

Abstract
The goal of many current studies of neovascularization is to define the phenotype of vascular cell populations of different origins and to determine how such cells promote assembly of vascular channel. Here, we describe a protocol to immunophenotype vascular cells by high-resolution imaging and by fluorescence-activated flow cytometry in an in vivo rodent model of pulmonary microvascular remodeling. Analysis of cells by this combined approach will characterize their phenotype, quantify their number and identify their role in the assembly of vascular channels.
INTRODUCTION
Currently, the phenotype and role of vascular cell populations of different origins in blood vessel formation and repair in adult tissues is of great interest. We have developed a protocol to study the kinetics of these cells using a rodent model of vascular remodeling (of vessels and/or capillaries) after hyperoxic acute lung injury (HALI1 and accompanying Nature Protocol by Jones et al.2). Briefly, in the model, injured small vessels (in rat lung) increase their wall thickness (10-fold) and disrupted capillary networks (in rat and mouse lung) subsequently reform1,3. Here, we describe high-resolution immunogold-labeling approaches for phenotyping and quantifying cells in situ in these vascular structures, and the use of fluorescence-activated flow cytometry to phenotype and quantify cells of interest circulating in blood or present in dissociated lung tissue. Both approaches will identify precursor vascular cell populations. The HALI model allows the cellular basis of this response to be analyzed3-5. Cells are readily characterized by their morphology and location as intravascular (circulating through the lung) or resident in vascular structures such as endothelial cells, pericytes, smooth muscle cells or perivascular fibroblasts, in high-resolution images (the gold standard to identify cell type). Antibodies to vascular growth factor ligands and receptors such as VEGFR-VEGF-R2 or PDGF-BB-PDGF-Rβ, or to cluster differentiation (CD) marker proteins such as CD11b or CD31, will further establish the phenotype of the cell populations targeted in high-resolution images or by fluorescence activated flow cytometry4-6. Immunophenotypic data obtained by fluorescence microscopy and flow cytometry are, at one level, suitable to characterize cell populations by their origin; however, these data lack sufficient resolution (fluorescence microscopy) or are unable (flow cytometry) to determine their precise location in situ and their contribution to vascular remodeling. The techniques of high-resolution imaging and flow cytometry can, by contrast, provide significant insight into the role of cells' remodeling vascular structures as well as determining their origin and phenotype. Thus, although the two methodologies can be employed separately to identify vascular precursors, we use both in this protocol because of the complementary results the data provide.
MATERIALS
REAGENTS
10× Dulbecco's phosphate-buffered saline (PBS; Gibco/Invitrogen, cat. no. 14200-075)
Ethanol, 95% (AAPER Alcohol & Chemical Co., cat. no. 04 H12QB)
Ethanol, 100% (AAPER Alcohol & Chemical Co., cat. no. 04 I13BA)
Unique acrylic resin (Unicryl), 4% mono-methacrylate esters/4% styrene kit (EMS, cat. no. 14660)
Toluidine blue (Ernest Fullam, cat. no. 50180)
Sodium borate (Fisher Scientific, cat. no. S-248)
Permount mounting medium (Fisher Scientific, cat. no. SP15-500)
Distilled/deionized water
Bovine serum albumin (BSA; Amersham, cat. no. RPN412)
Purified antibodies (e.g., anti-αSMA, Sigma, cat. no. A2547; anti-PDGF-BB, Oncogene Science, cat. no. PC21; anti-PDGF-Rβ, Oncogene Science, cat. no. PC17; anti-PDGF-AA, R&D Systems, cat. no. AB-221-NA; anti-PDGF-Rα, R&D Systems, cat. no. AF-307-NA; anti-CD11b, Chemicon, cat. no. CBL1512Z and BD Pharmingen, cat. no. 550282; anti-VEGF-R2, Calbiochem, cat. no. 676488; anti-CD31/PECAM-1/M-20, Santa Cruz Biotechnology, cat. no. SC-1506; anti-vWF (Factor VIII), Dako, cat. no. A0082)
Auroprobe AG10 (Amersham, cat. no. RPN 438)
IntenSE M silver enhancement kit (Amersham, cat. no. RPN 491
Uranyl magnesium acetate (Polysciences, cat. no. 01205)
Lead citrate (Polysciences, cat. no. 00378)
Collagenase type II (Worthington)
Peripheral blood (see REAGENT SETUP)
Single-cell suspension of enzymatically digested lung tissue (see REAGENT SETUP)
Phycoerythrin (PE)-labeled anti-rat CD11b mouse antibody (BD Pharmingen, cat. no. 555862 or similar products) or anti-mouse CD11b rat antibody (BD Pharmingen, cat. no. 553311 or similar products)
Purified anti-rat VEGF-R2 (931-997) rabbit antibody (Calbiochem, EMD Biosciences or similar products) or anti-mouse VEGF-R2-PE rat antibody (BD Pharmingen, cat. no. 555038 or similar products)
Purified anti-rat PDGF-Rβ (425-446) rabbit antibody (Calbiochem, EMD Biosciences or similar products) or anti-mouse PDGF-Rβ-PE rat antibody (eBioscience, cat. no. 12-1402 or similar products)
PE-Cy5-labeled anti-rat CD45 mouse antibody (BD Pharmingen, cat. no.559135 or similar products)
Fluorescein isothiocyanate (FITC)-labeled goat anti-rabbit IgG1 antibodies (Jackson ImmunoResearch Laboratories Inc., cat. no. 111-095-003 or similar products)
PE-and PE-Cy5-labeled isotype-matched (BD Pharmingen, cat. no. 555748, 555749 and 555750 or similar products)
Fc-receptor (e.g., CD16/CD32)-blocking antibody (Miltenyi Biotec, cat. no. 120-000-442 or similar products)
ACK lysis buffer (Cambrex Bio Science, cat. no. 10-548E)
10% (vol/vol) paraformaldehyde (methanol-free; Polysciences, cat. no. 04018-1)
25% (vol/vol) gluteraldehyde (Polysciences, cat. no. 01909) !CAUTION All fluorescent reagents are light sensitive. Refrigerate in a dark place. Paraformaldehyde is toxic.
EQUIPMENT
Fume hood
4 °C refrigerator and -20 °C freezer
PELCO UVC2 Cryochamber, with metal support rods and ultraviolet light (Ted Pella, cat. no. 6202)
Vacuum oven (Fisher Scientific, Model 280)
Ultracut E Reichert-Jung Microtome (preferably on a `floating' table; Kinetics System Inc., Vibraplane Model 1201)
Two Diatome Histo knives (one rough and one fine cut, 6-8 mm, 45° angle; Diatome)
Corning hotplate/stirrer (Corning, PC351)
Zeiss Axioplan brightfield microscope with ×10 eyepieces and ×10, ×25, ×40 objectives (system equipped with a SONY CCD-Iris color video DXC-107A and SPOT camera options; MVI Inc.)
Rotomix (Thermolyne, cat. no. 48200)
Transmission electron microscope (TEM; Philips/FEI Co., EM300)
CCD-300-RC high-sensitivity camera (Dage-MTI, advanced microscopy techniques)
EDTA collection tubes (e.g., Vacutainer; BD Biosciences, cat. no. 366450 or equivalent)
Water bath with shaker (e.g., Cox machine)
Seward Stomacher compact blender (Cole-Parmer, cat. no. EW-36850-00)
Rodents (a minimum of 3-4 rats or mice for each time point studied) that have breathed high oxygen or high oxygen followed by air, as described for our model of lung neovascularization1-5,7. For Steps 1-34 of this protocol, use fixed rodent lung tissue prepared as described2; for Steps 35-56, use freshly excised rodent lung tissue and/or blood ! CAUTION Experiments using live rodents must conform to local and national regulations.
Disposable transfer pipettes (Fisher Scientific, Fisherbrand cat. no. 13-711-7)
Dry ice pellets (Dry Ice Corp)
Zipper-seal bags, 5 inch × 8 inch (Fisher Scientific, cat. no. 01-816D)
Zipper-seal bags, 6 inch × 13 inch (Fisher Scientific, cat. no. 01-816B)
Plastic container (to hold BEEM cryocapsule holder in a bed of crushed ice)
Wood applicator sticks, 6 inch × 1.5 inch diameter (Fisher Scientific, cat. no. 01-340)
Formvar film/nickel grids, 200-square mesh, thin bar, high definition (EMS, cat. no. FF200-NI)
Dumont N-5 tweezers (EMS, cat. no. 72854-D)
Nunc 245 mm square bioassay dish (Fisher Scientific, cat. no. NNI 240835)
Tissue-culture Petri dish, 150 mm × 25 mm (Becton-Dickinson, cat. no. 3025)
Grid storage boxes (Leica Microsystems, cat. no. 705525)
5 cc Luer-Lok tip disposable syringe, no needle (Becton-Dickinson, cat. no. 309603)
Millex GP 33 mm sterile PES 0.22 mm Millipore syringe-driven filter unit (Fisher Scientific, cat. no. SLGP 033RS)
Parafilm, 4 inch × 250 ft (Fisher Scientific, cat. no. 13-374-12)
Staining box with rubber plate (knife station; Ted Pella, cat. no. 8092)
Teflon-coated forceps/chromosteel Fontax tweezers no. 5 (EMS, cat. no. 72702-F)
Single-edged industrial blades (Ted Pella, cat. no. 121-72)
Vacutainer tubes, sterile 4 ml (additive: K2EDTA, 7.2 mg; Becton-Dickinson, cat. no. 367861/4056428)
Shell vials, 15 mm OD×45 mm Hgt (EMS, cat. no. 72631-10)
BEEM cryocapsule holder (Ted Pella, cat. no. 6207)
BEEM capsules, size 00 (Ted Pella, cat. no. 130-1)
Metal vial holders (EMS, cat. no. 72630)
Precleaned superfrost glass slides (Fisher Scientific, Fisherbrand cat. no. 12-550-15)
Premium coverglass (Fisher Scientific, Fisher finest cat. nos. 12-548-5E and 12-548-B)
10 ml syringe (BD Biosciences, cat. no. 305559 or equivalent)
Stomacher bags (Cole-Parmer, cat. no. C-36850-91)
Blunt-end needles (16 gauge; StemCell Technologies, cat. no. 28110 or equivalent)
15 ml polystyrene conical centrifuge tube (BD Falcon, cat. no. 352099 or equivalent)
50 ml polystyrene conical centrifuge tube (BD Falcon, cat. no. 352098 or equivalent)
5 ml polystyrene round-bottomed test tube (BD Falcon, cat. no. 352054 or equivalent)
100 μm cell strainers (BD Falcon, cat. no. 352360)
40 μm cell strainers (BD Falcon, cat. no. 352340)
REAGENT SETUP
1× PBS
Make up to 10× PBS using distilled/deionized water.
3% (vol/vol) paraformaldehyde/0.1% (vol/vol) gluteraldehyde in 1× PBS
Prepare, store at 4 °C and bring to room temperature (RT; 18-25 °C) for use.
30%, 50%, 70% ethanol from 100% ethanol with distilled/deionized water
Store 95% and 100% ethanol at -20 °C and other ethanol solutions at 4 °C.
0.1% (vol/vol) toluidine blue in distilled/deionized water
Make up from a stock solution of 1% (wt/vol) toluidine blue in 1% sodium borate (wt/vol) and filter immediately before use through a PES 0.22 μm filter unit attached to a 5 ml disposable syringe.
1% (wt/vol) and 0.5% (vol/vol) BSA
Dissolve appropriate volume of powder in 1× PBS.
Auroprobe (AG10)
Dilute 1:50 with 1× PBS.
7.5% (wt/vol) uranyl magnesium acetate
Prepare with distilled/deionized water.
0.2% (wt/vol) lead citrate
Make up in distilled/deionized water.
0.5% (wt/vol) BSA and 2 mM EDTA in 1× PBS
Make up in distilled/deionized water using BSA powder, and 0.5 M EDTA and 10× PBS (Ca2+- and Mg2+-free) stock solutions.
2% paraformaldehyde
Make up from 10% stock solution in 1× PBS.
PROCEDURE
Preparation of lung tissue in Unicryl resin
1| From fixed rodent lung tissue (see EQUIPMENT) prepare 3 mm3 blocks and store overnight in fresh 3% paraformaldehyde/0.1% gluteraldehyde at 4 °C.
2| Put the shell vials with the tissue into metal vial holders and place in a plastic container surrounded with crushed ice.
▲ CRITICAL STEP To obtain consistent (and optimal) dehydration of tissue, the vials must remain in this container on crushed ice throughout the following steps.
3| Rinse the tissue for 3 × 10 min in cold 1× PBS on ice and leave overnight in 1× PBS at 4 °C.
4| Pipette off PBS, immerse tissue in cold 30% ethanol for 15 min at 4 °C. Repeat this procedure first using 50% and then 70% cold ethanol. The blocks in 70% ethanol should sink to the bottom of the vial; if not, place uncapped vials under vacuum for 20 min to extract trapped air and ensure complete penetration by the fluid.
5| Dehydrate tissue at 4 °C in 95% cold ethanol for 15 min. Repeat in fresh 95% ethanol for another 15 min at 4 °C.
6| Dehydrate tissue in 100% cold ethanol for 15 min at -20 °C. Repeat twice in fresh cold 100% ethanol for 15 min.
7| Infiltrate tissue in cold 100% Unicryl for 1 h at -20 °C. Repeat in fresh cold Unicryl for 1 h at -20 °C.
! CAUTION Store Unicryl at -20 °C in double Ziploc plastic bags for transfer between the hood and freezer. Similarly, after adding Unicryl to the tissue, place the complete plastic container with holders, vials and ice into double Ziploc plastic bags and seal the bags before transferring between the hood and the -20 °C refrigerator.
8| Infiltrate tissue in fresh 100% Unicryl overnight at -20 °C.
9| The next morning, layer crushed ice in the PELCO cryochamber, cover the wet ice with dry-ice pellets and allow the chamber to cool to -20 °C (this will take ~4 h).
10| Insert clean BEEM capsules into labeled BEEM cryocapsule holders, fill the capsules with fresh cold Unicryl and transfer the tissue blocks into the capsules with an applicator stick and (after ensuring that the blocks sink to the bottom of the capsule) close the cap.
11| Place the cryocapsule holders on the metal support rods inside the cryochamber, close the chamber top, check that the temperature is -20 °C and turn on the air-circulating fan and the UV light to polymerize the resin.
▲ CRITICAL STEP Monitor the temperature in the chamber throughout the day; when needed, add more ice to maintain the temperature at -20 °C. Overnight it can range from -20 °C to 10 °C, so always plan to add more ice early in the morning on the next day. Temperatures consistently lower than -20 °C will speed polymerization but lead to poor infiltration of the tissue by the resin.
12| After approximately 48 h, test a sample of resin blocks distributed throughout the chamber for sufficient hardness (open each capsule and tap with an applicator stick) and, when polymerization is complete, leave the capsules in the open chamber overnight to dissipate residual odor.
13| Cut the BEEM capsule away from the polymerized tissue block with a single-edge industrial blade, and label the block with a permanent ink marker.
■ PAUSE POINT These blocks are stable and can be stored at RT indefinitely.
Preparation of lung tissue sections
14| Under a stereomicroscope, use a single-edge industrial blade to remove excess resin from the surface of the block and (just) expose the tissue face.
15| As a prelude to cutting 90-nm-thick sections, collect 2-3 1-μm-thick sections on a glass slide, gently dry on a hotplate, stain with 0.1% toluidine blue, rinse with distilled/deionized water and mount under a coverslip using Permount. Check the tissue face by bright-field microscopy; when appropriate, that is, when the tissue face contains vessels or capillary networks of interest, proceed to cut 90-nm-thick sections.
16| Collect a series of 90-nm-thick sections onto Formvar-coated nickel grids with a (locking) Dumont tweezer, and place the tweezers with the grids in a closed Petri dish to air dry.
■ PAUSE POINT Store and catalogue these grids (with attached unstained tissue sections) in calibrated grid storage boxes until proceeding with immunogold labeling and high-resolution imaging. Plan on using these within weeks.
Immunogold labeling of lung tissue sections for high-resolution imaging
17| Place a strip of Parafilm on the rubber plate of a staining box and float Formvar-coated nickel grids (with tissue sections) face down in 1× PBS for 5 min (keep it covered to exclude dust particles).
▲ CRITICAL STEP Immediately before use, filter distilled/deionized water, PBS and staining solutions through Millex syringe-driven filters to minimize contamination by extraneous particulates. Transfer grids (with 90-nm-thick sections) using (locking) Dumont forceps, and rinse the forceps well between solutions to minimize cross-contamination.
18| Blot off excess PBS with filter paper strips and treat with 1% BSA in 1× PBS for 5 min to block endogenous/nonspecific tissue sites from reacting with the antibody.
19| Further block in 0.5% BSA in 1× PBS for 2 min.
20| Incubate in primary antibody (e.g., anti-αSMA, anti-PDGF-BB, anti-PDGF-Rβ, anti-CD11b and anti-VEGF-R2) diluted in 0.5% BSA in 1× PBS for 2 h at RT, or overnight at 4 °C (add a wet gauze pad to the staining box to make a humid chamber, if incubated overnight).
▲ CRITICAL STEP If incubated overnight at 4 °C, allow all staining boxes (with grids and sections) and reagents to reach RT before use.
21| Place all grids/sections in a clean drop of 1× PBS to halt the antibody reaction.
22| Pick up each grid/section with clean Dumont tweezers, wick off excess moisture from the grid with filter paper strips and rinse with 1× PBS; place each grid/section onto a clean drop of 1× PBS in the staining chamber, and rotate the chamber (on a rotomix) for 5 min. Repeat three times.
23| Wick off excess 1× PBS and incubate the grids/sections in 0.5% BSA in 1× PBS for 5 min.
24| Incubate in protein-A gold (pA-AU, Auroprobe AG10) diluted 1:50 in 1× PBS for 60 min at RT.
25| Wick off excess gold solution and rinse in filtered 1× PBS for 10 min. Repeat twice.
26| Collect each grid/section with a (locking) Dumont tweezer and place (the grid/section and tweezer) in a closed Petri dish to air dry. Grids with pA-AU-labeled antigenic sites can be treated with an optional silver aggregate procedure to enhance labeling (see Box 1).
▲ CRITICAL STEP To verify/validate the pA-AU result carry out the following steps: run appropriate negative control grids (concurrently) with Steps 17-26, for example; omit the antibody and incubate directly with the pA-AU to identify nonspecific adsorption of pA-AU to the section; incubate the section with IgG-rich plasma and the pA-AU complex to identify the ability of the pA conjugate to bind endogenous immunoglobulins; incubate the section with antigen-adsorbed antibody solution followed by the pA-AU complex to identify nonspecific antigen-antibody interactions; pretreat the antibody with a commercially available neutralizing peptide to block the reaction.
■ PAUSE POINT The dry grids can be left overnight.
Double-staining cell membranes to enhance (electron) density
27| Place each grid/section in a drop of 7.5% aqueous uranyl magnesium acetate for 20 min.
28| Rinse in distilled/deionized water and rotate for 10 min. Repeat twice.
29| Place each grid/section in a drop of 0.2% aqueous lead citrate for 5 min.
30| Rinse in distilled/deionized water and rotate for 10 min. Repeat twice.
31| Collect each grid/section with a (locking) Dumont tweezers and place in a closed Petri dish to air dry.
■ PAUSE POINT Stained grids/sections can be viewed repeatedly if carefully handled and stored in calibrated grid boxes to ensure reliable retrieval.
Analysis of immunogold-labeled cells
32| Examine the stained grids/sections by TEM (typically use a magnification of ×6,689 to ×9,818 to identify cell phenotype by morphology/organelles and position in a vessel or capillary wall, and magnifications from ×12,480 to ×19,636 to identify pA-AU-labeled antigenic sites).
33| Collect a library of high-resolution images of cells forming vessel or capillary walls.
34| Analyse images. Both images collected digitally or scanned from standard (3 inch × 4 inch) negatives are suitable for analysis; digital images facilitate quantitative analyses of antigenic sites (see Box 2 and Fig. 1), whereas the size (diameter) of a vessel, or of a capillary area of interest, is best determined from a standard negative.
Figure 1.
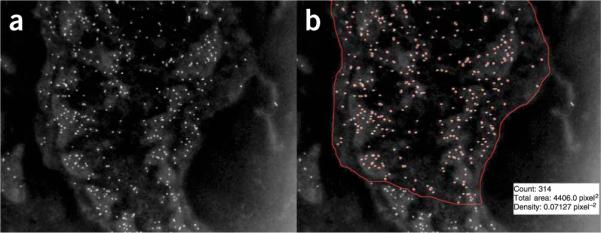
Quantification of gold particle density using image processing and analysis. In the inverted digital image collected by TEM (a) antigenic sites labeled with gold particles appear over the cell surface as bright dots of uniform size. The macro analysis of this image (b) detects, counts and outlines the particles, and also outputs the manually circumscribed area (red line enclosing the cell area). As shown for the fibroblast illustrated here (with pA-AU-labeled antigenic sites to PDGF-Rβ): the gold particle count is 314; the area analyzed is 4406.0 pixel2 and the density is 0.07127 pixel-2, which translates (from the number of nanometers per pixel) to an area of 3.90 μm2 and a calculated density of 80.0 μm-2.
Preparation of cell suspensions from blood and lung tissues
35| Draw (a minimum of) 1 ml of peripheral blood from rodents by cardiac puncture, and immediately transfer into EDTA collection tubes to prevent platelet degranulation. Keep the samples on wet ice at all times and process within 2-3 h of collection, as described in Steps 36-40.
36| Centrifuge fresh samples at 700g for 20 min at 4 °C (with no brake).
37| Gently remove the upper phase (plasma) with a 1 ml pipette tip (plasma can be stored in 0.25 ml aliquots for plasma protein measurements).
38| Resuspend the lower phase containing the blood cells using 5 ml of cold 1× PBS containing 0.5% (wt/vol) BSA and 1.5 mM EDTA.
39| Centrifuge samples at 700g for 20 min at 4 °C (with no brake).
40| Remove the upper phase and discard it; resuspend the cell pellet by gentle pipetting or vortex, and transfer 0.6 ml into a separate tube and keep on ice.
! CAUTION Lengthy storage of samples, or their maintenance at RT, is not recommended as it may affect cell phenotype.
41| Wash freshly excised rodent lungs in 1× PBS.
42| Cut the lung into small pieces (~1-2 mm in diameter) using scissors.
43| Homogenize the tissue by following option A or option B.
(A) Rocking
Transfer the lung pieces to a 50 ml conical tube and suspend in 20 ml of 2 mg ml-1 (prewarmed) collagenase type II solution in 1× PBS at 37 °C for 30 min.
Incubate for 45-75 min in a (rocking) water bath at 37 °C.
Aspirate off the collagenase and resuspend the homogenate in warm (37 °C) 1× PBS.
Using a 10 ml syringe with a 16-gauge blunt-end needle, homogenize the tissue by passing it through the needle (~20 times).
(B) Stomacher machine
Transfer the lung pieces to a 50 ml conical tube and suspend in 20 ml of 2 mg ml-1 (prewarmed) collagenase type II solution in 1× PBS at 37 °C for 30 min.
Incubate for 45 min in a (rocking) water bath at 37 °C.
Transfer the tissue suspension to a Stomacher bag and homogenize at low speed for 15 min using Stomacher machine, and transfer the tissue homogenate into a 50 ml tube.
44| Filter the tissue homogenate (through a 100 μm filter) into another 50 ml tube.
45| Centrifuge the samples at 700g for 20 min at 4 °C.
46| Resuspend the cells using 20 ml of cold 1× PBS.
47| Centrifuge the cells at 700g for 20 min at 4 °C.
48| Resuspend the cells using 5 ml of cold 1× PBS and keep on ice.
Immunostaining cells for multicolor flow cytometry
49| Immunostain cell suspensions obtained from lung samples or blood from rats (option A) or mice (option B).
▲ CRITICAL STEP It is recommended that antibody lots should be first tested for activity with positive control samples before experimental samples are assayed. For hematopoietic markers such as CD45 and CD11b, peripheral blood cells are the ideal test samples; staining with these markers should delineate clear positive and negative populations. For VEGF-R2, primary or immortalized endothelial cell lines can be used to test for activity; however, the degree of staining intensity will differ between adherent, purified, cultured and circulating cells. Fc blocking (when using the PDGF-Rβ antibody) is important to prevent nonspecific binding common to many cell types, including B lymphocytes, natural killer cells, granulocytes, monocytes, macrophages and platelets. Commercially available VEGF-R2 and PDGF-Rβ antibodies for mouse were conjugated to fluorophores, whereas the ones available for rat were not, necessitating additional steps (see Steps A(vi) and (vii)) for adding a fluorophore, that is, FITC-goat anti-rabbit IgG.
(A) For rat samples
Place 200 μl of blood or lung tissue cell samples into one isotype control and two sample 15 ml tubes. Add 1 mg ml-1 Fc-receptor-blocking agent; incubate the sample on ice for 10 min.
-
Add the following fluorophore-labeled antibodies:
Tube 1 (isotype control), 1 μl IgG-FITC, 1 μl IgG-PE, 1 μl IgG-PE-Cy5 and 1 μl IgG-allophycocyanin (APC);
Tube 2 (sample), 1 μl (purified) VEGF-R2 antibody, 1 μl CD11b-PE and 1 μl of CD45-PE-Cy5;
Tube 3 (sample), 1 μl (purified) PDGF-Rβ antibody, 1 μl CD11b-PE and 1 μl CD45-PE-Cy5.
-
For additional studies of lung cell phenotype, prepare extra tubes.
▲ CRITICAL STEP During the first run, it is important to run single-color controls to set up the compensation matrix. Cell viability can be tested using nuclear dyes, such as the DNA intercalator propidium iodide (replaces the PE antibody) or monomeric cyanine dyes (To-Pro-3, far-red channel). If using fluorescence-labeled antibodies from other manufacturers, the concentration should be optimized by testing serial concentrations to achieve similar staining patterns.
Vortex briefly and incubate for 30 min at 4 °C, or on ice in the dark.
Wash once with 5 ml of cold regular 1× PBS and centrifuge at 250g at 4 °C (with brake) for 5 min to remove the supernatant.
Resuspend the cells in tubes 2 and 3 in 200 μl of 1× PBS. Add 1 μl of FITC-goat anti-rabbit IgG.
Vortex briefly and incubate for 30 min at 4 °C, or on ice in the dark.
(B) For mouse samples
Place 200 μl of blood or digested lung tissue samples into one isotype control and two sample 15 ml tubes.
-
Add the following fluorophore-labeled antibodies:
Tube 1 (control), 1 μl IgG-FITC, 1 μl IgG-PE and 1 μl IgG-peridinin-chlorophyll-protein complex (PerCP);
Tube 2 (sample), 1 μl CD31-FITC, 1 μl VEGF-R2-PE, 1 μl CD45-PerCP, 1 μl CD11b-APC;
Tube 3 (sample), 1 μl CD31-FITC, 1 μl PDGF-Rβ-PE, 1 μl CD45-PerCP, 1 μl of CD11b-APC.
For additional studies of cell phenotype, prepare extra tubes. For example, use Stem cell antigen 1 (Sca1-PE, 1 μl per sample) to identify subsets of murine progenitor cells.
Vortex briefly and incubate for 30 min at 4 °C or on ice in the dark.
Wash once with 5 ml of cold 1× PBS and centrifuge at 250g at 4 °C (with brake) for 5 min to remove the supernatant.
50| Lyse RBCs in rat and mouse samples; first add 2 ml of ACK lysing buffer, vortex briefly and incubate at RT (18-25 °C) for 3 min. It may not be necessary to remove RBCs from tissue cell suspension but it would be helpful as it is often hard to completely avoid some RBC contamination.
▲ CRITICAL STEP Lysis should not be performed for more than 3-5 min as it may affect cell viability.
51| Wash once with 5 ml cold 1× PBS and centrifuge at 250g at 4 °C (with brake) for 5 min to remove the supernatant.
52| Resuspend the cell pellet in 400 μl 1× PBS and filter the samples through 40 μm cell strainer into 5 ml BD Falcon tubes and label the tubes with the staining combination information.
53| To assess viability, after washing, resuspend the pellet in 1 ml 1× PBS containing 20 μg ml-1 of viability dye, gently mix the suspension, incubate for approximately 20 min at 4 °C (in the dark) and run the samples on the flow cytometer in their staining solution. Use single-color control tubes with the viability dye only.
▲ CRITICAL STEP All tubes should be kept at 4 °C (or on ice) in the dark before acquisition on the flow cytometer and should be run within 1 h after the addition of the dye. If the samples are not run immediately, the samples should be fixed in 2% paraformaldehyde to ensure optimal results. Samples should not be exposed to light as bleaching of the fluorophores may occur.
Flow cytometry analysis of blood or lung tissue cells
54| Set the gate of the flow cytometer for the sample to be analyzed (the analysis of blood and lung cells will differ only in this step). For `blood samples,' set the gate on lymphocytes and monocytes (mononuclear cells) to avoid any RBC, platelet, cell debris and neutrophil contamination. Lymphocytes have low forward scatter (FSC) and side scatter (SSC), monocytes have high FSC and low SSC and neutrophils have high FSC and SSC characteristics. Collect at least 100,000-150,000 events in the mononuclear or cellular gate.
For `tissue cell suspensions,' gate on all cell size events. Individual cell populations cannot be defined using FSC and SSC. Therefore, set the gate on cell size events (see Fig. 2). This can be estimated by using the FSC characteristics of lymphocytes defined by the above peripheral blood samples. Collect 100,000-150,000 events in the mononuclear or cellular gate.
Figure 2.

Representative flow cytometry plots using FlowJo software to analyze cells from lung tissue or from rat peripheral blood. (a) FSC-SSC plot of lung cells-gate set on cell size events; (b) FSC-SSC plot of blood cells-gate set on mononuclear cells, excluding neutrophils, red blood cells, platelets and cell fragments; (c) Analysis of PDGF-Rβ and CD11b expression on cells from rat peripheral blood.
▲ CRITICAL STEP Save the settings for interchannel compensation and analysis and use for all samples, but set the gate individually for each sample on the mononuclear cell populations to exclude any contamination by neutrophil, platelet, RBC or cell debris (see Fig. 2). Flow-cytometric samples must be properly compensated; proper compensation techniques can be referenced using general flow cytometry textbooks or online sources (http://www.bdbiosciences.com/immunocytometry_systems/support/training/online/ITF/start.html and http://www.flowjo.com/v6/reference.html).
55| First, run the isotype-stained control sample with voltages set so that a majority of events are in the left lower quadrant. Next, run each single-color fluorochrome control. Signal that bleeds from one channel into an adjacent channel can be removed using the Cell-quest software during sample acquisition. Likewise, care must be taken so as not to overcompensate the samples. Alternatively, the data from single-color controls can be used to set up a compensation matrix at a later time using software programs such as Flowjo.
56| Identify endothelial cells as VEGF-R2+ and CD45- cells, mesenchymal cells as PDGF-Rβ+ and CD45- cells, and myeloid cells as CD11b+CD45+ cells. Small myeloid cell subsets (0.02%-0.10% of all blood mononuclear cells) may express VEGF-R2 and PDGF-Rβ. Calculate the number of cells for each population as percentage of the total number of mononuclear cells (Fig. 2).
? TROUBLESHOOTING
? TROUBLESHOOTING
Troubleshooting advice can be found in Table 1.
TABLE 1.
Troubleshooting table.
| Step | Problem | Possible reason(s) | Solution |
|---|---|---|---|
| Steps 1-34 | Incomplete hardening of the resin tissue blocks | Incomplete tissue dehydration and/or resin infiltration | Check the temperature of reagents for each step and extend timing as needed |
| Inadequate exposure of blocks to ultraviolet light or failure to keep the cryochamber temperature within correct range | Check that the ultraviolet light source of the chamber is intact and increase the frequency of temperature monitoring to maintain a range between -10 °C to -20 °C for 24-48 h | ||
| Nonspecific gold label (i.e., over non-tissue regions such as the lumen of a vessel or capillary) | Blocking serum is inappropriate for the antibody used | If appropriate, adjust the blocking serum | |
| Primary antibody is inappropriate for the tissue | Check that the antibody is appropriate, and the expiry date and dilution of the antibody and gold solution used | ||
| Gold label is low or absent | Antibody failed to bind antigenic sites | Run a dilution experiment to establish the optimal antibody concentration | |
| Gold solution failed to bind antibody-labeled sites | Complete a run on tissue that is a known positive control | ||
| Background debris (dirt) is high | Contaminated solutions | Add additional rinsing steps and ensure that all solutions are filtered immediately before use | |
| Steps 35-56 | Cell sample does not become red-transparent | Incomplete lysis of RBCs | Centrifuge at 300g at 4 °C (with brake) for 5 min to remove the supernatant and repeat the lysing step for 1-2 min at RT |
| Fluorescence signal is poor/inadequate | Instrument malfunction (i.e., a weak excitation signal is produced) | Use an alternate machine to determine if the problem is related to the hardware or reagents | |
| Antibody/reagents are inappropriate or used at wrong concentration | If an instrument is malfunctional, use another cytometer; if not, check and repeat the experimental and staining procedures | ||
| Inaccurate/shifting compensation matrix indicated by variable readings for nonspecific IgG-staining tubes for blood or tissue | Compensation matrix settings have changed | Run single-color controls, or alternating-color controls, to reset and confirm that the compensation matrix is accurate |
● TIMING
Steps 1-8: 48 h to dehydrate and infiltrate lung tissue blocks (36-48 per rodent group) with resin
Steps 9-13: 58 h to polymerize tissue blocks in Unicryl resin, label and archive them
Steps 14-16: 8 h to cut 3-4 tissue blocks, that is, 2 h to rough-cut 1 tissue block, and prepare 1-μm-thick sections on glass slides and (approximately 15-20) 90-nm-thick sections on Formvar-coated grids
Steps 17-20: 16 h to treat (approximately 15-20) 90-nm-thick sections with primary antibody
Steps 21-26: ~2-3 h to label 15-20 grids/sections with pA-AU; 3 h to treat approximately 15-20 grids/sections with silver halide (Box 1)
Steps 27-31: 4 h to enhance electron-density of cell membranes and organelles in 15-20 grids/sections
Steps 32-34: ~2-3 d to view multiple grids/sections by TEM, collect overviews and determine the appropriate analysis. Allow an additional ~3 h to collect 100 images for qualitative or quantitative analysis, and 2 min per cell to quantify antigenic sites using the macro program
Steps 35-40: ~2 h to prepare cell suspensions from blood
Steps 41-48: ~2 h to prepare cell suspensions from lung tissue
Steps 49-56: 5 h to immunostain cells and 30 min to analyze six tubes by flow cytometry after a compensation matrix has been established
ANTICIPATED RESULTS
Immunophenotype of cells remodeling vascular structures in situ in rodent lung
Cell types in normal lung, and the ones participating in vessel remodeling (in rat) and capillary remodeling (in rat and mouse), are identifiable in high-resolution images by their morphology1 (see also accompanying Nature Protocol by Jones et al.2). The presence of antigenic sites on these cells-detected by the panel of primary antibodies used, for example, VEGF-R2, αSMA, SMM-HC, PDGF-A, PDGF-B, PDGF-Rα, PDGF-Rβ (Fig. 3) or CD11b is visualized by the distribution of electron-dense particles4,5,7,8. This will characterize the immunophenotype of the cells forming vessel and capillary walls. For example, the precursor smooth muscle cells (i.e., interstitial fibroblasts) developing in vessel walls of the high-oxygen-injured lung are shown by this approach to acquire a characteristic `contractile' smooth muscle phenotype by expressing αSMA and SMM-HC7,8. Quantifying the density of gold particles over vascular and precursor cells (by a macro) detects shifts in the expression of specific proteins4,5. Studies of the ligand-tyrosine kinase receptor-mediated basis of the response, as shown by the use of antibodies to PDGF-A-PDGF-Rα or PDGF-B-PDGF-Rβ, demonstrate a shift in phenotype of these precursor cells, and of other adjacent vascular cells, in the high-oxygen-injured lung4,5 (Fig. 4). Based on quantitative data clearly demonstrating the development of high numbers of PDGF-Rβ+ vascular precursor cells5, the use of a specific PDGF-Rβ-blocking agent to prevent this response points to a potential therapeutic strategy. Similar studies can be performed for endothelial precursor cells responsible for vessel and capillary remodeling.
Figure 3.
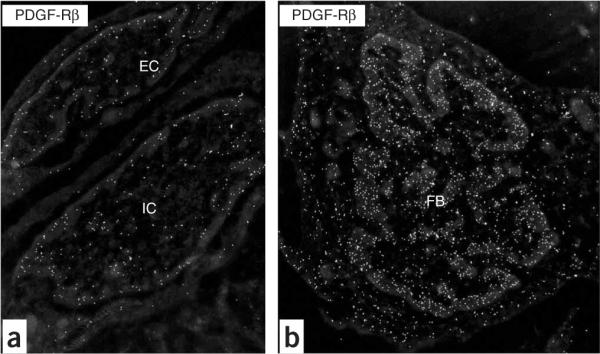
Inverted images of PDGF-Rβ+ vascular-associated cells in the rat lung after breathing high oxygen. (a) Endothelial cell (EC) and intermediate cell (IC) at D7; (b) mesenchymal cell, that is, fibroblast (FB) at D7. Note the condensation of heterochromatin at the edge of the nucleus. Original magnification: (a) ×19,636; (b) ×12,480.
Figure 4.

Schematic of vessel wall thickening by precursor cells in the rat after high oxygen (left); values for gold-labeled antigenic sites quantified by macro for each cell population at each time point (right). Note the distribution of sites in the normal lung (Control, D0) and the shift early (D4) and late (D28) in the hypertensive lung; although PDGF-AA expression predominates in vascular cells of the normal lung, a shift to PDGF-Rβ expression begins in the early stage of pulmonary hypertension and is pronounced in the late stage (reprinted with permission from ref. 4, Jones et al., 2006).
Immunophenotype of blood cells and lung tissue cell suspensions
In the complementary approach of flow cytometry we use in this protocol, the cell populations of interest-both in blood and lung tissue samples-are further characterized by multiple surface markers. For example, endothelial cells (VEGF-R2+CD45- cells) are detectable in the blood circulation in small numbers (ranging typically from 0.01% to 0.1% of blood mononuclear cells) but are abundant in cell suspensions from dissociated lung tissue. Multiple myeloid cell populations are detectable in blood and lung cell suspensions, including progenitor cells that are positive for VEGF-R2 and PDGF-Rβ (less than 0.1% of mononuclear cells, Fig. 2). The number and the phenotype of these cells change during HALI (see Fig. 2). The potential significance of these cell populations in adult neovascularization is well recognized; the HALI model provides data for kinetic studies of these cells within a highly predictable pattern of vessel and capillary remodeling. Finally, novel transgenic mice, and/or bone marrow transplantation models, can be integrated into the protocol described here to further study the function(s) of cells and molecules of interest. For example, mice that express the green fluorescence protein on activation of a promoter selective for a cell population (e.g., endothelial cells, fibroblasts or bone marrow-derived cells following restorative bone marrow transplantation) could be subjected to HALI, and cells analyzed based on recognition of endogenous green fluorescence protein expression (as opposed to immunophenotyping). Similar studies have been performed to analyze the basis of neovascularization in tumor models by flow cytometry6,9-11.
BOX 1 | OPTIONAL STEP TO SILVER ENHANCE pA-AU-LABELED ANTIGENIC SITES.
Mix equal parts of solutions A and B from the silver enhancement kit immediately before use.
Place 50 μl of the mixed silver solution for each grid/section on a strip of Parafilm; place each grid face down on these drops of mixed silver solution for 5 min (adjust this time to control the intensity of the silver halide).
Quickly move the grid/section from the silver solution drops to adjacent filtered/de-ionized water drops.
Rinse (jet wash) each grid/section with approximately 5 ml of de-ionized water.
Place each grid/section face down on a clean drop of water and rotate for 10 min. Repeat twice.
Collect each grid with a (locking) Dumont tweezers, and place in a closed Petri dish to air-dry.
▲ CRITICAL STEP Treat all grids/sections with silver solution for the same amount of time, as the size of the developing gold-silver aggregate is time-dependent; use only Teflon forceps for each step of the silver procedure as regular Dumont tweezers will react with the silver solution.
BOX 2 | OPTIONAL STEP TO QUANTIFY DENSITY OF pA-AU-LABELED ANTIGENIC SITES PER UNIT AREA OF CELL USING DIGITAL IMAGE ANALYSIS.
Determine the number of images required for quantitative analyses, based on the frequency of a given cell type and, as individual cells or entire cell populations may be positive or negative, the number of cells positive for the specific protein(s) of interest. Note that this distribution may shift with the length of time of breathing high oxygen (i.e., Day 4 versus Day 28) or of breathing air after high oxygen (1 week versus 4 weeks).
Characterize cells of interest by their morphology; vessel walls, for example, will include endothelial cells, intermediate cells, smooth muscle cells or perivascular fibroblasts, and capillary walls will include endothelial cells and pericytes.
-
Assess the density of antigenic sites associated with a given cell using the NIH-supported, public domain, java-based, application ImageJ 1.38, which can be downloaded at http://www.rsb.info.nih.gov/ij/.
The protocol notation refers to `menu'/`command'. So, for example, the `Sharpen' command is found under the `Process' menu. To automate the process, these steps can readily be incorporated into a macro with a single command. As we are interested in the antigen density on a given cell, we first make a selection using the free selection tool in ImageJ to enclose the area of interest. Once this is done, the processing and analysis can be performed. Figure 1 shows a representative raw image (Fig. 1a) and the results of processing and analysis (Fig. 1b).
Process/Sharpen.
Process/Filters/Maximum.
Process/Filters/Minimum.
Process/Find Edges.
Image/Adjust/Threshold—set threshold; move bottom slider to 255 and top slider to 230 (these values may have to be adjusted for images with different brightness or contrast levels).
Process/Binary/Fill Holes.
Process/Binary/Erode.
-
Analyze Particles; set options in the dialog box as follows:
Size: 2—75
Circularity: 0.00-1.00
Show: choose any option you like here—this only affects how the results are indicated on the image.
Check the `Summarize', `Clear Results' and `Include Holes' boxes.
Click "OK"; the particles will then be counted along with the area of the selected region.
The data can be viewed by selecting Window/Summary. Results listed include the file name, the number of particles (`Count'), the selected area (`Total Area'), the `Average Particle Size' and the `Area Fraction' (the number of pixels associated with particles divided by the selected area). The relevant parameter for most studies is the gold particle density, which can be calculated as the number of particles divided by the selected area. Length scales can be calibrated within ImageJ (by choosing Analyze/Set Scale), or postanalysis if the number of nanometers per pixel is known (by multiplying the area in pixels by N2, where N is the number of nanometers per pixel).
ACKNOWLEDGMENTS
Supported by NIH R01HL 070866 (to RJ), NIH R01CA 115767 (to RKJ) and by a Career Development Award to DGD (from the American Association for Cancer Research-Genentech BioOncology).
References
- 1.Jones R. Ultrastructural analysis of contractile cell development in lung microvessels in hyperoxic pulmonary hypertension. Fibroblasts and intermediate cells selectively reorganize nonmuscular segments. Am. J. Pathol. 1992;141:1491–1505. [PMC free article] [PubMed] [Google Scholar]
- 2.Jones RC, Capen D, Petersen B, Jain RK, Duda DG. A protocol for a lung neovascularization model in rodents. Nat. Protoc. 2008;3:378–387. doi: 10.1038/nprot.2007.536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jones RC, Jacobson M. Angiogenesis in the hypertensive lung: response to ambient oxygen tension. Cell Tissue Res. 2000;300:263–284. doi: 10.1007/s004419900100. [DOI] [PubMed] [Google Scholar]
- 4.Jones R, Capen D, Jacobson M, Munn L. PDGF and microvessel wall remodeling in adult rat lung: imaging PDGF-AA and PDGF-Rα molecules in progenitor smooth muscle cells developing in experimental pulmonary hypertension. Cell Tissue Res. 2006;326:759–769. doi: 10.1007/s00441-006-0177-5. [DOI] [PubMed] [Google Scholar]
- 5.Jones R, Capen D, Jacobson M. PDGF and microvessel wall remodeling in adult lung: imaging PDGF-Rβ and PDGF-BB molecules in progenitor smooth muscle cells developing in pulmonary hypertension. Ultrastruct. Pathol. 2006;30:267–281. doi: 10.1080/01913120600820336. [DOI] [PubMed] [Google Scholar]
- 6.Duda DG, et al. Evidence for incorporation of bone marrow-derived endothelial cells into perfused blood vessels in tumors. Blood. 2006;107:2774–2776. doi: 10.1182/blood-2005-08-3210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jones R, Jacobson M, Steudel W. Alpha-smooth-muscle actin and microvascular precursor smooth-muscle cells in pulmonary hypertension. Am. J. Respir. Cell Mol. Biol. 1999;20:582–594. doi: 10.1165/ajrcmb.20.4.3357. [DOI] [PubMed] [Google Scholar]
- 8.Jones R, Steudel W, White S, Jacobson M, Low R. Microvessel precursor smooth muscle cells express head-inserted smooth muscle myosin heavy chain (SM-B) isoform in hyperoxic pulmonary hypertension. Cell Tissue Res. 1999;295:453–465. doi: 10.1007/s004410051251. [DOI] [PubMed] [Google Scholar]
- 9.De Palma M, et al. Tie2 identifies a hematopoietic lineage of proangiogenic monocytes required for tumor vessel formation and a mesenchymal population of pericyte progenitors. Cancer Cell. 2005;8:211–226. doi: 10.1016/j.ccr.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 10.De Palma M, Venneri MA, Roca C, Naldini L. Targeting exogenous genes to tumor angiogenesis by transplantation of genetically modified hematopoietic stem cells. Nat. Med. 2003;9:789–795. doi: 10.1038/nm871. [DOI] [PubMed] [Google Scholar]
- 11.Duda DG, et al. Differential transplantability of tumor-associated stromal cells. Cancer Res. 2004;64:5920–5924. doi: 10.1158/0008-5472.CAN-04-1268. [DOI] [PubMed] [Google Scholar]