

SUMMARY
Sonic Hedgehog (Shh) has dual roles in vertebrate development, as it promotes progenitor cell proliferation and induces tissue patterning. Here we show mitogenic and patterning functions of Shh can be uncoupled from one another. Using a genetic approach to selectively inhibit Shh-proteoglycan interactions in a mouse model, we show binding of Shh to proteoglycans is required for proliferation of neural stem/precursor cells but not for tissue patterning. Shh-proteoglycan interactions regulate both spatial and temporal features of Shh signaling. Proteoglycans localize Shh to specialized mitogenic niches and also act at the single cell level to regulate the duration of Shh signaling, thereby promoting a gene expression program important for cell division. As activation of the Shh pathway is a feature of diverse human cancers, selective stimulation of proliferation by Shh-proteoglycan interactions may also figure prominently in neoplastic growth.
Development of complex tissues requires concomitant growth and cell fate specification. One mechanism for achieving spatial and temporal coordination of size and form is to utilize the same signaling molecules for both processes. The morphogen Hedgehog (Hh), and its mammalian counterparts Sonic, Desert and Indian Hedgehog (Shh, Dhh and Ihh), are critical in the growth and patterning of developing embryos1. One model for morphogen activity postulates Hedgehog proteins disperse from a localized source and form a gradient that patterns fields of responsive cells 2. Controlled ligand distribution may also localize stem cell proliferation to specialized niches3,4. By influencing patterning and proliferation, Hedgehog proteins could coordinate form and size to allow scaling of developing organisms.
Here we asked whether proteoglycans differentially regulate Shh-dependent proliferation and patterning. Genetic evidence in Drosophila indicates proteoglycans are required for Hh dispersal and gradient formation5,6. Biochemical evidence indicates proteoglycans bind to Hh and the receptor component Ihog to affect Hh activity7,8. In Drosophila, proteoglycans critical for Hh functions are heparan sulfate proteoglycans (HSPGs), and the core protein components are glypicans (GPI-linked proteins) 6,9–12. Dally-like glypicans are required for Hedgehog and Wingless dispersal and responses, modulating signaling of both ligands13. However, how HSPGs affect Hedgehog signaling and biological responses are not yet understood.
Proteoglycans (PGs) have also been implicated in Hedgehog-pathway signaling in mammalian systems11,12,14. Loss of glypican GPC3 causes an overall growth increase, reflecting changes in Shh and/or IGF signaling11, while mutations in HSPG synthesizing enzymes cause dramatic defects that may reflect changes in FGF, Wnt, and/or Shh responses15,16. To investigate the physiologic role of Shh-proteoglycan interactions, we took the alternative approach of mutating Shh itself. Shh contains an N-terminal Cardin-Weintraub motif that mediates Shh-proteoglycan interactions (KRRHPKK). As mutations in this motif (R34A/K38A, designated ShhAla) reduce high-affinity Shh-proteoglycan interactions without altering Shh’s affinity for its receptor Patched (Ptc)14, we can use this mutation to investigate Shh-HSPG interactions, without confounding effects due to other growth factors that bind proteoglycans.
To identify on an organismal level responses that require Shh-proteoglycan interactions we generated mice in which wild type Shh is replaced with ShhAla. While Shh is needed for growth and patterning of diverse tissues, we find proteoglycan interactions selectively affect Shh-induced proliferation rather than Shh-induced patterning. Proteoglycans localize Shh ligand to mitogenic niches in developing brain to promote proliferation of neural stem/precursor cells. Proteoglycans also function at the level of individual responding cells; cell-associated proteoglycans bind Shh and alter ligand perdurance. In this way, Shh-proteoglycan interactions stimulate expression of bmi-1, D-type Cyclins and other genes implicated in proliferation and neoplastic growth17, 18. These studies indicate that HSPGs selectively promote mitogenic responses to Shh.
RESULTS
HSPG binding motif mutations affect Shh-PG interactions
We identified a conserved Cardin-Weintraub motif in Shh that is responsible for high affinity binding of Shh to HSPGs14. To determine whether mutations in this motif can be used to identify functions of Shh-proteoglycan interactions, we quantitatively evaluated Shh binding to heparin. We assessed binding of equal amounts of wild type Shh or ShhAla to heparin sulfate-coated plates using alkaline phosphatase tagged Shh isoforms. We measured binding in the absence, or presence of increasing amounts of soluble heparin sulfate. Overall binding of mutant Shh was significantly less than wild type. Furthermore, higher concentrations of soluble heparin sulfate were required to abolish binding of wild type Shh to immobilized heparin than concentrations needed to abolish binding of ShhAla (Fig. 1a). Binding of ShhAla to endogenous proteoglycans was also strikingly impaired. Wild type Shh, but not ShhAla, binds endogenous proteoglycans in CNS tissue sections (14, Fig. 1b). Pre-treatment of sections with heparinases, which remove heparan sulfates, or PI-PLC, which removes GPI-linked molecules, prevented binding of wild type Shh to tissue proteoglycans (Fig. 1b, Supplementary Fig. 1). As glypicans are HSPGs with a GPI-linkage, these results suggest that wild type Shh binds glypican proteoglycans in the developing cerebellum and that the ShhAla mutation interferes with binding to these endogenous glypicans in developing cerebellum. We next introduced the ShhAla mutation into a full length Shh expression vector and produced protein in HEK cells. The ShhAla precursor undergoes proteolytic cleavage to generate a mature isoform of the correct size (Fig. 1c) that, like wild type Shh, is palmitoylated (Fig. 1d)19. We previously demonstrated that ShhAla binds Ptc with similar affinity as wild type ligand 14. Together, these data show ShhAla can be used as a specific reagent to determine functions of Shh-proteoglycan interactions.
Figure 1. ShhAla specifically alters proteoglycan binding.
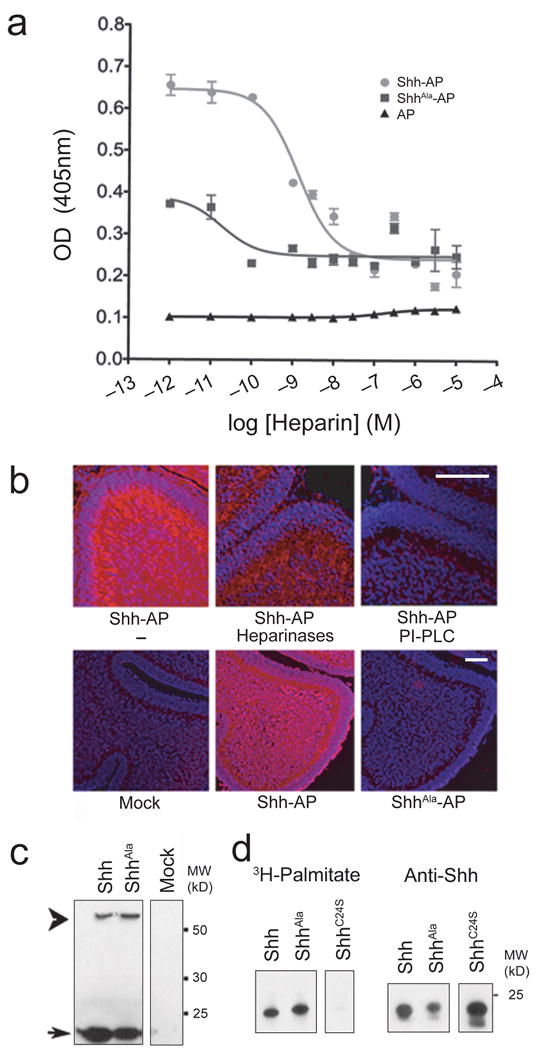
(a) ShhAla shows reduced binding to heparin-coated plates. Shh-AP (circles), ShhAla-AP (squares), or AP (triangles) were incubated with heparin-coated plates in the presence of increasing concentrations of soluble heparin.
(b) Shh binds GPI-linked proteoglycans in cerebellar sections; ShhAla does not. P6 sections treated with vehicle control (−), Heparinases or PI-PLC, incubated with Shh-AP, ShhAla-AP or vehicle controls, and processed for binding of AP-tagged ligand. Scale bar, 100 μm.
(c) ShhAla is processed to mature isoform. Lysates of HEK293 transfected with Shh or ShhAla were analyzed by immunoblot with anti-Shh. Immature 45kD (arrowhead) and mature 20kD isoform (arrow) are seen.
(d) ShhAla is palmitoylated. HEK293 cells expressing Shh, ShhAla or ShhC24S were labeled with 3H-palmitate, analyzed for palmitoylation, and probed with anti-Shh. Shh and ShhAla are palmitoylated. ShhC24S is not.
Using homologous recombination, we generated knock-in mice in which endogenous Shh was replaced with ShhAla (Supplementary Fig. 2). With this targeting strategy, a loxP site remained within the intron between exons 1 and 2. Because intronic elements can affect Shh expression, we also generated a line of control animals wherein the only alteration is this loxP site (ShhCtl); ShhCtl/Ctl animals are indistinguishable from wild type. Furthermore, the ShhAla mutation itself does not alter the expression of Shh protein in vivo: Shh levels are indistinguishable in Shh+/+, ShhAla/+ and ShhAla/Ala tissues (Supplementary Fig. 2e, f). This novel genetic approach enables us to determine the role of Shh binding to proteoglycans without interference from proteoglycan-dependent modulation of other growth factor pathways.
Shh-PG interactions affect growth but not patterning
Shh−/− animals are embryonic lethal and display holoprosencephaly and limb patterning -defects20. Hypomorphic alleles of Shh usually alter size and pattern19. In contrast, homozygous ShhAla/Ala mice only exhibited growth defects and showed no patterning defects. While Shh is critical in generating and patterning diverse organs, mutant animals were viable and fertile, with all organs present and correctly localized. In contrast, ShhAla/Ala mice displayed multiple growth abnormalities. Overall size was reduced in mutant animals, with particular differences in size of the brain, spinal cord and eyes. Body weight of ShhAla/Ala mice was 11% less than wild type or ShhCtl/Ctl animals, and brain weight was reduced by 13% (p=0.0018). There was, however, disproportionate hypoplasia of cerebellum and olfactory bulb (Fig. 2a). In contrast to these clear differences in tissue growth, when we examined areas where Shh is critical for pattern formation1,21,22, we found ShhAla/Ala mice displayed normal digit number and shape, and the spinal cord laminae were correctly formed (Fig. 2a,b, Supplementary Fig. 3). Furthermore, while holoprosencephaly and cyclopia are cardinal features of Shh−/− mice20, ShhAla/Ala animals displayed normal separation of cerebral ventricles and two well-spaced (but small) eyes (Fig. 2a). Together these findings indicate that Shh-proteoglycan interactions mediated by the Cardin-Weintraub motif are specifically required for growth regulation but not for patterning activities.
Figure 2. ShhAla/Ala and ShhAla/− mice exhibit defects in growth, with normal patterning.

(a) Skeletal morphology, body and brain patterning are normal in ShhAla/Ala mice, but size of adult ShhAla/Ala mice is 11% less than wild type, and sizes of olfactory bulbs and cerebella are 30 and 31% reduced (arrows) respectively. Sagittal view of cerebellum shows normal patterning with reduced size in ShhAla/Ala. Scale bar, 1 mm. ShhAla/Ala animals also display well-spaced eyes.
(b) Spinal cord patterning is normal in ShhAla/Ala mice. In situ hybridization for Shh (expressed in notochord and floor plate), Nkx2.2 (motor neuron precursors), Nkx6.1 (ventral spinal cord), Isl1 (dorsal root ganglia and motor neurons), and Dbx1 (V0 interneurons) in E10.5 littermates. Scale bar, 100 μm.
(c) Growth defects are seen in ShhAla/−, without defects in patterning. Olfactory bulb and cerebellum are reduced in size (red arrows). Scale bar, 1 mm. Eyes of ShhAla/− mice are well spaced.
In mice, one copy of Shh is sufficient for normal development as Shh+/− mice exhibit normal patterning and growth20. One copy of ShhAla is also sufficient for patterning, as ShhAla/− mice were viable with normal limbs and digits. However, even more dramatic changes in growth were evident in ShhAla/− than in ShhAla/Ala animals (Fig. 2c). The overall size of ShhAla/− mice, and in particular, cerebellar and olfactory bulb size were greatly reduced compared to Shh+/− animals. However, even when one copy of ShhAla is present, no defects were seen in tissue patterning (Fig. 2c). The characterization of ShhAla/Ala and ShhAla/− mice underscores the selectivity of the phenotype, and indicates Shh-proteoglycan interactions particularly regulate tissue growth.
Shh-PG interactions promote stem/precursor proliferation
To determine why proteoglycan interactions are critical for tissue growth, we focused on the cerebellum, as cerebella of ShhAla/Ala mice are one third smaller than their wild type littermates. As Shh regulates cell division and cell death in the nervous system23,24, we asked whether the decrease in cerebellar size in ShhAla/Ala and ShhAla/− mice reflects abnormal proliferation or apoptosis. In early postnatal life, granule cell precursors (GCPs) undergo extensive proliferation in the external granule cell layer (EGL) then exit the cell cycle and migrate inwards past the Purkinje cell layer to reside in the internal granule cell layer (IGL). Previous studies have shown Purkinje cell-synthesized Shh promotes GCP proliferation23,25. In ShhAla/Ala mice, GCP proliferation in the EGL is approximately 30% less than in wild type, as detected by percentage of EGL cells positive for M-phase marker phospho-Histone H3 and by S-phase labeling using bromodeoxyuridine (BrdU) (Fig. 3a–c). This decreased proliferation, which was seen throughout the cerebellum (Fig. 3d), indicates Shh-proteoglycan interactions promote mitogenesis of cerebellar precursors during early postnatal life across the rostral-caudal axis of the developing hindbrain.
Figure 3. Reduced proliferation of ShhAla/Ala cerebellar granule precursors is seen in the EGL of developing mice.

(a) p-Histone H3 (p-H3)-positive or BrdU (BrdU)-positive (both in red) cells are fewer in the EGL of P3 ShhAla/Ala mice as compared to Shh+/+ littermates. DAPI is in grey or blue. Scale bar, 100 μm (left), 50 μm (right).
(b) p-Histone H3 mitotic indices are reduced in ShhAla/Ala mice (white bars) at multiple postnatal ages (Shh+/+, black bars) (*p<0.001). Reduced proliferation in ShhAla/Ala mice is also seen in one wild type and mutant littermate pair at P9 (1.37% versus 1.07% for Shh+/+ and ShhAla/Ala, respectively). Error bars are +/− s.e.m.
(c) BrdU proliferation indices are less in ShhAla/Ala mice (white bars) as compared to wild type (black bars) at multiple postnatal ages (*p<0.001). While no difference is seen at intermediate time points (P6 and P9), this, in part, reflects the cerebellar size difference between ShhAla/Ala and Shh+/+ mice. There is still a significant difference in the number of proliferating cells/EGL length at P6 (3.6% in Shh+/+ versus 2.7% in ShhAla/Ala). Error bars are +/− s.e.m.
(d) p-Histone H3 mitotic indices in P3 wild type (black bars) and ShhAla/Ala mice (white bars) in the anterior, middle and posterior cerebellum are not statistically different from their respective totals in (b), demonstrating that lobes throughout the cerebellum are affected in the mutant. Error bars are +/− s.e.m.
Ext1 and Ext2 are required for HSPG chain elongation, and so are necessary for generating glycan chains that interact with Shh. While we previously identified developmental changes in expression of ext1 and ext2 in BALB/c mice during the early postnatal period14, the 129/C57BL6 strain used here exhibited consistent expression of ext1 and ext2 in the postnatal period when proliferation is reduced in ShhAla/Ala (Supplementary Fig. 4). Many other genes required for synthesizing and modifying glycans are also appropriately expressed at this time26. Thus, Shh and proteoglycans are expressed appropriately to regulate GCP proliferation, which is reduced when Shh-proteoglycan interactions are impaired.
The subventricular zone (SVZ) adjacent to the lateral ventricles and the subgranular layer (SGL) of the hippocampus represent two additional locations where there is significant Shh-regulated postnatal proliferation of neural stem/precursor cells3,4,27, while the embryonic spinal cord represents a site where Shh regulates prenatal expansion of neural stem/precursors28. Neural precursor proliferation in the spinal cord was reduced in ShhAla/Ala embryos as compared to Shh+/+ littermates (Fig. 4a,b). Proliferation of neural stem/precursors in the SVZ and SGL of adult mutants was also decreased compared to age- and sex-matched ShhCtl/Ctl (Fig. 4c,d). As stem/precursor cells in the SVZ generate olfactory bulb neurons, decreased SVZ proliferation may contribute to the olfactory bulb hypoplasia (Fig. 2, Supplementary Fig. 5). Together, these findings demonstrate that Shh-proteoglycan interactions promote neural stem/precursor cell proliferation in multiple locations in both the developing and adult CNS, and thus highlight the widespread importance of proteoglycans in mitogenesis.
Figure 4. Embryonic and adult neural stem/precursor proliferation is reduced in ShhAla/Ala mice.

(a) More proliferating neural stem/precursors are seen in the spinal cord of Shh+/+ mice (left) than in their ShhAla/Ala (right) E10.5 littermates. p-Histone H3 is in red and DAPI in blue. Scale bar, 100 μm.
(b) A reduction in the total number of p-Histone H3 positive (p-H3+) cells in the spinal cord of ShhAla/Ala embryos is seen compared to the number in ShhCtl/Ctl littermates (*p<0.05). A significant reduction in the total p-H3+ cells per unit spinal cord area is also seen in ShhAla/Ala E10.5 embryos (data not shown). Error bars are +/− s.e.m.
(c) Fewer p-H3+ cells are present in the SVZ of adult ShhAla/Ala mice (*p<0.05). Error bars are +/− s.e.m.
(d) Fewer BrdU positive (BrdU+) cells are seen in the hippocampal SGL of adult mutant mice (*p≤0.05). Error bars are +/− s.e.m.
Surprisingly, apoptosis was decreased in the EGL of ShhAla/Ala mutants compared with wild type littermates (Supplementary Fig. 6). As the Shh pathway can stimulate or suppress apoptosis under different conditions24,29, it is possible that pro-apoptotic effects of Shh require proteoglycan interactions, while anti-apoptotic effects do not. Alternatively, decreased apoptosis in ShhAla/Ala mice may be an indirect consequence of the mutation. In either case, the increased survival did not mitigate the proliferation decrease, and the net result was a dramatically smaller cerebellum (30% smaller in ShhAla/Ala and 50% smaller in ShhAla/−).
PGs delineate the Shh mitogenic niche in the EGL
There are two potential mechanisms to explain the effects of proteoglycans on Shh-induced proliferation. One is that proteoglycan interactions localize Shh to a mitogenic niche. Previous studies have suggested that proteoglycans regulate Hh dispersal5,9,12,13 and localize Ihh in developing bone30. Thus, without proteoglycan binding, Shh might not collect in the EGL, resulting in decreased proliferation. A second is that binding of Shh to proteoglycans on the surface of GCPs modulates Shh-dependent intracellular signal cascades7,13. As these models are not mutually exclusive, we investigated both.
We previously developed an assay to evaluate the nature and location of mitogenic niches in neural tissues31. In this assay, GFP-labeled precursors are introduced onto organotypic slices and incorporate into the slices. We then incubate the slice culture with BrdU to label proliferating cells, and measure the proliferative index of GFP+ precursors in distinct microenvironments. Precursors exposed to the cerebellar EGL proliferate extensively due to mitogenic effects of Shh31. To determine whether Shh-proteoglycan interactions are important in delineating this niche, we introduced wild type GFP-labeled precursors or DiO-labeled ShhAla/Ala precursors onto slices cultured from wild type or ShhAla/Ala littermates. While wild type or ShhAla/Ala precursor proliferation is enhanced when cells are introduced onto a wild type EGL, the EGL of ShhAla/Ala mice does not provide a mitogenic niche for wild type precursors (Fig. 5a). To determine whether decreased proliferation is due to impaired proteoglycan interactions, we added heparan sulfates to cultures of wild type precursors on wild type slices. Excess glycans can compete with, and so diminish the actions of endogenous proteoglycans30. Excess heparan sulfates phenocopied the ShhAla mutation, as there was decreased proliferation of cells introduced onto the EGL (Fig. 5b). Together these data indicate that interactions between Shh’s Cardin-Weintraub motif and endogenous proteoglycans promote neural precursor proliferation and establish the EGL mitogenic niche.
Figure 5. ShhAla cannot specify a mitogenic niche.

(a) Granule cell precursors (GCPs) from GFP+ mice were cultured on P6 Shh+/+ or ShhAla/Ala cerebellar slices, or DiO-labeled ShhAla/Ala GCPs were cultured on wild type slices. Proliferation of introduced GCPs was analyzed by BrdU incorporation. The proliferative index is the percent (%) of GFP- or DiO-positive cells in each layer that are BrdU+ (*p<0.05 versus EGL of WT slice; **p<0.05 vs IGL of WT slice; no significant difference between EGL and IGL proliferation on ShhAla/Ala slices is seen). Error bars are +/− s.e.m.
(b) EGL mitogenic niche in ShhAla/Ala slices (light gray bar) is phenocopied by added exogenous heparan sulfates (HS) (dark gray bar) (*p<0.05). Error bars are +/− s.e.m.
(c) Shh immunostaining of wild type and ShhAla/Ala P6 cerebella shows a reduction in Shh staining in the EGLa of mutant mice (arrowhead) compared to wild type cerebella (arrows) (52%±11% of wild type (p=0.01)). EGLa: external granule cell layer, outer proliferative zone, EGLb: external granule cell layer, inner post-mitotic zone; ML: molecular layer; PCL: Purkinje cell layer; IGL: internal granule cell layer. Scale bar, 50 μm
In slice overlay cultures, proliferation of introduced precursor cells was decreased in the EGL but increased in the IGL of ShhAla/Ala slices (Fig. 5a). These data suggest that proteoglycan binding enables Shh to accumulate in the EGL rather than in other layers. To test this, we carried out immunohistochemistry for Shh. Consistent with previous studies31, 32 in wild type animals, Shh immunostaining was detected in Purkinje cells, and in the IGL and EGL (Fig. 5c, Supplementary Fig. 7). In ShhAla/Ala animals, Shh immunostaining was decreased in the outer EGL (EGLa) where proliferation occurs (Fig. 5c). These findings indicate proteoglycans localize Shh to a mitogenic niche within the outer EGL. Electron microscopy studies have identified a proteoglycan matrix in EGLa just under the pia and adjacent to proliferating GCPs33. Thus one mechanism by which Shh-HSPG interactions stimulate precursor proliferation is by localizing ligand to the EGL mitogenic niche.
Proteoglycan interactions modulate responses to Shh
While the data above indicate Shh-proteoglycan interactions are important for localizing Shh to proliferative zones and so establishing a mitogenic niche, it is also possible proteoglycans on neural stem/precursors bind Shh, thereby specifying proliferative responses. To address this possibility, we asked whether proteoglycans on GCPs affect Shh-induced responses at the level of an individual responding cell. Consistent with previous studies14,23,25,34,35, wild type Shh induces robust GCP proliferation. In dissociated cell cultures, the proliferative response evoked by wild type Shh was much greater than that evoked by equivalent concentrations of ShhAla (Fig. 6a). In contrast, no consistent effects on survival were seen when dissociated GCP cultures were stimulated with either wild type Shh or ShhAla (Fig. 6b). Thus, the decreased proliferation observed in ShhAla/Ala mice is a direct consequence of the Cardin-Weintraub mutation, while decreased apoptosis may be an indirect result of the mutation. These data demonstrate Shh interacts with proteoglycans on individual GCPs, thereby inducing cells to proliferate, and suggest that the decreased proliferation observed in ShhAla/Ala mice results both from altered localization of ligand and from changes in intracellular signaling cascades.
Figure 6. Shh-proteoglycan interactions promote proliferation in dissociated cell cultures of GCPs, but are not needed for survival.

(a) The proliferative response to Shh (gray) (as assessed by BrdU incorporation) is greater than that of GCPs to ShhAla (black) (*p≤0.05, **p<0.01). Error bars are +/− s.e.m.
(b) No consistent effects on survival (as assessed by activated caspase 3 immunostaining) are seen in GCPs stimulated with Shh (gray) or ShhAla (black). Error bars are +/− s.e.m.
Shh responses are mediated by the transcription factors Gli1, Gli2 and Gli336. Therefore, we analyzed expression of these factors in ShhAla/Ala mice. While Gli transcription factors can function either as transcriptional activators or repressors37, Gli1 only functions as an activator, and only one isoform of Gli1 has been reported38. Gli1 protein levels were equivalent in P3 ShhAla/Ala and Shh+/+ animals (Fig. 7a). Gli2 can function as activator and repressor, and distinct isoforms have been identified that subserve these different functions38,39. In cerebella of mutant animals, the ratio of Gli2 activator (GliAct) to the shorter Gli2 repressor (Gli2Rep) was reduced by 50.9% (Fig. 7b). These data suggest overall Gli2-dependent transcription is likely to be altered in mutants. Previous studies have indicated Gli3 functions primarily as a repressor40. We did not detect any differences in Gli3Rep levels, and we did not detect Gli3Act in cerebellar tissue of either wild type or mutant animals (Fig. 7c). Thus Gli2 isoforms represent the Shh-dependent transcription factor(s) that are clearly altered in ShhAla/Ala cerebellum. As Gli2 activity is required to mediate expansion of GCPs41, changes in Gli2 may potentially explain the observed ShhAla/Ala phenotype.
Figure 7. Shh-proteoglycan interactions modulate transcriptional activity through the regulation of Gli2 isoforms and signaling kinetics.

(a) Gli1 protein levels in P3 ShhAla/Ala and Shh+/+ cerebella are equivalent. At left, anti-Gli1 western blot of ShhAla/Ala and Shh+/+ cerebellar lysates. At right, quantification of blotting results, ShhAla/Ala (white bar), Shh+/+ (black bar). Error bars are +/− s.e.m.
(b) The ratio of Gli2 activator (Gli2Act) to repressor (Gli2Rep) is reduced in P3 ShhAla/Ala cerebella compared to Shh+/+. At left, anti-Gli2 western blot. At right, quantification of results, ShhAla/Ala (white bar), Shh+/+ (black bar) (*p<0.01). Error bars are +/− s.e.m.
(c) Gli3 repressor protein levels are unchanged in P3 ShhAla/Ala cerebella, as compared to Shh+/+. At left, anti-Gli3 western blot. At right, quantification of results, ShhAla/Ala (white bar), Shh+/+ (black bar). Error bars are +/− s.e.m.
(d) The ratio of Gli2 activator (Gli2Act) to repressor (Gli2Rep) is greater in cells stimulated with Shh compared to ShhAla. At top, anti-Gli2 western blot. At bottom, quantification of results, Shh (left bar), ShhAla (right bar) (*p<0.01). Error bars are +/− s.e.m.
(e) Kinetics of signaling differ for GCPs stimulated with ShhAla as compared to GCPs stimulated with Shh. At top, anti-Gli2Act western blot of Shh- or ShhAla-stimulated GCPs (or blot against tubulin as a loading control). Numbers above blots are hours in culture. At bottom, anti-Gli2Rep western blot of Shh- or ShhAla-stimulated GCPs (or blot against tubulin as a loading control).
Since the Gli2 activator/repressor ratio is altered in vivo in ShhAla/Ala cerebella and Gli2 has been shown to play a critical role in GCP proliferation41, we examined in greater detail Gli2 protein levels in GCPs acutely stimulated with wild type or mutant Shh. We found that GCPs stimulated with either Shh or ShhAla exhibited an increase in Gli2Act protein (9.7% increase and 34.7% increase for Shh- and ShhAla-stimulated GCPs respectively). However, consistent with in vivo results, the Gli2Act/Gli2Rep ratio in GCPs acutely stimulated with ShhAla was lower than that seen in GCPs stimulated with Shh (Fig. 7d). Furthermore, the temporal profiles of Gli2Act and Gli2Rep differed when GCPs were stimulated with ShhAla rather than wild type Shh (Fig. 7e, Supplementary Fig. 8) (i.e average Gli2Act levels declined by 3.7% and 31.4% between 24 and 32 hours post-stimulation by Shh and ShhAla respectively). These data indicate Shh interacts with proteoglycans on GCPs to alter the nature and timing of Gli2-dependent transcription.
PGs alter Shh-dependent gene expression and Shh kinetics
Changes in the Gli2 activator/repressor ratio are likely to alter expression of Shh-responsive genes. A number of Shh-responsive genes have been identified, including genes involved in proliferation and tissue patterning17,34,35, 42–44. We analyzed Shh-responsive gene expression when dissociated wild type GCPs were stimulated acutely with equal concentrations of wild type Shh or ShhAla. Shh-dependent genes can be separated into three clusters that are differentially modulated by proteoglycan interactions (Fig. 8a). One cluster, including gli1 and ptc1, are similarly induced by ShhAla and wild type Shh. A second cluster of target genes (gli2 and N-myc) is induced to a greater extent by ShhAla than by wild type Shh. Surprisingly, expression of N-myc and gli2, genes implicated in proliferation, are in the gene set that is better induced by ShhAla. We note that Shh regulates both Gli2 and N-myc via transcriptional and post-transcriptional mechanisms 39, 45, and so Gli2 and N-myc activity need not correspond to RNA levels. Induction of a third cluster, including gli3, Cyclin D1, Cyclin D2, and bmi-1, was diminished when cells were stimulated with ShhAla rather than wild type ligand. Cyclins D1 and D2 are cell cycle regulators at the G1/S transition that are induced in response to Shh stimulation35 and have been particularly implicated in Shh-induced stem/precursor cell proliferation35. The polycomb protein, Bmi-1, is a Shh target that is critical for self-renewal of stem cells and for cancer cell proliferation17,18,44. Together, these data indicate the gene expression program induced by Shh is modulated by proteoglycans of responding cells. Moreover, Shh target genes that are altered when cells are stimulated by ShhAla include many implicated in proliferation and oncogenesis.
Figure 8. Proteoglycan interactions modulate Shh perdurance and differentially affect Shh-dependent gene expression.

(a) The gene pattern induced by ShhAla stimulation of wild type P6 GCPs is different than that of GCPs stimulated by equivalent amounts of Shh (*p<0.05). Error bars are +/− s.e.m.
(b) GCP proteoglycans modulate Shh ligand perdurance. At left, (top) anti-Shh western blot of Shh- or ShhAla-stimulated C3H10T1/2s (or blot against actin as loading control). At right, (top) anti-Shh western blot of Shh- or ShhAla-stimulated GCPs (or blot against tubulin as loading control). Numbers above blots are hours in culture. At bottom, quantification of results from one representative experiment. Shh (solid), ShhAla (dashed).
(c) Expression of Shh target genes in P1-2 ShhAla/Ala cerebella (P1-3 ShhAla/Ala cerebella for gli2) relative to Shh+/+ littermates demonstrates that Shh-proteoglycan interactions differentially affect gene subsets (*p<0.01, **p<0.05). Error bars are +/− s.e.m.
Previous studies have demonstrated distinct responses to Shh can be elicited depending on ligand concentration22. To determine whether a shift in the dose response curve explains why ShhAla induces an altered program of gene expression, we performed dose response experiments using Shh or ShhAla protein. C3H10T1/2 differentiation as assessed by alkaline phosphatase induction (Supplementary Fig. 9a) is a standard assay for Shh activity; we found no shift in the ShhAla dose response curve compared to Shh. Furthermore, maximal efficacy of ShhAla was not reduced compared to Shh. We next measured gli1 and CyclinD2 mRNA levels in GCPs stimulated across a wide range of Shh and ShhAla doses (Supplementary Fig. 9b). Whereas gli1 expression did not differ significantly in response to equivalent doses of Shh and ShhAla throughout the dose range, CyclinD2 induction by ShhAla was diminished regardless of dose. These data indicate the altered biological activity of ShhAla does not reflect a shift in the dose response curve. As recent studies have highlighted the importance of Shh signaling kinetics in determining Shh responses46, we asked whether proteoglycan interactions alter the ability of Shh to signal over prolonged periods of time. Equivalent amounts of Shh or ShhAla were added to either C3H10T1/2 or GCP cultures. After twenty-four hours of stimulation, we removed ligand from the media and analyzed Shh perdurance in the stimulated cells over the ensuing two days. In both systems, ShhAla levels declined more precipitously than wild type (Fig. 8b). Taken together, these data indicate that proteoglycans alter the kinetics of signaling, promoting a gene expression signature important for Shh-dependent precursor proliferation.
To determine whether changes in Shh-dependent gene expression explain the in vivo phenotype of ShhAla/Ala mice, we used quantitative RT-PCR to compare Shh-responsive gene expression in wild type and mutant cerebella. gli1 and ptc1 levels were equivalent in wild type and ShhAla/Ala cerebella. However, expression of a second set of genes including Cyclin D1, Cyclin D2, and bmi-1 was significantly reduced in ShhAla/Ala animals compared to wild type littermates (Fig. 8c). Decreased expression of CyclinD1, D2 and bmi-1 do not reflect a generalized decrease in cell cycle-associated proteins, as CyclinE expression was unchanged. A third set of Shh-regulated genes (gli2 and N-myc) was expressed at higher levels in ShhAla/Ala than in wild type mice. The observed changes in vivo do not reflect alterations in the distribution of cells expressing Shh-responsive genes as indicated by in situ hybridization studies for gli1, gli2 and gli3 (Supplementary Fig. 10). A critical feature of these data is that proteoglycan interactions do not uniformly increase or decrease Shh-dependent gene expression in vivo. Furthermore, clusters that are unchanged, increased, or decreased in ShhAla/Ala animals exhibit a striking similarity to gene expression patterns in GCPs acutely stimulated with wild type versus mutant Shh.
Taken together, these studies indicate Shh-proteoglycan interactions selectively promote neural stem/precursor proliferation by two mechanisms. First proteoglycans localize the ligand within developing tissue and so establish the mitogenic niche, and second they alter ligand perdurance, preferentially activating intracellular cascades that culminate in mitogenesis and precursor renewal.
DISCUSSION
Using a genetic approach, we find that Shh-proteoglycan interactions are required for proliferative, rather than patterning responses to Shh. We generated mutant mice that express Shh that cannot bind proteoglycans but can bind to Ptc. Mutants exhibit a selective deficit in neural stem/precursor cell proliferation. We identified two distinct activities of proteoglycans in Shh-dependent proliferation: these molecules localize Shh to mitogenic niches and they also trigger a gene expression program important for cell division and stem cell renewal.
Proteoglycans are tremendously diverse; each proteoglycan represents a complex, yet poorly understood readout of the glycogenes, the genes encoding core proteins, sugar transporters, glycosyltransferases, sulfatases, and acetylating enzymes. As proteoglycans interact with many growth factors and other molecules, analyses of mutants that interfere with individual steps of proteoglycan synthesis are difficult to interpret. To identify the functions of proteoglycans for Shh responses in vivo, we took an alternative approach. Mutations in the Cardin-Weintraub motif of Hh proteins interfere with proteoglycan binding, but do not alter Ptc affinity, lipid modifications, or expression level. Therefore ShhAla/Ala and ShhAla/− mutants provide a unique genetic approach for identifying functions of Shh-proteoglycan interactions.
We find proteoglycans are specifically needed for proliferative responses to Shh, but are dispensable for most of its patterning activities. One intriguing finding is that proteoglycans localize Shh to proliferative zones, and also function at the single cell level to determine the nature of the response. Previous studies using mutations in the HSPG synthesizing enzymes of the ext gene family, mutations in the glypican Dally-like or deletion of the Cardin-Weintraub motif have indicated that HSPGs are critical for appropriate localization of Hh proteins 11–13, 47. Here we show that Shh that cannot bind to HSPGs do not accumulate in the EGL mitogenic niche. Thus, direct interactions of Shh with proteoglycans are important for Shh dispersal from the Purkinje cell layer and/or its sequestration in the EGL. Altered Shh localization in mutant animals impairs the establishment of mitogenic niches in developing brain.
In addition to their function in localizing Shh, we find that proteoglycans on receiving cells modulate the Shh response. Previous studies have indicated that proteoglycans function in Hh-responding cells7,13. Surprisingly, some groups have found that glypicans on receiving cells are needed for full-strength signaling while others have found that responding cell glypicans compete with Ptc1 for Hh interaction, thereby inhibiting Hh activity11,13. Our studies suggest proteoglycan interactions differentially affect intracellular signaling cascades downstream of Smoothened, and so selectively regulate Shh target gene subsets, thereby modifying the response.
As Shh target gene expression is primarily regulated by the Gli transcription factors36, we examined levels and isoforms of Gli1, Gli2 and Gli3 proteins in wild type and mutant animals. Whereas there were no discernable differences between Gli1 and Gli3 protein levels of wild type and mutant cerebella, clear differences were seen in Gli2 proteins. Gli2 is the major transcription factor driving Shh-induced proliferation41,48 and proliferation is reduced in multiple locations pre- and post-natally in ShhAla/Ala animals. Gli2 proteins can function as either transcriptional activators or repressors; unprocessed Gli2 functions as a weak transcriptional activator37,39, while Gli2 cleavage and removal of the C-terminal activator domain generates a transcriptional repressor39. Therefore both Gli2 protein levels and the ratio between unprocessed and processed forms are likely to be critical for the net Gli2-dependent transcriptional response. In vivo, gli2 mRNA is lower in wild type than in ShhAla/Ala cerebella, but the Gli2 activator/repressor ratio is two-fold greater in wild type. Similarly, gli2 mRNA levels are lower in GCPs acutely stimulated with Shh than ShhAla, but GCPs acutely stimulated with Shh had a larger Gli2 activator/repressor ratio than those stimulated with ShhAla.
Gli2 expression and processing are both Shh-regulated39,48. The ability of proteoglycans to affect the levels and proportions of the Gli2 isoforms in vivo and in vitro would alter Shh-dependent transcriptional activation and repression, and so could account for increases and decreases in Shh-target gene expression. One set of Shh-regulated genes, including classical targets ptc1 and gli1, are similarly induced by Shh that can and cannot bind proteoglycans. A second set, including gli2 and N-myc, are preferentially increased by Shh that cannot bind to proteoglycans. Induction of a third set of Shh-regulated genes, including bmi-1, and D-type Cyclins, genes implicated in stem cell maintenance and in tumor biology, requires proteoglycan interactions. Differences in Shh target genes are observed in vivo and in vitro and are not due to shifts in dose response curves, but may be explained by changes in ligand signaling kinetics. Taken together, analysis of the mutant phenotype indicates that Shh-HSPG interactions particularly affect overall Gli2 transcriptional activity, thereby promoting proliferative responses.
Changes in ligand intracellular localization or ligand perdurance provide two possible mechanisms whereby Shh-proteoglycan interactions could selectively modulate signaling. Shh signaling kinetics are important in determining morphogen responses46. Here we find that Shh-proteoglycan interactions affect Shh perdurance in responding cells and alter the temporal profile of Shh signaling. It has been proposed that signal duration is proportional to Shh concentration46, our data indicate that proteoglycan interactions alter that relationship. In this way, proteoglycan interactions can modulate the pattern of gene induction and the biological response to Shh. Proliferation without appropriate patterning is a cardinal feature of tumor biology. The Hedgehog pathway may promote oncogenesis when it stimulates proliferation without patterning. It is intriguing that glypican overexpression can augment growth in cancers that depend on Shh activity, including rhabdomyosarcoma, prostate cancer, and pancreatic carcinoma49,50. Similarly, D-type Cyclins and bmi-1, gene targets that require Shh-HSPG interactions, play central roles in tumor stem cell biology17,18.
In summary, we have demonstrated that Shh-proteoglycan interactions are selectively critical for Shh-dependent mitogenesis, functioning in two ways. First, proteoglycans localize Shh to establish mitogenic niches. Second, proteoglycans on responsive cells selectively promote intracellular pathways that lead to precursor proliferation. Both mechanisms contribute to the phenotype of the ShhAla/Ala mice. Thus, Shh-proteoglycan interactions promote proliferation within mitogenic niches by localizing Shh to these proliferative zones and by modulating intracellular signaling cascades and transcriptional programs.
METHODS
Section binding assay
To evaluate proteoglycan binding in situ14, cryosections were treated with PBS, vehicle control, 500 mU/ml of heparinases (Sigma), or 500 mU/ml of PI-PLC for 1 h (37°C), then O/N (4°C). Shh:AP or ShhAla:AP was added (1 h, RT), sections were washed and immunostained with anti-alkaline phosphatase.
Size measurements
6 month old animals were weighed, then sacrificed. Total brain and cerebella were weighed. Olfactory bulb dimensions (OB) were measured from 10 μm thick coronal cryosections and overall volume was calculated. At least 3 pairs of ShhAla/Ala and age-matched ShhCtl/Ctl were assessed.
Shh staining quantification
Quantification was done on mid-sagittal cerebellar sections from three wild type-ShhAla/Ala littermate pairs, stained in parallel for Shh. Staining was analyzed in primary and secondary fissures using NIH Image J software. In each fissure, three lines from the pia through the IGL were drawn perpendicular to the pia, and an intensity plot was determined for each line (plot profile function).
Proliferation and apoptosis quantification in vivo
Analyses of p-Histone H3 staining, BrdU labeling, and TUNEL were performed on cerebella from 3–5 matched wild-type and mutant littermate pairs. Proliferation or apoptotic index was calculated by dividing the number of positive cells by the number of DAPI-stained cells in the EGL. Comparable locations (midpoint of primary, secondary and tertiary fissures within vermis) were assessed. In coronal sections of adults, every 9th section of the SGL or SVZ was stained with p-Histone H3, BrdU, and activated caspase 3. At least 3 pairs of adult ShhAla/Ala animals and age-matched controls were analyzed. SGL was the inner half of the thickness of the dentate gyrus, extending two cell widths into the hilus.
Supplementary Material
Acknowledgments
Supported by NIH (JAC, RAS, SB), DFCI Mahoney Center for Neuro-Oncology (JAC), Musella Foundation (KJN), Quan Fellowship (RMW), Children’s Hospital MRDDRC and Harvard NeuroDiscovery Center. We thank Jennifer Despinoy for excellent assistance; C Stiles, D Rowitch, M Greenberg and the Segal lab for helpful discussions; D Rowitch, Q Ma, P Chuang, D Paul, S O’Gorman, P Silver and A McMahon for reagents.
Footnotes
References
- 1.Capdevila J, Izpisua Belmonte JC. Patterning mechanisms controlling vertebrate limb development. Annu Rev Cell Dev Biol. 2001;17:87–132. doi: 10.1146/annurev.cellbio.17.1.87. [DOI] [PubMed] [Google Scholar]
- 2.Hooper JE, Scott MP. Communicating with Hedgehogs. Nature reviews. 2005;6:306–317. doi: 10.1038/nrm1622. [DOI] [PubMed] [Google Scholar]
- 3.Machold R, et al. Sonic hedgehog is required for progenitor cell maintenance in telencephalic stem cell niches. Neuron. 2003;39:937–950. doi: 10.1016/s0896-6273(03)00561-0. [DOI] [PubMed] [Google Scholar]
- 4.Palma V, et al. Sonic hedgehog controls stem cell behavior in the postnatal and adult brain. Development. 2005;132:335–344. doi: 10.1242/dev.01567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bellaiche Y, The I, Perrimon N. Tout-velu is a Drosophila homologue of the putative tumour suppressor EXT-1 and is needed for Hh diffusion. Nature. 1998;394:85–88. doi: 10.1038/27932. [DOI] [PubMed] [Google Scholar]
- 6.Desbordes SC, Sanson B. The glypican Dally-like is required for Hedgehog signalling in the embryonic epidermis of Drosophila. Development. 2003;130:6245–6255. doi: 10.1242/dev.00874. [DOI] [PubMed] [Google Scholar]
- 7.Lum L, et al. Identification of Hedgehog pathway components by RNAi in Drosophila cultured cells. Science. 2003;299:2039–2045. doi: 10.1126/science.1081403. [DOI] [PubMed] [Google Scholar]
- 8.Yao S, Lum L, Beachy P. The ihog cell-surface proteins bind Hedgehog and mediate pathway activation. Cell. 2006;125:343–357. doi: 10.1016/j.cell.2006.02.040. [DOI] [PubMed] [Google Scholar]
- 9.Han C, Belenkaya TY, Khodoun M, Tauchi M, Lin X. Distinct and collaborative roles of Drosophila EXT family proteins in morphogen signalling and gradient formation. Development. 2004;131:1563–1575. doi: 10.1242/dev.01051. [DOI] [PubMed] [Google Scholar]
- 10.Bornemann DJ, Duncan JE, Staatz W, Selleck S, Warrior R. Abrogation of heparan sulfate synthesis in Drosophila disrupts the Wingless, Hedgehog and Decapentaplegic signaling pathways. Development. 2004;131:1927–1938. doi: 10.1242/dev.01061. [DOI] [PubMed] [Google Scholar]
- 11.Capurro MI, et al. Glypican-3 inhibits Hedgehog signaling during development by competing with patched for Hedgehog binding. Dev Cell. 2008;14:700–711. doi: 10.1016/j.devcel.2008.03.006. [DOI] [PubMed] [Google Scholar]
- 12.Hacker U, Nybakken K, Perrimon N. Heparan sulphate proteoglycans: the sweet side of development. Nature reviews. 2005;6:530–541. doi: 10.1038/nrm1681. [DOI] [PubMed] [Google Scholar]
- 13.Gallet A, Staccini-Lavenant L, Therond PP. Cellular trafficking of the glypican Dally-like is required for full-strength Hedgehog signaling and wingless transcytosis. Dev Cell. 2008;14:712–725. doi: 10.1016/j.devcel.2008.03.001. [DOI] [PubMed] [Google Scholar]
- 14.Rubin JB, Choi Y, Segal RA. Cerebellar proteoglycans regulate sonic hedgehog responses during development. Development. 2002;129:2223–2232. doi: 10.1242/dev.129.9.2223. [DOI] [PubMed] [Google Scholar]
- 15.Pallerla SR, Pan Y, Zhang X, Esko JD, Grobe K. Heparan sulfate Ndst1 gene function variably regulates multiple signaling pathways during mouse development. Dev Dyn. 2007;236:556–563. doi: 10.1002/dvdy.21038. [DOI] [PubMed] [Google Scholar]
- 16.Inatani M, Irie F, Plump AS, Tessier-Lavigne M, Yamaguchi Y. Mammalian brain morphogenesis and midline axon guidance require heparan sulfate. Science. 2003;302:1044–1046. doi: 10.1126/science.1090497. [DOI] [PubMed] [Google Scholar]
- 17.Molofsky AV, et al. Bmi-1 dependence distinguishes neural stem cell self-renewal from progenitor proliferation. Nature. 2003;425:962–967. doi: 10.1038/nature02060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pardal R, Molofsky AV, He S, Morrison SJ. Stem cell self-renewal and cancer cell proliferation are regulated by common networks that balance the activation of proto-oncogenes and tumor suppressors. Cold Spring Harb Symp Quant Biol. 2005;70:177–185. doi: 10.1101/sqb.2005.70.057. [DOI] [PubMed] [Google Scholar]
- 19.Chen MH, Li YJ, Kawakami T, Xu SM, Chuang PT. Palmitoylation is required for the production of a soluble multimeric Hedgehog protein complex and long-range signaling in vertebrates. Genes Dev. 2004;18:641–659. doi: 10.1101/gad.1185804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chiang C, et al. Cyclopia and defective axial patterning in mice lacking Sonic hedgehog gene function. Nature. 1996;383:407–413. doi: 10.1038/383407a0. [DOI] [PubMed] [Google Scholar]
- 21.Harfe BD, et al. Evidence for an expansion-based temporal Shh gradient in specifying vertebrate digit identities. Cell. 2004;118:517–528. doi: 10.1016/j.cell.2004.07.024. [DOI] [PubMed] [Google Scholar]
- 22.Briscoe J, Ericson J. The specification of neuronal identity by graded Sonic Hedgehog signalling. Semin Cell Dev Biol. 1999;10:353–362. doi: 10.1006/scdb.1999.0295. [DOI] [PubMed] [Google Scholar]
- 23.Wechsler-Reya RJ, Scott MP. Control of neuronal precursor proliferation in the cerebellum by Sonic Hedgehog. Neuron. 1999;22:103–114. doi: 10.1016/s0896-6273(00)80682-0. [DOI] [PubMed] [Google Scholar]
- 24.Charrier JB, Lapointe F, Le Douarin NM, Teillet MA. Anti-apoptotic role of Sonic hedgehog protein at the early stages of nervous system organogenesis. Development. 2001;128:4011–4020. doi: 10.1242/dev.128.20.4011. [DOI] [PubMed] [Google Scholar]
- 25.Wallace VA. Purkinje-cell-derived Sonic hedgehog regulates granule neuron precursor cell proliferation in the developing mouse cerebellum. Curr Biol. 1999;9:445–448. doi: 10.1016/s0960-9822(99)80195-x. [DOI] [PubMed] [Google Scholar]
- 26.Yabe T, Hata T, He J, Maeda N. Developmental and regional expression of heparan sulfate sulfotransferase genes in the mouse brain. Glycobiology. 2005;15:982–993. doi: 10.1093/glycob/cwi090. [DOI] [PubMed] [Google Scholar]
- 27.Ahn S, Joyner AL. In vivo analysis of quiescent adult neural sterm cells responding to Sonic hedgehog. Nature. 2005;437:894–897. doi: 10.1038/nature03994. [DOI] [PubMed] [Google Scholar]
- 28.Jeong J, McMahon AP. Growth and pattern of the mammalian neural tube are governed by partially overlapping feedback activities of the hedgehog antagonists patched 1 and Hhip1. Development. 2005;132:143–154. doi: 10.1242/dev.01566. [DOI] [PubMed] [Google Scholar]
- 29.Yamamoto Y, Stock DW, Jeffery WR. Hedgehog signalling controls eye degeneration in blind cavefish. Nature. 2004;431:844–847. doi: 10.1038/nature02864. [DOI] [PubMed] [Google Scholar]
- 30.Hilton MJ, Tu X, Cook J, Hu H, Long F. Ihh controls cartilage development by antagonizing Gli3, but requires additional effectors to regulate osteoblast and vascular development. Development. 2005;132:4339–4351. doi: 10.1242/dev.02025. [DOI] [PubMed] [Google Scholar]
- 31.Choi Y, Borghesani PR, Chan JA, Segal RA. Migration from a mitogenic niche promotes cell-cycle exit. J Neurosci. 2005;25:10437–10445. doi: 10.1523/JNEUROSCI.1559-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gritli-Linde A, Lewis P, McMahon AP, Linde A. The whereabouts of a morphogen: direct evidence for short- and graded long-range activity of hedgehog signaling peptides. Developmental Biology. 2001;236:364–386. doi: 10.1006/dbio.2001.0336. [DOI] [PubMed] [Google Scholar]
- 33.Hausmann B, Sievers J. Cerebellar external granule cells are attached to the basal lamina from the onset of migration up to the end of their proliferative activity. J Comp Neurol. 1985;241:50–62. doi: 10.1002/cne.902410105. [DOI] [PubMed] [Google Scholar]
- 34.Kenney AM, Cole MD, Rowitch DH. Nmyc upregulation by sonic hedgehog signaling promotes proliferation in developing cerebellar granule neuron precursors. Development. 2003;130:15–28. doi: 10.1242/dev.00182. [DOI] [PubMed] [Google Scholar]
- 35.Kenney AM, Rowitch DH. Sonic hedgehog promotes G(1) cyclin expression and sustained cell cycle progression in mammalian neuronal precursors. Mol Cell Biol. 2000;20:9055–9067. doi: 10.1128/mcb.20.23.9055-9067.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ulloa F, Briscoe J. Morphogens and the control of cell proliferation and patterning in the spinal cord. Cell Cycle. 2007;6:2640–2649. doi: 10.4161/cc.6.21.4822. [DOI] [PubMed] [Google Scholar]
- 37.Sasaki H, Nishizaki Y, Hui C, Nakafuku M, Kondoh H. Regulation of Gli2 and Gli3 activities by an amino-terminal repression domain: implication of Gli2 and Gli3 as primary mediators of Shh signaling. Development. 1999;126:3915–3924. doi: 10.1242/dev.126.17.3915. [DOI] [PubMed] [Google Scholar]
- 38.Aza-Blanc P, Lin HY, Ruiz i Altaba A, Kornberg TB. Expression of the vertebrate Gli proteins in Drosophila reveals a distribution of activator and repressor activities. Development. 2000;127:4293–4301. doi: 10.1242/dev.127.19.4293. [DOI] [PubMed] [Google Scholar]
- 39.Pan Y, Bai CB, Joyner AL, Wang B. Sonic hedgehog signaling regulates Gli2 transcriptional activity by suppressing its processing and degradation. Mol Cell Biol. 2006;26:3365–3377. doi: 10.1128/MCB.26.9.3365-3377.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Blaess S, Corrales JD, Joyner AL. Sonic hedgehog regulates Gli activator and repressor functions with spatial and temporal precision in the mid/hindbrain region. Development. 2006;133:1799–1809. doi: 10.1242/dev.02339. [DOI] [PubMed] [Google Scholar]
- 41.Corrales JD, Rocco GL, Blaess S, Guo Q, Joyner AL. Spatial pattern of sonic hedgehog signaling through Gli genes during cerebellum development. Development. 2004;131:5581–5590. doi: 10.1242/dev.01438. [DOI] [PubMed] [Google Scholar]
- 42.Yoon JW, et al. Gene expression profiling leads to identification of GLI1-binding elements in target genes and a role for multiple downstream pathways in GLI1-induced cell transformation. J Biol Chem. 2002;277:5548–5555. doi: 10.1074/jbc.M105708200. [DOI] [PubMed] [Google Scholar]
- 43.Duman-Scheel M, Weng L, Xin S, Du W. Hedgehog regulates cell growth and proliferation by inducing Cyclin D and Cyclin E. Nature. 2002;417:299–304. doi: 10.1038/417299a. [DOI] [PubMed] [Google Scholar]
- 44.Leung C, et al. Bmi1 is essential for cerebellar development and is overexpressed in human medulloblastomas. Nature. 2004;428:337–341. doi: 10.1038/nature02385. [DOI] [PubMed] [Google Scholar]
- 45.Kenney AM, Widlund HR, Rowitch DH. Hedgehog and PI-3 kinase signaling converge on Nmyc1 to promote cell cycle progression in cerebellar neuronal precursors. Development. 2004;131:217–228. doi: 10.1242/dev.00891. [DOI] [PubMed] [Google Scholar]
- 46.Dessaud E, et al. Interpretation of the sonic hedgehog morphogen gradient by a temporal adaptation mechanism. Nature. 2007;450:717–720. doi: 10.1038/nature06347. [DOI] [PubMed] [Google Scholar]
- 47.Vyas N, et al. Nanoscale organization of hedgehog is essential for long-range signaling. Cell. 2008;133:1214–1227. doi: 10.1016/j.cell.2008.05.026. [DOI] [PubMed] [Google Scholar]
- 48.Galvin KE, Ye H, Wetmore C. Differential gene induction by genetic and ligand-mediated activation of the Sonic hedgehog pathway in neural stem cells. Dev Biol. 2007;308:331–342. doi: 10.1016/j.ydbio.2007.05.031. [DOI] [PubMed] [Google Scholar]
- 49.Kleeff J, et al. The cell-surface heparan sulfate proteoglycan glypican-1 regulates growth factor action in pancreatic carcinoma cells and is overexpressed in human pancreatic cancer. J Clin Invest. 1998;102:1662–1673. doi: 10.1172/JCI4105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Williamson D, et al. Role for amplification and expression of glypican-5 in rhabdomyosarcoma. Cancer Res. 2007;67:57–65. doi: 10.1158/0008-5472.CAN-06-1650. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.