

Abstract
Background
Sphingosine-1-phosphate (S-1-P) is a bioactive sphingolipid released from activated platelets, which stimulates migration of vascular smooth muscle cells (VSMC) in vitro. S-1-P will activate akt, which can regulate multiple cellular functions including cell migration. Akt activation is downstream of phosphatidyl-inositol 3′ kinase (PI3-K) and Phosphoinositide-dependent protein kinase-1 (PDK1).
Objective
To examine the regulation of akt signaling during smooth muscle cell migration in response to S-1-P.
Methods
Murine arterial SMCs were cultured in vitro. Linear wound and Boyden microchemotaxis assays of migration were performed in the presence of S-1-P with and without an akt inhibitor (aktI). Assays were performed for PI3-K, PDK1, akt and GSK3β activation in the presence of various inhibitors and after transfection with the Gβγ inhibitor. βARKCT.
Results
S-1-P induced time dependent PI3-K, PDK1 and akt activation. The migratory responses in both assays to S-1-P were blocked by akt inhibitor (aktI). Activation of akt and dephosphorylation of its downstream kinase, GSK3 β, were inhibited by aktI. Inhibition of PI3-K with LY294002 significantly reduced both PI3-K and akt activation. Inhibition of G βγ inhibited akt activation through a reduction in both PI3-K and PDK1 activation. While inhibition of the ras with manumycin A had no effect, inhibition of rho with C3 limited both PI3K and akt activation. PDK1 responses were unchanged by inhibition of GTPases. Inhibition of reactive oxygen species generation with N-acetylcysteine and of EGFR with AG1478 inhibited PDK1 activation in response to S-1-P.
Conclusion
S-1-P mediated migration is akt dependent. S-1-P mediated akt phosphorylation is controlled by G βγ dependent, PI3-K activation, which requires the GTPase rho and Gβγ. PDK1 activation requires Gβγ reactive oxygen species generation and EGFR activation.
Keywords: S-1-P, vascular smooth muscle cell, cell signaling, PI3-K, PDK1, akt, migration
INTRODUCTION
Atherosclerosis remains a significant public problem and changes in the vessel wall and in smooth muscle cell (VSMC) biology are keys to plaque formation and development. One key VSMC function in plaque development is cell migration. S-1-P is a ligand for a family of G-protein-coupled receptors (S-1-P1, S-1-P2 and S-1-P3) that are highly regulated and differentially expressed in various tissues. S-1-P1 and S-1-P3 are considered the major receptors for S-1-P-stimulated VSMC migration, while S-1-P2 seems to suppress VSMC motility (1). S-1-P1/S-1-P3 receptors promote, whereas S-1-P2 receptors antagonize, phenotypic modulation in vivo after vascular injury (2). S-1-P activates mitogen activated protein kinases (MAPK) in VSMC. Through a Gαi mediated, ras-dependent mechanism, S-1-P activates MEK1/2 and ERK1/2; MKK3/6 and p38MAPK are activated by a Gαi mediated phosphatidyl-inositol 3′ kinase (PI3-K) -dependent mediated pathway in vascular smooth muscle cells (3, 4). S-1-P can also activate mTOR through a PI3-K mediated pathway (5) and S-1-P can activates the small GTPase, rho, and induces the generation of reactive oxygen species, which both modulate MAPK activation and cell migration in VSMC and are known to interact with PI3-K mediated pathways (6, 7). Most of the aforementioned mechanisms involve PI3-K, which in turn can activate akt (a.k.a. Protein kinase B). Akt activation is downstream of PI3-K and Phosphoinositide-Dependent protein Kinase-1 (PDK1) (Fig 1A). Therefore, akt is a key intracellular kinase that has been shown to regulate multiple cellular functions including cell migration. The aim of this study is to examine the mechanisms and regulation of akt signaling during VSMC migration in response to S-1-P.
Figure 1.
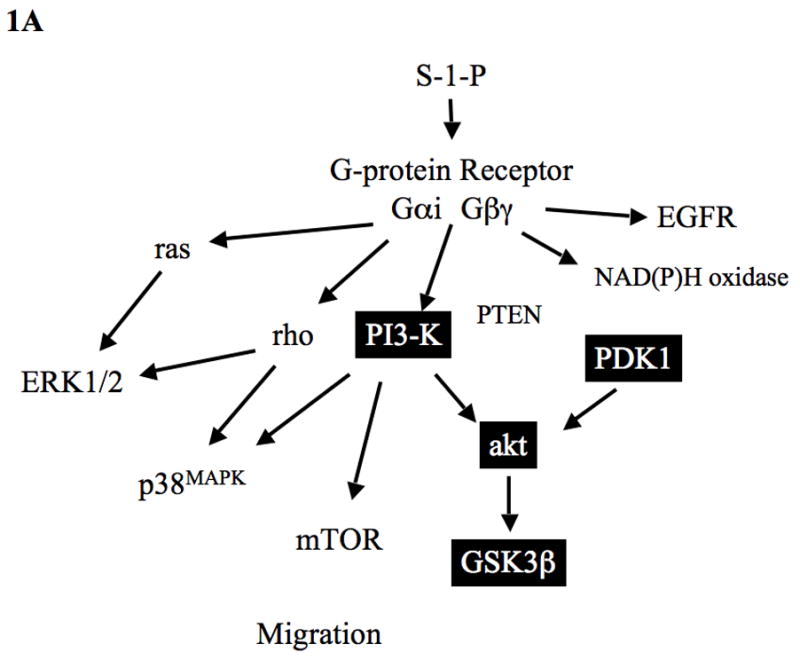
(A) Summary of S-1-P signaling in VSMC: S-1-P has been shown to interact with PI3-K and akt in other tissues. Activation akt leads to dephosphorylation of GSK3β. PI3-K activity is modulated by its inhibitor PTEN. A role for PDK1 has not been defined. S-1-P activation of PI3-K involves the Gαi and Gβγ proteins and can lead to activation of MAPK and mTOR. S-1-P can mediate rho activation and modulate MAPK activity. Other data has shown that S-1-P can induce the production of oxygen free radicals through NAD(P)H oxidase and can activate the EGF receptor (EGFR).
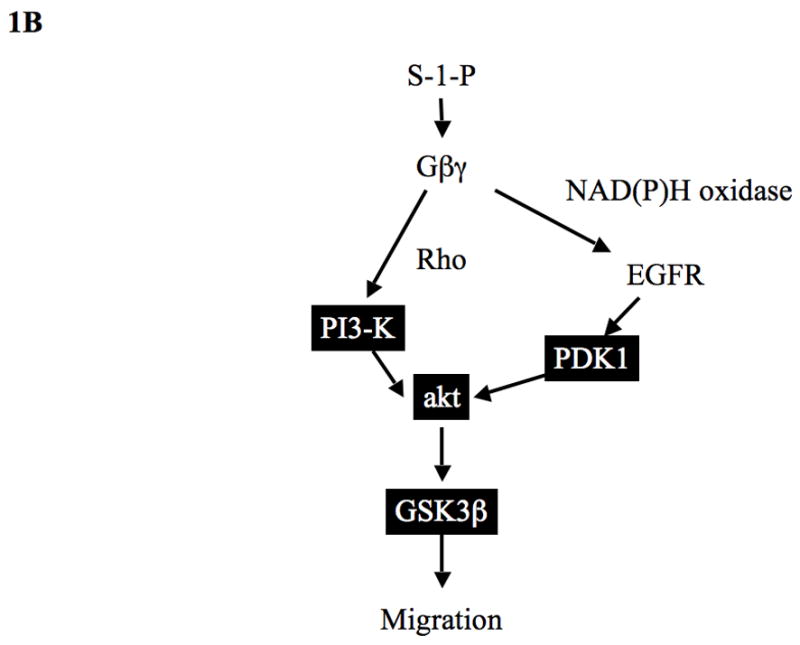
(B) Summary of S-1-P PI3-K/PDK1/akt activation pathways in VSMC: S-1-P mediated akt phosphorylation is controlled by Gβγ dependent, PI3-K activation, which requires the GTPase rho and by PDK1 activation, which requires Gβγ, generation of reactive oxygen species and activation of EGFR.
MATERIALS AND METHODS
Experimental Design
Murine arterial VSMCs were cultured in vitro. Linear wound and Boyden microchemotaxis assays of migration were performed in the presence of S-1-P with and without an akt inhibitor. Assays were performed for activation of PI3-K, PDK1, akt and GSK3β activation in the presence of various inhibitors and after transfection with the Gβγ inhibitor βARKCT. Additional experiments were performed to determine the role of oxygen free radical generation and the epidermal growth factor receptor (EGFR) in akt activation.
Materials
D-erythro-sphingosine-1-phosphate was purchased from Avanti Polar-Lipids (Alabaster, AL). C3 (rho inhibitor), LY294002 (PI3-K inhibitor), N-acetyl cysteine (free radical scavenger, NAC) and akt inhibitor (Calbiochem #124008; C29H59O10P) were purchased from Calbiochem (La Jolla, CA). Manumycin A (ras inhibitor, MA), AG1478 (EGFR inhibitor) and di-phenylene-iodonium (NAD(P)H oxidase inhibitor, DPI) was purchased from Sigma Chemical Co. (St. Louis, MO). Peroxidase-conjugated anti-rabbit IgG antibody (raised in goat) and peroxidase-conjugated anti-mouse IgG antibody (raised in goat) were purchased from Jackson ImmunoResearch Laboratories Inc. (West Grove, PA). Phospho-akt (ser472) and Phospho GSK3β (ser9) and phospho EGFR (tyr1068) were purchased from Cell Signaling (Beverley, MA). Empty adenovirus and adenoviral βARKCT were purchased from Weigen Inc. (Worchester, MA). Dulbecco’s Minimum Essential medium (DMEM) and Dulbecco’s phosphate buffered saline (dPBS) were purchased from Cellgro (Manassas, VA).
Wound Assay
The wound assay was performed with VSMC as previously described (8, 9). Trials with each reagent or inhibitor were performed in six separate dishes, and the results were averaged. Cells were then allowed to migrate over 24h at 37°C in DMEM with or without S-1-P (100nM). In a second series of experiments, migration in response to S-1-P was examined in the presence and absence of the inhibitors.
Boyden Chamber
Chemotaxis was measured using a 48-well Boyden chamber (Neuroprobe Inc. Gaithersburg, MD) and polycarbonate filters (Neuroprobe, Inc., 10 μm pore size, 25 × 80 mm, PVP free) with VSMC as previously described (8, 9). S-1-P (100nM) was added to the lower wells. For trials with the inhibitors, the inhibitor was added to 2 ml of the cell suspension 1h prior to addition of cells to the upper wells. Trials included eight or twelve wells per reagent or inhibitor per trial, and were repeated no fewer than three times.
Western Blotting
Cells were allowed to grow to 80% confluence and starved for 48h. Cells were then stimulated with S-1-P alone and in the presence of pharmacologic inhibitors (see Results) and harvested at time points from 0 to 30 minutes. Western blotting was performed as previously described (5). Activation of PI3-K was determined by phosphorylation of its p85 subunit, that of PDK1 by phosphorylation of its active site, that of akt by phosphorylation of S473 and T308, and that of GSK3β by determination of its dephosphorylated state at Ser9. Activation of EGFR was determined by phosphorylation of the Tyr 1068 site. Total protein was determined using antibodies against each intact kinase.
Adenoviral Infection
Adenoviral vectors were constructed by Welgen, Inc. using purified plasmid encoding βARKCT, obtained from Guthrie cDNA Resource center (Sayre, PA). VSMC were plated at 70% confluence in 100mm dishes and allowed to grow overnight. Recombinant adenovirus was then added at the appropriate concentrations (βARKCT; 100 MOI) in a reduced volume of media (1.5 – 2 ml). After 48h incubation, the media was changed and cells grown for an additional 24h. The cells were then used for assays. Empty vector served as control. Transfection was performed as previously described (6).
Data and Statistical Analysis
All data are presented as the mean ± standard error of the mean (s.e.m.), and statistical differences between groups were tested with a Kruskal-Wallis nonparametric test with post hoc Dunn’s multiple comparison correction, where appropriate. A p-value less than 0.05 was regarded as significant. Non-significant p-values were expressed as p=ns.
RESULTS
Cell Migration
To determine if akt was important in cell migration, we examined VSMC migration in the wound and Boyden chamber assays. S-1-P induced a concentration dependent cell migration in both migration assays and these responses were blocked by both the PI3-K inhibitor LY294002, and the akt inhibitor (Fig 2). These findings suggest that PI3-K and akt are involved in S-1-P mediated migration.
Figure 2.
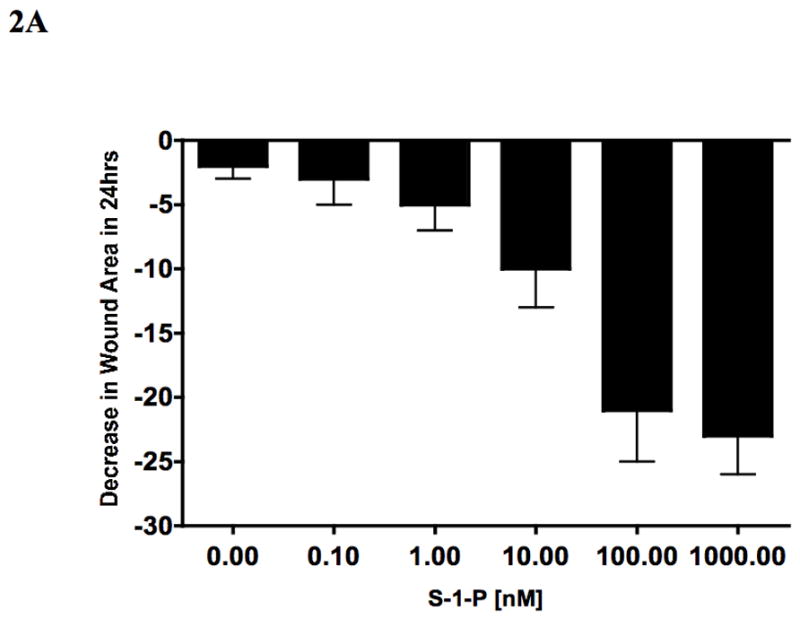
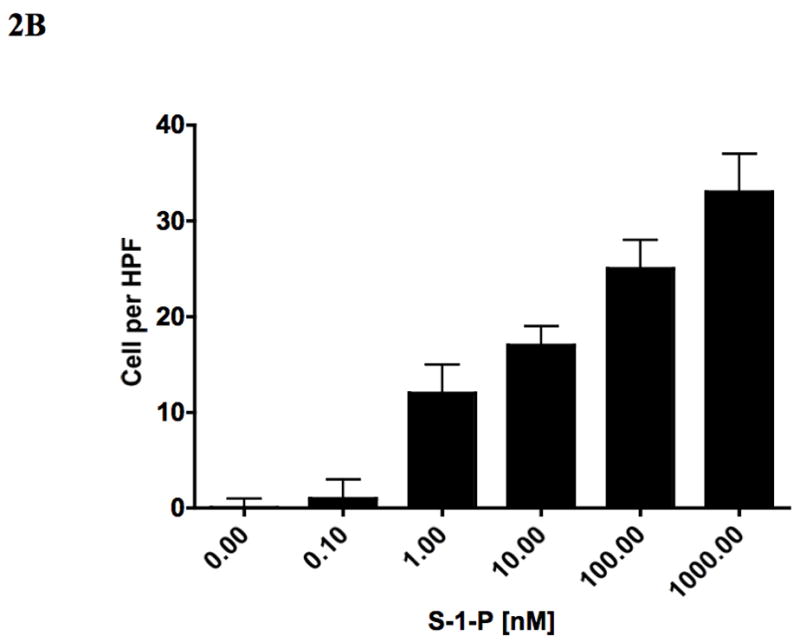
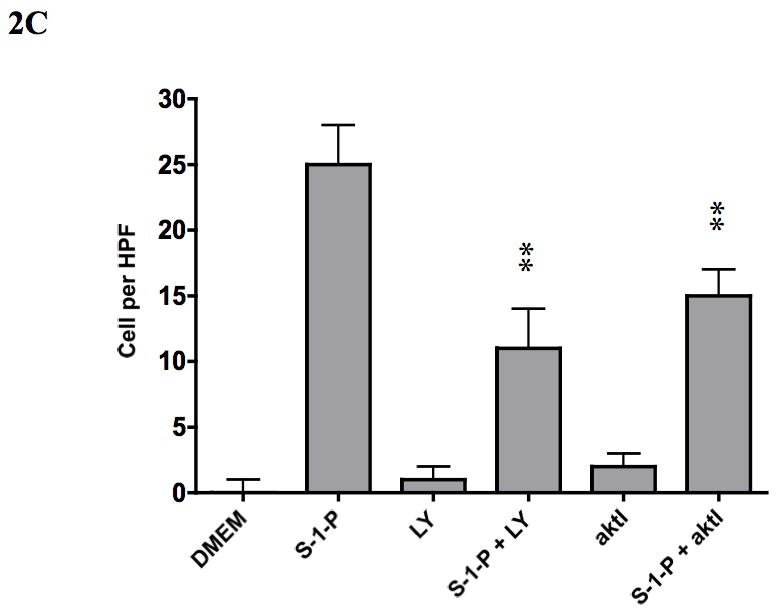
VSMC migration in response to S-1-P is does dependent (0.1–1000nM) in both the wound assay (A) and the Boyden chamber assays (B) of migration. This response was inhibited by the PI3K inhibitor LY294002 and akt inhibitor (aktI, Calbiochem #124008; C29H59O10P) (C). Values are the mean±SEM (n=6, **p<0.01 compared to control).
Akt activation pathways
To assess if S-1-P can activate both the kinases upstream of akt (PI3-K and PDK1), we examined its effects on PI3-K, PDK1 and akt phosphorylation and GSK3β dephosphorylation by western blotting in VSMC. S-1-P induced a time dependent phosphorylation of phosphatidyl-inositol 3′ kinase (PI3-K) and phosphoinositide-dependent protein kinase-1 (PDK1) and activated akt (Fig 3A). There was the expected concomitant dephosphorylation of GSK3β, one of the down stream kinases of akt (Fig 3A). There are two phosphorylation sites on akt, S473 and T308, and both are required for maximal activation of the kinase (Fig 3A). There was time dependent activation of both activation sites of akt in response to S-1-P (Fig 3B). Activation of akt and concomitant lack of dephosphorylation of GSK3β were inhibited by the akt inhibitor (Fig 3C). Inhibition of PI3-K with the PI3-K inhibitor, LY294002, significantly reduced both PI3-K and akt activation (Fig 3C). Activation of the PDK1 was unaffected by pre-incubation with the PI3-K inhibitor, LY294002 and the akt inhibitor suggesting that it is not down stream of PI3-K(Fig 3C).
Figure 3.
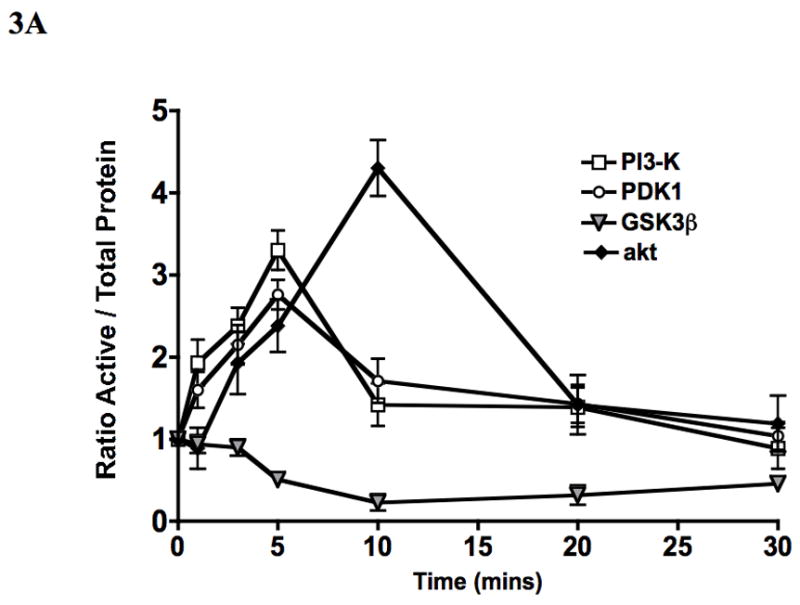
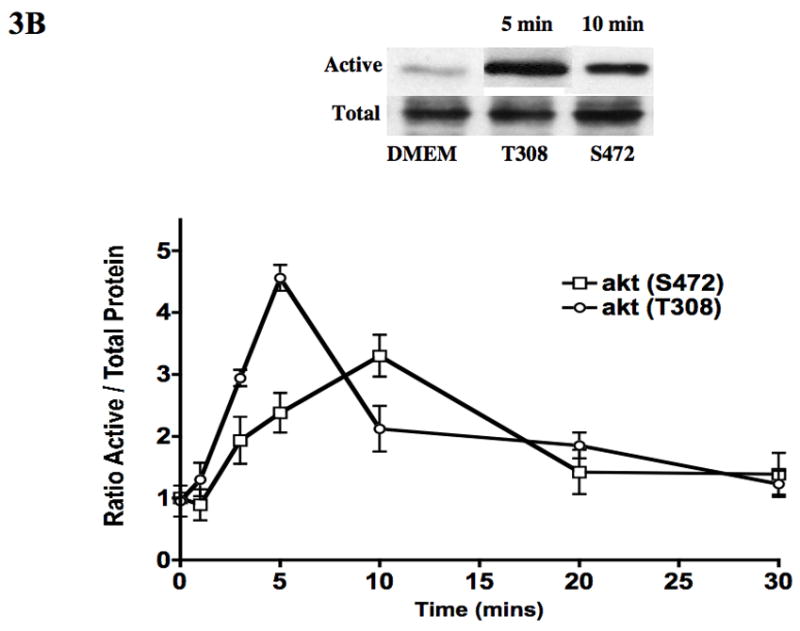
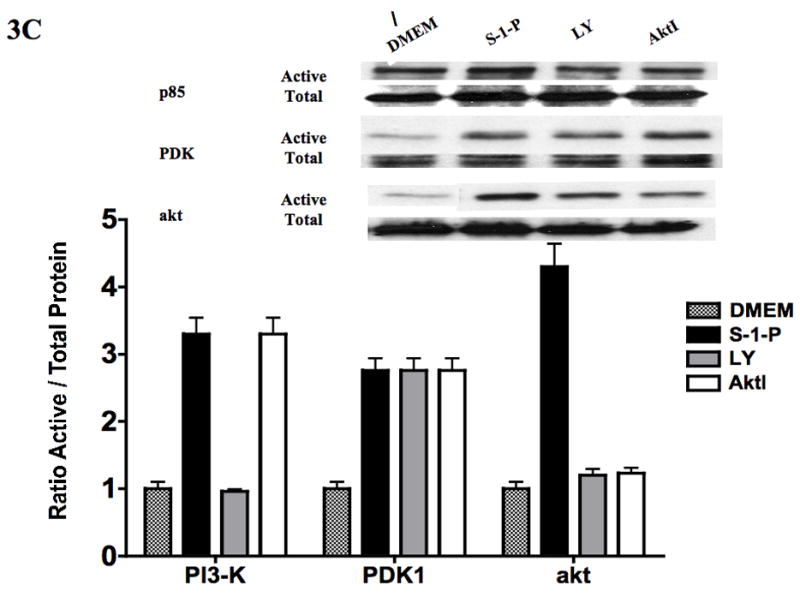
S-1-P (100nM) induced time dependent PI3-K, PDK1 and akt phosphorylation and dephosphorylation of GSK3β (A) as detected by changed in protein phosphorylation. There was time dependent activation of both the activation sites of akt S472 and T308 in response to S-1-P (B). Activation of PI3-K was inhibited by the PI3K inhibitor LY294002 and akt by the akt inhibitor (aktI) (C). Values are the mean±SEM of the ratio of the phosphorylation of the respective protein relative to the total unphosphosylated protein (n=6, *p<0.05 compared to control). Representative Western blots are shown above the graphs.
Upstream activation of Akt pathways
S-1-P is known to mediate its responses via G-proteins (Gαi and Gβγ) and the small GTPases (ras and rho). Gβγ is a potent, G-protein dimer formed after G-protein coupled receptor activation and Gβγ has been linked to PI3-K activation. Inhibition of the G-protein, Gβγ, with adenovirus contianing βARKCT, decreased akt activation in response to S-1-P through a reduction in both PI3-K and PDK1 activation (Fig 4). An empty adenovirus had no effect. To determine the role of the small GTPases ras and rho, we incubated VSMC with inhibitors of ras (manumycin A) and rho (C3). While inhibition of ras with manumycin A (10mM) had no effect, inhibition of rho with C3 at a concentration that inhibits rho activation, limited both PI3-K and akt activation. PDK1 responses were unchanged by inhibition of the ras and rho GTPases (Fig 4).
Figure 4.
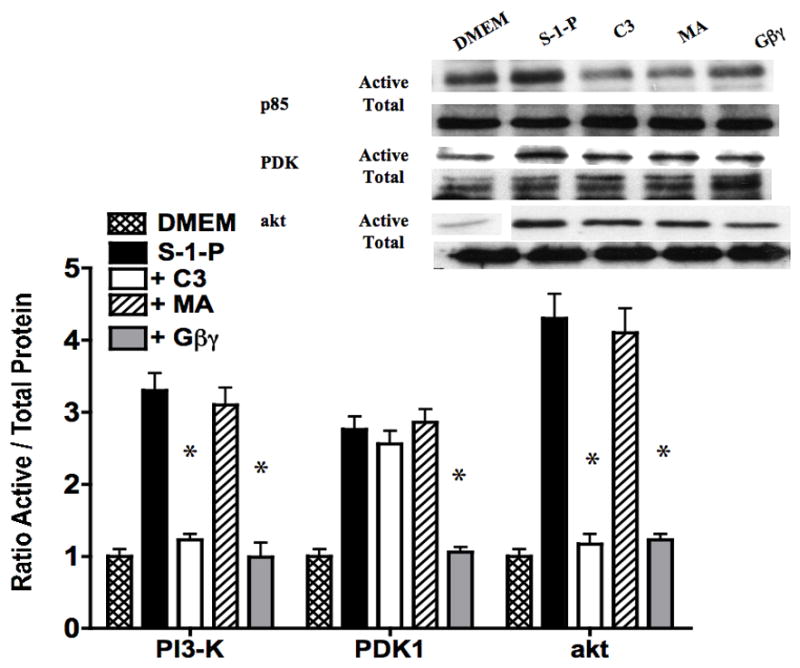
Inhibition of the ras with manumycin A had no effect had no effect on S-1-P mediated PI3K and akt phosphorylation, while inhibition of rho with C3 limited both PI3K and akt activation. PDK1 responses were unchanged by inhibition of GTPases, ras and rho. Inhibition of the G-protein, Gβγ, with βARKCT, inhibited akt activation in response to S-1-P through a reduction in both PI3-K and PDK1 activation. Values are the mean±SEM of the ratio of the phosphorylation of the respective protein relative to the total unphosphosylated protein (n=6, *p<0.05 compared to control). Representative Western blots are shown above the graphs.
G-protein coupled receptors have also been shown to activate epidermal growth factor receptor (EGFR) and to generate reactive oxygen species, both of which are important intermediate mediators of transmembrane signaling. We chose to examine these alternative pathways of activation. Inhibition of oxygen free radical signaling with the general oxygen free radical scavenger, NAC, and of NAD(P)H oxidase with di-phenylene-iodonium (DPI) inhibited PDK1 activation. Inhibition of EGFR with AG1478 (10nM) also inhibited PDK1 activation in response to S-1-P (Fig 5). Activation of EGFR by S-1-P was inhibited by both NAC and DPI. Activation of PI3-K was unchanged by these conditions and akt activation was blunted (Fig 5).
Figure 5.
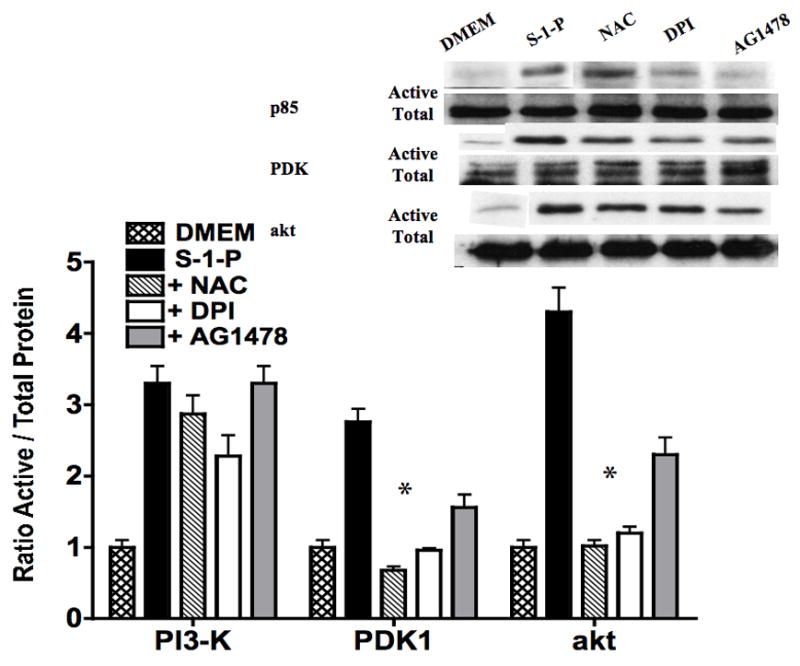
S-1-P mediated activation of PDK1 and akt was inhibited by the presence of the oxygen free radical scavenger N-acetylcysteine and the NAD(P)H oxidase inhibitor DPI. Inhibition of EGFR with AG1478 (10nM) also inhibited PDK1 and akt activation in response to S-1-P. Values are the mean±SEM of the ratio of the phosphorylation of the respective protein relative to the total unphosphosylated protein (n=6, *p<0.05 compared to control). Representative Western blots are shown above the graphs.
DISCUSSION
This report demonstrates that S-1-P mediated migration is akt dependent and that the S-1-P mediated akt phosphorylation is controlled by Gβγ dependent, PI3-K activation, which also requires the GTPase rho and by PDK1 activation, which requires Gβγ, generation of reactive oxygen species and activation of EGFR. The novel findings are the role of NAD(P)H oxidase rho and EGFR in the activation of akt. The present study has extended our previous observations on the role of PI3-K pathway in cell migration in response to S-1-P, in that we have shown its role in MAPK activation, rho activation and mTOR regulation during VSMC migration (3–7). The phospho-inositol-3′-kinase (PI3-K) family of kinases is a lipid kinase family which phosphorylate the inositol ring 3′-OH group in inositol phospholipids to generate the second messenger phosphatidylinositol 3,4,5, triphosphate (PIP3). PIP3 lipids can then activate ras, PKA, akt, PKC, PKG, p70S6 kinase, serum- and glucocorticoid-inducible kinases (SGK) (10). PI3-K is linked to both receptor-linked tyrosine kinase and G-protein coupled receptors. It is known that Gβγ will activate PI3-K after ligand receptor interaction and that leads to MAPK activation (11). Interruption of Gβγ signaling leads to attenuated VSMC migration and MAPK activation (6, 12, 13). The current data supports these contentions but also suggests that Gβγ may modulate PDK1, a second kinase in the PI3-K network and that interference in the activity of both kinases alters akt activation at one or both of its phosphorylation sites. PI3-K can also interact with the small GTPase, ras, which leads to ERK1/2 pathway. Unsustained early activation of ERK1/2 is required for VSMC migration. This report confirms that ras is downstream of PI3-K and that Gβγ and rho are upstream of PI3-K activation. As ras can activate raf, which in turn can activate MEK1/2, PI3-K can modulate the ERK1/2 pathway and thus cell migration (3).
Activation of PI3-K can also promote cytoskeletal re-arrangements and cell motility by the activation of guanine exchange factors such as Vav, Tiam1 and sos (14). There is also a reported interaction with rho and PI3-K during this cytoskeletal re-arrangement. We had previously shown that S-1-P can activate rho and that both rho and rho kinase can modulate ERK1/2 and p38MAPK activation during S-1-P induced cell migration (7). The present data suggests that PI3-K activation has a requirement for rho and not the GTPase protein, ras. Maximal PI3-K activation requires the cytosolic protein to be on the membrane and for its substrates to be in the same compartment for activation. Interference with rho may interfere with substrate and/or kinase localization and thus, interfere with maximal PI3-K and akt activation. However, there is one caveat. PI3-K actions are tightly modulated by PTEN. PTEN (phosphatase and tensin homologue deleted in chromosome 10) is a phosphatase, whose main physiologic lipid substrate is PIP3, the product of PI3-K. PI-3,4,5-P3 allows for akt translocation to the membrane and this enhances its activity. The regulation of PTEN by rho and Gβγ is not defined. The local balance between PI3K and PTEN activity can allow for distinct activation pathways in various compartments/regions of the cell. Interference in PTEN signaling could also produce the results we have demonstrated.
PDK1, a 63 kDa protein kinase, plays a pivotal role as a master kinase in receptor tyrosine kinase signaling (e.g. EGFR). It consists of an N-terminal kinase domain and a C-terminal pleckstrin homology (PH) domain. The PH domain of PDK1 binds to the PI3-K products PIP3 and PIP2, which target PDK1 to the plasma membrane where it phosphorylates Akt at T308. The presence of PIP3 and PIP2 are required for PDK1 activation. PDK1 is activated by phosphorylation in the activation loop S241 (15, 16) and this phosphorylation has been shown to be mediated by dimerization and transphosphorylation (17). Little is defined on the regulation of PDK1 activation except that it is integral to receptor-linked tyrosine kinases and that compartmentalization to the membrane, cytosol or nucleus allow for the many differential effects attributed to PDK1 (18). The present study supports the contention that PDK1 is activated by receptor-linked tyrosine kinases and in the current paradigm by EGFR. Gβγ and reactive oxygen species have been implicated in this study but it is likely that these are related to mechanism of EGFR transactivation by S-1-P and not involved in the direct action on PDK1.
Akt is downstream of PI3-K and PDK1. There are three family members of the akt family: each of which has a PH domain followed by a kinase domain (19). Within this region is the T308 phosphorylation site, which is required for akt activation. Following the kinase domain is the C-terminal tail containing the second regulatory phosphorylation site, S473. Both T308 and S473 sites must be phosphorylated for maximal akt activation (20). We saw that both sites could be activated by S-1-P and that interruption of either PI3-K or PDK1, blunted akt activation. T308 is phosphorylated by PDK1, while S473 is phosphorylated by PDK2, both of which require PI3-K (18). There are multiple targets of akt : Glycogen synthase kinase-3 (GSK-3β), Phosphodiesterase-3B, mTOR, insulin receptor substrate -1(IRS-1), Forkhead family member: FKHR, cyclin dependent inhibitors, p21CIP1/WAF1 and p27KIP1, and possibly Raf-1 allowing it to regulate multiple processes (21). We chose to examine GSK3β in this report and have previously examined mTOR in VSMC stimulated with S-1-P (5). GSK3β is normally in a phosphorylated state and its active role in cell signaling occurs with dephosphorylation. GSK-3β inhibition by akt prevents the phosphorylation of the cytoplasmic signaling molecule, β-catenin, slowing its degradation and permitting its translocation to the nucleus. This process also involves end-binding protein, APC (Adenomatosis Polyposis Coli). Once in the nucleus, β-catenin combines with various transcription factors to induce the expression of several genes (e.g. cyclin D1, which induces cell cycle progression via regulation of retinoblastoma hyperphosphorylation). In a similar manner, decreased cyclin D1 phosphorylation by GSK3β promotes the stabilization of this protein (22). Akt can also phosphorylate p21 and inhibits its anti-proliferative effects by retaining it within the cytoplasm (23). A similar mechanism has recently been described for p27 (24, 25). There is an interaction between the ERK1/2 pathways and the akt pathways at the level of p21. ERK1/2 will increase p21 phosphorylation, while akt through GSK3β will inhibit p21 activation. p21 has also been shown to modulate Rho kinase. This modulation of Rho kinase will affect the cytoskeleton at the level of myosin light chain kinase and LIM kinase. Sun et al have also demonstrated a role for p27-dependent and p27-independent mechanisms in smooth muscle cell migration (26). Therefore, the akt dependent migration in response to S-1-P may be modulated by several mechanisms, which lead to alterations in cytoskeletal structure and decreased motility.
We showed a role for EGFR in PDK1 activation. S-1-P has been shown to transactivation of the EGFR and PDGFR-β through src or reactive oxygen species mediated pathways (27). In a previous report, we demonstrated that S-1-P can induce the generation of reactive oxygen species through a Gβγ PLC-β mediated pathway (6). At present, the triple membrane passing signaling (TMPS) mechanism of G-protein coupled induced EGFR activation is a widely accepted model of EGFR transactivation (28). In this model, there is a sequence of three transmembrane signaling events: receptor activation followed by metalloproteinase activation to release a tethered ligand and subsequent activation of the EGFR by the release ligand, such as heparin binding EGF. Activation of this pathway has been shown to involve one or more of the following signaling molecules: src, intracellular calcium, and protein kinase C (28). An alternative mechanism may be inactivation of protein tyrosine phosphatases due to the generation of reactive oxygen species by the NAD(P)H oxidase complex. Activation of PDK1 appears to be dependent on reactive oxygen species generation and EGFR suggesting that EGFR transactivation is occurring through the alternative mechanism.
Conclusion
S-1-P mediated migration is akt dependent. S-1-P mediated akt phosphorylation is controlled by Gβγ dependent, PI3-K activation, which requires the GTPase rho and Gβγ. PDK1 activation requires reactive oxygen species generation and EGFR activation. Inhibition of either PI3K-akt will disrupt migration most likely through disruption of events at the leading edge and microtubule reorganization.
Acknowledgments
Funded by the U.S. Public Health Service HL086968 and HL67746 awarded to MG Davies
Footnotes
Presented at the 3rd Academic Surgical Congress (Huntington Beach, CA; Feb 2008)
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ryu Y, Takuwa N, Sugimoto N, Sakurada S, Usui S, Okamoto H, et al. S-1-P, a platelet derived lysopholipid mediator, negatively regulates cellular rac activity and cell migration in VSMC. Circ Res. 2002;90:325–332. doi: 10.1161/hh0302.104455. [DOI] [PubMed] [Google Scholar]
- 2.Wamhoff BR, Lynch K, Macdonald TL, Owens G. Sphingosine-1-Phosphate Receptor Subtypes Differentially Regulate Smooth Muscle Cell Phenotype. Arterioscler Thromb Vasc Biol. 2008 doi: 10.1161/ATVBAHA.107.159392. epub. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tanski WJ, Roztocil E, Davies MG. Sphingosine-1-phosphate induces Gαi-coupled, PI3K/ras dependent smooth muscle cell migration. J Surg Res. 2002;108(1):98–106. doi: 10.1006/jsre.2002.6529. [DOI] [PubMed] [Google Scholar]
- 4.Fegley AJ, Tanski WJ, Roztocil E, Davies MG. Sphingosine-1-phosphate stimulates smooth muscle cell migration through Gαi- and PI3-kinase-dependent p38MAPK activation. J Surg Res. 2003;113:32–41. doi: 10.1016/s0022-4804(03)00120-3. [DOI] [PubMed] [Google Scholar]
- 5.Tanski WJ, Nicholl SM, Kim D, Fegley AJ, Roztocil E, Davies MG. Sphingosine-1-Phosphate-induced smooth muscle cell migration involves the Mammalian Target of Rapamycin. J Vasc Surg. 2005;39:91–98. doi: 10.1016/j.jvs.2004.08.058. [DOI] [PubMed] [Google Scholar]
- 6.Roztocil E, Nicholl SM, Davies MG. S-1-P induced VSMC migration via activation of NAD(P)H oxidase requires Gα12/13 protein mediated phospholipase C activation. J Vasc Surg. 2007;46:1253–1259. doi: 10.1016/j.jvs.2007.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Galaria II, Fegley AJ, Nicholl SM, Roztocil E, Davies MG. Differential regulation of ERK1/2 and p38MAPK by components of the rho signaling pathway during sphingosine-1-phosphate-induced smooth muscle cell migration. J Surg Res. 2004;22(2):173–9. doi: 10.1016/j.jss.2004.05.012. [DOI] [PubMed] [Google Scholar]
- 8.Nicholl SM, Roztocil E, Davies MG. Urokinase (uPA) induced smooth muscle cell responses require distinct signaling pathways: a role for the epidermal growth factor receptor. J Vasc Surg. 2005;41:672–81. doi: 10.1016/j.jvs.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 9.Tanski WJ, Fegley AJ, Roztocil E, Davies MG. Domain dependent actions of urokinase on smooth muscle cell responses. J Vasc Surg. 2004;39:214–22. doi: 10.1016/s0741-5214(03)01031-0. [DOI] [PubMed] [Google Scholar]
- 10.Wymann MP, Zvelebil M, Laffargue M. Phosphoinositide 3 kinase signalling - which way to target? Trends Pharmacol Sci. 2003;24:366–376. doi: 10.1016/S0165-6147(03)00163-9. [DOI] [PubMed] [Google Scholar]
- 11.Luttrell LM, Hawes BE, Anbiesen T, Luttrell DK, Lansing TJ, Lefkowitz RJ. Role of c-Src tyrosine kinase in G protein-coupled receptor- and Gβγ subunit-mediated activation of mitogen-activated protein kinases. J Biol Chem. 1996;271:19443–19450. doi: 10.1074/jbc.271.32.19443. [DOI] [PubMed] [Google Scholar]
- 12.Koch WJ, Hawes BE, Allen LF, Lefkowitz RJ. Direct evidence that Gi coupled receptor stimulation of mitogen activated protein kinase is mediated by Gβγ activation of p21ras. Proc Natl Acad Sci USA. 1994;91:12706–12710. doi: 10.1073/pnas.91.26.12706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Koch WJ, Hawes BE, Inglese J, Luttrell LM, Lefkowitz RJ. Cellular expression of the carboxyl terminus of a G-protein coupled receptor kinase attenuated Gβγ mediated signalling. J Biol Chem. 1994;269:6193–6197. [PubMed] [Google Scholar]
- 14.Vara JAF, Casado E, deCastro J, Cejas P, Belda-Iniesta C, Gonzalez-Baron M. PI3K/akt signalling pathway and cancer. Cancer Treatment Reviews. 2004;30:193–204. doi: 10.1016/j.ctrv.2003.07.007. [DOI] [PubMed] [Google Scholar]
- 15.Casamayor A, Morrice NA, Alessi DR. Phosphorylation of Ser-241 is essential for the activity of 3-phosphoinositide-dependent protein kinase-1: Identification of five sites of phosphorylation in vivo. Biochem J. 1999;342(Pt 2):287–292. [PMC free article] [PubMed] [Google Scholar]
- 16.Wick MJ, Wick KR, Chen H, He H, Dong L, Quon MJ, et al. Substitution of the autophosphorylation site Thr516 with a negatively charged residue confers constitutive activity to mouse 3-phosphoinositide-dependent protein kinase-1 in cells. J Biol Chem. 2002;277(1):6632–16638. doi: 10.1074/jbc.M112402200. [DOI] [PubMed] [Google Scholar]
- 17.Wick MJ, Ramos FJ, Chen H, Quon MJ, Dong LQ, Liu F. Mouse 3-phosphoinositide-dependent protein kinase-1 undergoes dimerization and trans-phosphorylation in the activation loop. J Biol Chem. 2003;278:42913–42919. doi: 10.1074/jbc.M304172200. [DOI] [PubMed] [Google Scholar]
- 18.Vanhaesebroeck B, Alessi DR. The Pi3K-PDK1 connection: more than just a road to PKB. Biochem J. 2000;346:581–576. [PMC free article] [PubMed] [Google Scholar]
- 19.Coffer PJ, Woodgett JR. Moleular cloning and characterization of a novel putative protein-serine kinase related cAMP-dependent and protein knase. Eur J Biochem. 1991;201:475–481. doi: 10.1111/j.1432-1033.1991.tb16305.x. [DOI] [PubMed] [Google Scholar]
- 20.Alessi DR, Andjelkovic M, Caudwell B, et al. Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J. 1996;15:6541–51. [PMC free article] [PubMed] [Google Scholar]
- 21.Blume-Jensen P, Hunter T. Oncogenic kinase signalling. Nature. 2001;411:355–365. doi: 10.1038/35077225. [DOI] [PubMed] [Google Scholar]
- 22.Diehl JA, Cheng M, Roussel MF, Sherr CJ. Glycogen synthase kinase 3β regulates cyclin D1 proteolysis and subcellular localization. Gene Dev. 1998;12:3499–511. doi: 10.1101/gad.12.22.3499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhou BP, Liao Y, Xia W, et al. Cytoplasmic localization of p21CIP1/WAF1 by akt-induced phosphorylation in HER-1/neu overexpressing cells. Nat Cell Biol. 2001;3:245–52. doi: 10.1038/35060032. [DOI] [PubMed] [Google Scholar]
- 24.Shin I, Yakes FM, Rojo F, et al. PKB/Akt mediates cell cycle progression by phosphorylation of p27kip1 at threonine 157 and modulation of its cellular localization. Nat Med. 2002;8:1145–52. doi: 10.1038/nm759. [DOI] [PubMed] [Google Scholar]
- 25.Liang J, Zubovitz J, Petrocelli T, et al. PKB/akt phosphorylates p27, impairs nuclear import of p27 and opposes p27-mediated G1 arrest. Nat Med. 2002;8:1136–1144. doi: 10.1038/nm761. [DOI] [PubMed] [Google Scholar]
- 26.Sun J, Marx SO, Chen HJ, et al. Role of p27(kip1) in vascular smooth muscle cell migration. Circulation. 2001;103:2967–2972. doi: 10.1161/01.cir.103.24.2967. [DOI] [PubMed] [Google Scholar]
- 27.Tanimoto T, Lungu AO, Berk BC. S-1-P transactivates the PDGFβ receptor and EGFR in VSMC. Circ Res. 2004;94:1050–1058. doi: 10.1161/01.RES.0000126404.41421.BE. [DOI] [PubMed] [Google Scholar]
- 28.Wetzker R, Bohmer FD. Transactivation joins multiple tracks to the ERK/MAPK cascade. Nat Rev Mol Cell Biol. 2003;4(8):651–7. doi: 10.1038/nrm1173. [DOI] [PubMed] [Google Scholar]