expression pattern matches the higher chemoresistance of cancer cells induced by HMVEC-L co-culture and chemosensitization by MTDH knockdown in both conditions. To investigate the functional importance of ALDH3A1 in MTDH-mediated chemoresistance, we engineered the LM2 cell line to express an inducible shRNA against ALDH3A1 in order to produce conditional knockdown of ALDH3A1. LM2 cells were more sensitive to chemotherapeutic agent paclitaxel, doxorubicin and 4-hydroxycylcophosphamide (4-HC) when ALDH3A1 knockdown was induced by addition of doxycycline, while release of ALDH3A1 repression restored the chemoresistance of LM2 cells (Figure 5D).
We also tested the chemoresistance function of MET. In human patients, enhanced expression or activation of MET was observed in nearly all tumor types. In most cases, its expression is associated both with resistance to radio- and chemo-therapy, and with poor prognosis(Birchmeier et al., 2003). In experimental models, exogenous hepatocyte growth factor (HGF) or overexpression of MET induces resistance to ionizing radiation and many chemotherapeutics, including doxorubicin, cisplatin, etoposide, camptothecin, paclitaxel, TNF and gefitinib in diverse human cancer cells from different tumor types, as well as in endothelial cells (Engelman et al., 2007; Wei and Au, 2005). MET knockdown in LM2 cells lead to a significant reduction of chemoresistance to doxorubicin, an effect that is similar to but weaker than that of MTDH knockdown (Figure 5E). When MET and ALDH3A1 were simultaneously knocked down in LM2 cells, the chemo-sensitizing effects reached a level comparable to that of MTDH knockdown (Figure 5E). We further tested the ability of ALDH3A1 and MET to rescue the chemoresistance phenotype in MTDH knockdown cells. Constitutive overexpression of ALDH3A1 or MET in the MTDH knockdown cells was able to partially recover the chemoresistance of LM2 cells to paclitaxel, doxorubicin and 4-HC (Figure 5F). Together, these results suggest that ALDH3A1 and MET are among MTDH downstream genes that collectively contribute to its role in broad-spectrum chemoresistance.
MTDH correlates with poor prognosis in clinical samples
To evaluate the clinical importance of MTDH in breast cancer, we stained the tissue microarray used in the previous FISH analysis by an antibody against MTDH. Among the 170 samples on the tissue microarray, 47% expressed MTDH in a moderate to high level (Figure 6A). Overexpression of MTDH is not linked to any specific breast tumor subtypes in terms of HER2 status, triple maker status (ER/PR/HER2), or the basal epithelial cell maker CK5/6 status (Figure S14), but is significantly associated with a higher risk of metastasis (log rank, p = 0.0058) and shorter survival time (p = 0.0008). Univariate survival analysis using the Cox proportional hazard model also suggested that a high MTDH expression is strongly associated with a higher hazard ratio (HR) and worse clinical outcomes (HR = 3.7, p = 0.01 for metastasis; HR = 8.3, p = 0.005 for cancer-related death). Of note, immunohistochemical analysis of CCNE2 protein expression (encoded by the only gene present in both poor-prognosis signatures identified by van’t Veer et al. and Wang et al.) in the same breast tumor tissue array did not reveal any significant correlation with metastasis (Figure S15). Interestingly, CCNE2 is located in very close proximity to the recurrent 8q22 genomic gain (Figure S15 and Table S5). It is possible that the recurrent presence of CCNE2 in multiple poor-prognosis signatures is due to its close physical linkage to 8q22.
Figure 6. MTDH is associated with poor prognosis of human breast tumors.

A,. Typical MTDH immunostaining images of a human breast cancer tissue microarray. B, MTDH protein levels are positively correlated with the FISH 8q22 DNA copy numbers. C, High MTDH protein level is associated with early metastasis in cancer patients. D, High MTDH expression is also linked to worse cancer-specific survival. E, A schematic model for the dual role of MTDH in breast cancer progression. In poor-prognosis tumors, 8q22 genomic gain leads to overexpression of MTDH, which in turn activate two parallel programs to promote chemoresistance and metastasis. Elevated expression of chemoresistance genes ALDH3A1, MET, HMOX1 and HSP90, as well as repression of apoptosis inducing genes TRAIL and BNIP3 promote the survival and outgrowth of cancer cells in the primary site as well as secondary organs in the face of physiological stress and chemotherapeutic challenges. MTDH additionally promotes metastasis by mediating tumor cell adhesion through interactions with unknown receptors and by activating pro-metastasis genes and suppressing metastasis-suppressive genes. Some of the molecular mediators of the MTDH function may play a role in both functional categories. For example, MET can promote both metastasis and chemoresistance, and endothelial adhesion can further enhance MTDH-mediated chemoresistance.
We further analyzed the correlation of MTDH protein levels with 8q22 DNA copy numbers using the samples with both successful immunostaining and FISH results. While the data showed that all but one of the tumors with 8q22 gain express abundant (medium or high) level of MTDH protein (Figure 6B, chi-square test p<0.001), a substantial fraction (12%) of samples with normal DNA copy numbers also have a high level of MTDH protein. Therefore, alternative mechanisms distinct from 8q22 gain may also result in MTDH activation in breast tumors. Nevertheless, survival analysis of the tumor samples in our tissue microarray and in the three published datasets consistently showed that MTDH activated by genomic gain or other means leads to similar clinical outcomes (Figure S16).
To further analyze the prognostic significance of MTDH, we performed Cox hazard ratio analysis of MTDH expression with the tissue samples stratified by other common clinicopathological paramters including ER, PR, HER2, and p53 status as well as the sizes of primary tumors at the time of cancer diagnosis (Table 2). MTDH expression level retained its prognostic significance in these analyses, suggesting that it is a prognostic factor independent of other clinicopathological factors. Indeed, a multivariate Cox analysis combining all of the above parameters with MTDH expression showed that the hazard of metastasis was still significantly higher with MTDH expressed (p = 0.023) even when all the other factors were considered.
Table 2.
Cox hazard ratios for metastasis in breast cancer based on MTDH expression levels in tissue array analysis
| Pathological Parameter | MTDH hazard ratio in stratified analysis * |
Multivariate Analysis # |
||
|---|---|---|---|---|
| HZ (95% CI) | p | HZ (95% CI) | p | |
| MTDH | 3.70 (1.36-10.0) | 0.010 | 4.28 (1.32-13.9) | 0.015 |
| ER | 3.91 (1.30-11.8) | 0.015 | 3.13 (0.59-16.5) | 0.180 |
| PR | 3.84 (1.27-11.6) | 0.016 | 0.682 (0.17-3.76) | 0.800 |
| HER2 | 3.20 (1.20-9.99) | 0.026 | 4.79 (1.75-19.6) | 0.030 |
| P53 | 4.09 (1.34-12.5) | 0.012 | 0.60 (0.19-1.94) | 0.400 |
| ER+PR+HER2 | 2.47 (1.25-9.56) | 0.017 | 3.15 (0.51-19.3) | 0.200 |
| Tumor size | 4.21 (1.28-12.5) | 0.007 | 2.38 (0.87-6.52) | 0.090 |
Univariate analysis of MTDH expression alone (the first row), or stratified by the indicated second parameter (all other rows).
Multivariate analysis of all the parameters including MTDH, ER, PR, HER2, P53 and tumor size.
DISCUSSION
In this study, we used the ACE algorithm to unveil functionally significant cytogenetic events directly linked to altered gene expression in poor-prognosis tumors. High-throughput genomic profiling methods such as aCGH and SNP arrays have facilitated the recent discovery of several cancer genes (Garraway et al., 2005; Kim et al., 2006). As an addition to the repertoire of integrative genomic analysis tools, ACE is particularly useful when cytogenetic data are not available, or it can be used as a complementary strategy to fine map results obtained from cytogenetic analyses and help narrow down the list of genes for functional analysis. A further advantage of ACE is that it can detect regional epigenetic alterations that can not be discerned by the aCGH or the SNP array approach (Figure S1). Additionally, ACE provides a direct link between cytogenetic events and gene activity changes, thereby facilitating the search for functionally important candidate genes. Given the large amount of archived gene expression data available in public domains and the difficulty in obtaining matched cancer samples, ACE will be a useful data-mining tool to complement the direct copy number detection methods to help shed light on the functional mechanism of cancer progression.
Our ACE analysis of breast cancer, together with clinical and functional studies of MTDH, strongly suggested that MTDH is a metastasis gene with great prognostic potential and therapeutic value. Brown et al previously used phage display to identify MTDH as a homing receptor that mediates the adhesion of the 4T1 murine mammary tumor cell line to lung endothelial cells and that promotes lung metastasis (Brown and Ruoslahti, 2004). In that study, only the mouse 4T1 cell line and the biologically irrelevant HEK 293T cell line were used to analyze the lung-targeting function of MTDH. The involvement of MTDH in human cancer, however, has not been previously reported. In this study, we used an extensive collection of human breast tumor samples to demonstrate that elevated MTDH protein level is an important prognostic factor independent of other clinicopathological factors. Our results indicated that a substantial proportion of human breast tumors exhibit MTDH genomic copy gains with a subsequent increase in MTDH expression, which is clearly associated with poor survival and higher risk of progression. We further firmly validated the functional importance of MTDH in systemic metastasis using a well-established model for human breast cancer metastasis. The importance of MTDH in cancer metastasis might not be limited to promoting lung-specific spread of breast tumor cells. Although MTDH was previously reported to enhance murine mammary tumor cell adhesion to lung endothelial cells (Brown and Ruoslahti, 2004), we showed that MTDH also enhances the affinity of human breast cancer cells to other endothelial cell types, consistent with its function to increase systemic metastasis in vivo of a mildly metastatic cell line. However, MTDH overexpression alone in non-metastatic breast cancer cell lines, including BT20, ZR-75-1 and ZR-75-30, was not sufficient to allow these cells to gain metastasis ability in animal assays (data not shown). This is consistent with recent studies showing that simultaneous overexpression of multiple metastasis genes are often required to achieve high metastatic capabilities (Gupta et al., 2007; Kang et al., 2003; Minn et al., 2005). Nevertheless, our in vitro, in vivo and clinical studies firmly established MTDH as an appealing target for therapeutic intervention of metastatic diseases.
Current standard treatment for breast cancer uses the combination of surgery to remove localized disease and chemotherapy to eliminate systemic spreading. However, relapsed breast cancers almost invariably acquire resistance to chemotherapy and are often inoperable. Thus, over 90% of breast cancer related deaths are not due to cancer at the primary site, but rather due to the spread of chemoresistant cancer cells from breast to secondary vital organs, such as lung, bone, liver and brain. Metastasis and chemoresistance remain two major obstacles and challenges to curative therapy. Our study uncovered a role for MTDH in chemoresistance of cancer cells. Thus, MTDH may be among an important class of genes that play dual roles in metastasis and chemoresistance (Figure 6E). This may explain why some of the metastasis genes are selected for in the primary tumor — presumably as a consequence of their ability to endow cancer cells with enhanced tolerance to therapeutic and physiological stresses that human tumors may endure — but do not confer an apparent growth advantage in animal tumorigenesis assays (Bernards and Weinberg, 2002). In addition to promoting chemoresistance of primary tumors, MTDH may also increase the risk of metastatic recurrence by enhancing the survival of metastatic lesions against chemotherapy (Figure 6E), although this has not been proven directly in our current study due to difficulty to segregate the two functions (metastasis and chemoresistance) of MTDH in the in vivo metastasis assays. Ongoing studies in our laboratory have also validated the dual-functions of MTDH in metastasis and chemoresistance in prostate cancer (Hu et al., manuscript in preparation), suggesting a potential broader functional involvement of MTDH in the progression of a variety of cancers.
Microarray profiling of MTDH-knockdown cells revealed several genes, including ALDH3A1 and MET, that collectively contribute to the multi-drug chemoresistance function of MTDH. Several other genes identified by the microarray experiment may also contribute to the pro-metastasis function of MTDH. For example, genes that are downregulated by MTDH inhibition include several previously reported metastasis-promoting genes such as MET, ADAMTS1 and CTGF (Kang et al., 2003; Lorenzato et al., 2002). Conversely, several genes that have been reported to suppress metastasis, including GPR56, TIMP3 and TRAIL (Lin et al., 2002; Manka et al., 2005; Xu et al., 2006), were overexpressed in the MTDH-knockdown line (Fig 5A and Table S7). The mechanism of regulation of these downstream genes by MTDH and the identification of the functional partners of MTDH will be important questions to be addressed by future studies.
In conclusion, we used a combination of computational biology, in vivo and in vitro functional metastasis assays, and extensive clinical correlation analysis to identify the 8q22 poor-prognosis genomic gain that harbors the dual functional metastasis gene MTDH. Overexpression of MTDH occurs in up to 40% of breast cancer patients and promotes metastatic seeding as well as chemoresistance of breast tumors. There are several potential applications of this study in the clinical management of human breast cancer. Genomic gain and overexpression of MTDH can become a powerful prognosis marker independent from other well-established markers for breast cancer. Molecular targeting of the dual-function metastasis gene MTDH may not only prevent the seeding of breast cancer cells to lung and other vital organs but also sensitize cancer cells to chemotherapy, thereby stopping the deadly spread of breast cancer.
EXPERIMENTAL PROCEDURES
Development of ACE (Analysis of CNAs by Expression) algorithm
ACE detects genetic alterations in three steps: calculating neighborhood scores (NS) for each chromosomal locus as an indicator of CNA likelihood at that locus, estimating the significance of the NS, and defining the regions of gain and loss.
The expression score (ES) for each gene is first calculated according to the correlation of its expression with the phenotypes in comparison. In this manuscript, paired t-statistics (for ovarian cancer cell lines) or independent t-statistics (for other datasets) were used to score gene expression. In general, other metrics can also be used. Consider the genes 1, 2, ⋯, N on a chromosome ordered by their physical positions. We define the NS at locus i as the weighted sum of the ES of this chromosome:
where wji is the weight of gene j. Because the linkage strength between two loci becomes weaker as the distance increases, the weight wji decreases when locus j is farther way from the locus i. The contribution from each gene is weighted by a Gaussian function.
where c is a constant to normalize all NS into a range of [-1, 1]. The variation parameter 2σ2 controls the weight decay rate and is arbitrarily set to 100 in the analyses presented here. An analysis using varying 2σ2 values from 20 to 200 showed similar results with slight shifts at the boundaries of detected regions. For each locus, only the genes in its physical proximity will have measurable influence on its NS because of weight decay. Positive and negative NS suggest genomic gain and loss, respectively. To evaluate the significance of the NS, the gene positions (or sample class labels if the sample size is large enough) are permuted 1,000 times, and each time the NS are recomputed. The p values of observed NS are then computed using the distribution of permuted NS and adjusted to FDR-q values by the Benjamini-Hochberg procedure. In all the CNA analyses presented in this manuscript, we defined a region of genomic gain as one with at least 20 continuous positive NS of FDR-q<0.01, or a region of genomic loss when such NS are all negative. In the epigenetic analysis, we used a cutoff of 5 continuous NS as epigenetic regulation usually has a smaller functioning range.
ACE has been implemented as a software tool to analyze expression data obtained from various array platforms and is available at Supplemental Data online.
Tumorigenesis and metastasis assays in nude mice
All animal work was done in accordance with the guidelines of the IACUC of Princeton University under approved protocols. 2 × 105 cells were washed in PBS and injected intravenously to female athymic Ncr-nu/nu mice to study the lung metastasis activity as previously described (Minn et al., 2005). For bone metastasis analysis, 1 × 105 cells were injected to the left ventricle of the animal heart as described (Kang et al., 2003). Non-invasive bioluminescence imaging was performed to quantify the metastasis burden at the target organs using the IVIS 200 Imaging System (Caliper Life Sciences) as previously described (Minn et al., 2005).
To study primary tumorigenesis, cancer cells harvested from culture were resuspended in PBS at a concentration of 1 × 107 cells/ml. An incision was made in the abdomen and the skin was recessed to locate the #4 mammary fat pad, into which 105 cells (10 μl) were injected under a dissection microscope. The primary tumor volume was monitored weekly as previously described (Minn et al., 2005).
Human tumor samples
Tumor specimens were obtained from the Cancer Institute of New Jersey with informed consent from all subjects in accordance with the Institutional Review Boards of Princeton University and the University of Medicine and Dentistry of New Jersey. Details on the characterization of each tumor specimen can be found in Supplemental Experimental Procedures.
Statistical analysis
The Kaplan–Meier method was used to estimate survival curves for the patients and animals. Log rank test and Wilcoxon test were used to compare the differences between curves. Two-sided Wilcoxon rank test was performed to analyze the bioluminescent imaging results in the in vivo studies. A two-sided independent student’s t-test without equal variance assumption was performed to analyze the results of luciferase assays and clonogenic assays.
Supplementary Material
Supplemental Data include Supplemental Experimental Procedures, nine tables, sixteen figures and the ACE software package and can be found with this article online at XXX.
Acknowledgments
We thank J. Massagué, D. Botstein, S. Tavazoie, Y. Shi, O. Matveeva for insightful discussions and technical suggestions, L. M. Staudt for the pRSMX vector and C.J. Kirkpatrick for STA cell line, L. Cong, J. Friedman and L. Goodell at Tissue Analytic Service Core of Cancer Institute of New Jersey for assistance with immunohistochemistry analysis of tissue arrays, Y. Xiao, K. Jones and C. Lee at Dana Farber Cancer Institute Cytogenetic Core for FISH analysis. Y.K. was funded by a Department of Defense Era of Hope Scholar Award, and grants from the NIH (R01CA134519), American Cancer Society, the Susan G. Komen Foundation and the New Jersey Commission on Cancer Research. G.H. is supported by postdoctoral fellowships from the New Jersey Commission on Cancer Research.
Footnotes
ACCESSION NUMBER Microarray data reported herein have been deposited at the NCBI Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/) with the accession GSE9187.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ballestrero A, Nencioni A, Boy D, Rocco I, Garuti A, Mela GS, Van Parijs L, Brossart P, Wesselborg S, Patrone F. Tumor necrosis factor-related apoptosis-inducing ligand cooperates with anticancer drugs to overcome chemoresistance in antiapoptotic Bcl-2 family members expressing jurkat cells. Clin Cancer Res. 2004;10:1463–1470. doi: 10.1158/1078-0432.ccr-1365-02. [DOI] [PubMed] [Google Scholar]
- Bergamaschi A, Kim YH, Wang P, Sorlie T, Hernandez-Boussard T, Lonning PE, Tibshirani R, Borresen-Dale AL, Pollack JR. Distinct patterns of DNA copy number alteration are associated with different clinicopathological features and gene-expression subtypes of breast cancer. Genes Chromosomes Cancer. 2006;45:1033–1040. doi: 10.1002/gcc.20366. [DOI] [PubMed] [Google Scholar]
- Bernards R, Weinberg RA. A progression puzzle. Nature. 2002;418:823. doi: 10.1038/418823a. [DOI] [PubMed] [Google Scholar]
- Bertram J, Palfner K, Hiddemann W, Kneba M. Increase of P-glycoprotein-mediated drug resistance by hsp 90 beta. Anti-cancer drugs. 1996;7:838–845. doi: 10.1097/00001813-199611000-00004. [DOI] [PubMed] [Google Scholar]
- Birchmeier C, Birchmeier W, Gherardi E, Vande Woude GF. Met, metastasis, motility and more. Nat Rev Mol Cell Biol. 2003;4:915–925. doi: 10.1038/nrm1261. [DOI] [PubMed] [Google Scholar]
- Britt DE, Yang DF, Yang DQ, Flanagan D, Callanan H, Lim YP, Lin SH, Hixson DC. Identification of a novel protein, LYRIC, localized to tight junctions of polarized epithelial cells. Experimental cell research. 2004;300:134–148. doi: 10.1016/j.yexcr.2004.06.026. [DOI] [PubMed] [Google Scholar]
- Brown DM, Ruoslahti E. Metadherin, a cell surface protein in breast tumors that mediates lung metastasis. Cancer Cell. 2004;5:365–374. doi: 10.1016/s1535-6108(04)00079-0. [DOI] [PubMed] [Google Scholar]
- Chin L, Gray JW. Translating insights from the cancer genome into clinical practice. Nature. 2008;452:553–563. doi: 10.1038/nature06914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark EA, Golub TR, Lander ES, Hynes RO. Genomic analysis of metastasis reveals an essential role for RhoC. Nature. 2000;406:532–535. doi: 10.1038/35020106. [DOI] [PubMed] [Google Scholar]
- Croker AK, Goodale D, Chu J, Postenka C, Hedley BD, Hess DA, Allan AL. High aldehyde dehydrogenase and expression of cancer stem cell markers selects for breast cancer cells with enhanced malignant and metastatic ability. Journal of cellular and molecular medicine. 2008 doi: 10.1111/j.1582-4934.2008.00455.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, Lindeman N, Gale CM, Zhao X, Christensen J, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science (New York, NY) 2007;316:1039–1043. doi: 10.1126/science.1141478. [DOI] [PubMed] [Google Scholar]
- Fan C, Oh DS, Wessels L, Weigelt B, Nuyten DS, Nobel AB, van’t Veer LJ, Perou CM. Concordance among gene-expression-based predictors for breast cancer. The New England journal of medicine. 2006;355:560–569. doi: 10.1056/NEJMoa052933. [DOI] [PubMed] [Google Scholar]
- Fidler IJ. The pathogenesis of cancer metastasis: the ‘seed and soil’ hypothesis revisited. Nature reviews. 2003;3:453–458. doi: 10.1038/nrc1098. [DOI] [PubMed] [Google Scholar]
- Garraway LA, Widlund HR, Rubin MA, Getz G, Berger AJ, Ramaswamy S, Beroukhim R, Milner DA, Granter SR, Du J, et al. Integrative genomic analyses identify MITF as a lineage survival oncogene amplified in malignant melanoma. Nature. 2005;436:117–122. doi: 10.1038/nature03664. [DOI] [PubMed] [Google Scholar]
- Ginestier C, Hur MH, Charafe-Jauffret E, Monville F, Dutcher J, Brown M, Jacquemier J, Viens P, Kleer CG, Liu S, et al. ALDH1 Is a Marker of Normal and Malignant Human Mammary Stem Cells and a Predictor of Poor Clinical Outcome. Cell stem cell. 2007;1:555–567. doi: 10.1016/j.stem.2007.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta GP, Massague J. Cancer metastasis: building a framework. Cell. 2006;127:679–695. doi: 10.1016/j.cell.2006.11.001. [DOI] [PubMed] [Google Scholar]
- Gupta GP, Nguyen DX, Chiang AC, Bos PD, Kim JY, Nadal C, Gomis RR, Manova-Todorova K, Massague J. Mediators of vascular remodelling coopted for sequential steps in lung metastasis. Nature. 2007;446:765–770. doi: 10.1038/nature05760. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- Kang DC, Su ZZ, Sarkar D, Emdad L, Volsky DJ, Fisher PB. Cloning and characterization of HIV-1-inducible astrocyte elevated gene-1, AEG-1. Gene. 2005;353:8–15. doi: 10.1016/j.gene.2005.04.006. [DOI] [PubMed] [Google Scholar]
- Kang Y. Functional genomic analysis of cancer metastasis: biologic insights and clinical implications. Expert Rev Mol Diagn. 2005;5:385–395. doi: 10.1586/14737159.5.3.385. [DOI] [PubMed] [Google Scholar]
- Kang Y, Siegel PM, Shu W, Drobnjak M, Kakonen SM, Cordon-Cardo C, Guise TA, Massague J. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell. 2003;3:537–549. doi: 10.1016/s1535-6108(03)00132-6. [DOI] [PubMed] [Google Scholar]
- Kim M, Gans JD, Nogueira C, Wang A, Paik JH, Feng B, Brennan C, Hahn WC, Cordon-Cardo C, Wagner SN, et al. Comparative oncogenomics identifies NEDD9 as a melanoma metastasis gene. Cell. 2006;125:1269–1281. doi: 10.1016/j.cell.2006.06.008. [DOI] [PubMed] [Google Scholar]
- Lin T, Huang X, Gu J, Zhang L, Roth JA, Xiong M, Curley SA, Yu Y, Hunt KK, Fang B. Long-term tumor-free survival from treatment with the GFP-TRAIL fusion gene expressed from the hTERT promoter in breast cancer cells. Oncogene. 2002;21:8020–8028. doi: 10.1038/sj.onc.1205926. [DOI] [PubMed] [Google Scholar]
- Lorenzato A, Olivero M, Patane S, Rosso E, Oliaro A, Comoglio PM, Di Renzo MF. Novel somatic mutations of the MET oncogene in human carcinoma metastases activating cell motility and invasion. Cancer Res. 2002;62:7025–7030. [PubMed] [Google Scholar]
- Manka D, Spicer Z, Millhorn DE. Bcl-2/adenovirus E1B 19 kDa interacting protein-3 knockdown enables growth of breast cancer metastases in the lung, liver, and bone. Cancer research. 2005;65:11689–11693. doi: 10.1158/0008-5472.CAN-05-3091. [DOI] [PubMed] [Google Scholar]
- Mellor HR, Harris AL. The role of the hypoxia-inducible BH3-only proteins BNIP3 and BNIP3L in cancer. Cancer metastasis reviews. 2007;26:553–566. doi: 10.1007/s10555-007-9080-0. [DOI] [PubMed] [Google Scholar]
- Minn AJ, Gupta GP, Siegel PM, Bos PD, Shu W, Giri DD, Viale A, Olshen AB, Gerald WL, Massague J. Genes that mediate breast cancer metastasis to lung. Nature. 2005;436:518–524. doi: 10.1038/nature03799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naylor TL, Greshock J, Wang Y, Colligon T, Yu QC, Clemmer V, Zaks TZ, Weber BL. High resolution genomic analysis of sporadic breast cancer using array-based comparative genomic hybridization. Breast Cancer Res. 2005;7:R1186–1198. doi: 10.1186/bcr1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park H, Kim Y, Lim Y, Han I, Oh ES. Syndecan-2 mediates adhesion and proliferation of colon carcinoma cells. The Journal of biological chemistry. 2002;277:29730–29736. doi: 10.1074/jbc.M202435200. [DOI] [PubMed] [Google Scholar]
- Pollack JR, Sorlie T, Perou CM, Rees CA, Jeffrey SS, Lonning PE, Tibshirani R, Botstein D, Borresen-Dale AL, Brown PO. Microarray analysis reveals a major direct role of DNA copy number alteration in the transcriptional program of human breast tumors. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:12963–12968. doi: 10.1073/pnas.162471999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramaswamy S, Ross KN, Lander ES, Golub TR. A molecular signature of metastasis in primary solid tumors. Nat Genet. 2003;33:49–54. doi: 10.1038/ng1060. [DOI] [PubMed] [Google Scholar]
- Ried T, Just KE, Holtgreve-Grez H, du Manoir S, Speicher MR, Schrock E, Latham C, Blegen H, Zetterberg A, Cremer T, et al. Comparative genomic hybridization of formalin-fixed, paraffin-embedded breast tumors reveals different patterns of chromosomal gains and losses in fibroadenomas and diploid and aneuploid carcinomas. Cancer research. 1995;55:5415–5423. [PubMed] [Google Scholar]
- Sreerama L, Sladek NE. Primary breast tumor levels of suspected molecular determinants of cellular sensitivity to cyclophosphamide, ifosfamide, and certain other anticancer agents as predictors of paired metastatic tumor levels of these determinants. Rational individualization of cancer chemotherapeutic regimens. Cancer chemotherapy and pharmacology. 2001;47:255–262. doi: 10.1007/s002800000208. [DOI] [PubMed] [Google Scholar]
- Steeg PS. Tumor metastasis: mechanistic insights and clinical challenges. Nature medicine. 2006;12:895–904. doi: 10.1038/nm1469. [DOI] [PubMed] [Google Scholar]
- Tanaka S, Akaike T, Fang J, Beppu T, Ogawa M, Tamura F, Miyamoto Y, Maeda H. Antiapoptotic effect of haem oxygenase-1 induced by nitric oxide in experimental solid tumour. Br J Cancer. 2003;88:902–909. doi: 10.1038/sj.bjc.6600830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van ’t Veer LJ, Dai H, van de Vijver MJ, He YD, Hart AA, Mao M, Peterse HL, van der Kooy K, Marton MJ, Witteveen AT, et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature. 2002;415:530–536. doi: 10.1038/415530a. [DOI] [PubMed] [Google Scholar]
- van de Vijver MJ, He YD, van’t Veer LJ, Dai H, Hart AA, Voskuil DW, Schreiber GJ, Peterse JL, Roberts C, Marton MJ, et al. A gene-expression signature as a predictor of survival in breast cancer. The New England journal of medicine. 2002;347:1999–2009. doi: 10.1056/NEJMoa021967. [DOI] [PubMed] [Google Scholar]
- Wang Y, Klijn JG, Zhang Y, Sieuwerts AM, Look MP, Yang F, Talantov D, Timmermans M, Meijer-van Gelder ME, Yu J, et al. Gene-expression profiles to predict distant metastasis of lymph-node-negative primary breast cancer. Lancet. 2005;365:671–679. doi: 10.1016/S0140-6736(05)17947-1. [DOI] [PubMed] [Google Scholar]
- Wei Y, Au JL-S. Role of tumor microenvironment in mediating chemoresistance. Cancer Growth and Progression 2005 [Google Scholar]; Meadows G, editor. Integration/Interaction of Oncologic Growth. Vol. 15. Kluwer Academic Publishers; pp. 285–321. [Google Scholar]
- Xu L, Begum S, Hearn JD, Hynes RO. GPR56, an atypical G protein-coupled receptor, binds tissue transglutaminase, TG2, and inhibits melanoma tumor growth and metastasis. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:9023–9028. doi: 10.1073/pnas.0602681103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Mani SA, Donaher JL, Ramaswamy S, Itzykson RA, Come C, Savagner P, Gitelman I, Richardson A, Weinberg RA. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell. 2004;117:927–939. doi: 10.1016/j.cell.2004.06.006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Data include Supplemental Experimental Procedures, nine tables, sixteen figures and the ACE software package and can be found with this article online at XXX.