

Abstract
The tumor suppressor C/CAAT enhancer binding protein alpha (C/EBPα) is a transcription factor involved in cell cycle control and cellular differentiation. A recent study showed that C/EBPα is frequently downregulated in Head and Neck Squamous Cell carcinoma (HNSCC) by DNA methylation in an upstream regulatory region. Here we investigated how DNA methylation in the upstream regulatory region disrupts the transcriptional regulation of C/EBPα in HNSCC. The results reveal that aberrant methylation correlates with Methyl Binding Domain protein binding and repressive histone modifications. This methylated region contains previously uninvestigated AP2α binding sites. AP2α suppresses C/EBPα promoter activity and protein expression. Interestingly, silencing AP2α by shRNA increases the anti-proliferative isoform of C/EBPα (p42C/EBPα). Furthermore, growth analysis revealed that these two isoforms yield very different proliferative properties in HNSCC.
Keywords: DNA methylation, AP2α, C/CAAT enhancer binding protein alpha (C/EBPα), 5-aza-2′-deoxycitidine (5-aza-dC), alternative translation, p30C/EBPα, p42C/EBPα, Head and Neck Squamous Cell Carcinoma (HNSCC)
Introduction
C/EBPα is a transcription factor involved in cell cycle regulation and cellular differentiation in hepatocytes and adipocytes (1). It promotes anti-proliferation through inhibition of CDK2, 4, and 6 and repression of S-phase gene transcription (2). C/EBPα is alternatively translated via leaky translation, creating p42C/EBPα and p30C/EBPα proteins (3). Although both isoforms are able to bind to C/CAAT elements in target promoters, only the larger isoform contains the anti-mitotic activity (2). In this way, the truncated isoform behaves as a dominant negative isoform.
Recent studies suggest putative tumor suppressor function of C/EBPα not only in leukemia (4) but also in solid tumors, such as lung cancer (5). C/EBPα was found to be downregulated in 78% (31/40) of HNSCC samples in a microarray profiling study (6). Furthermore, there was a significant correlation between C/EBPα downregulation and poor prognosis patients with extensive lymph node metastasis(6). Subsequently, tumor suppressor activity in HNSCC was demonstrated, and epigenetic alterations were shown to play a major role in altering C/EBPα expression in tumor samples (7).
AP2α has been previously shown to act as a transcriptional suppressor for C/EBPα promoter activity in adipocytes, hepatocytes, and keratinocytes by binding to the core promoter (8–10). An inverse correlation between AP2α and C/EBPα expression is required for adipocyte differentiation: AP2α expression decreases and C/EBPα expression increases during differentiation (8). Decreased C/EBPα expression has been observed in HNSCC, correlating with decreased cellular differentiation (7). Therefore, AP2α may also provide transcriptional suppression of C/EBPα in HNSCC. In this study, we demonstrate with C/EBPα promoter assays and ChIP analysis that upstream AP2α binding inhibits SP1 binding and suppresses C/EBPα transcription in HNSCC. Also, AP2α silencing using stable shRNA reveals restored promoter activity and increased p42 C/EBPα protein expression.
Materials and Methods
Cell lines
The human HNSCC cell lines used in the study (SCC11B, 17AS, 22B, and 25) were maintained in DMEM with 10% FBS and 1% Streptomycin/Penicillin antibiotics. The HaCat immortalized keratinocytes (11) were maintained in keratinocyte growth medium with 10% FBS and 1% Streptomycin/Penicillin antibiotics.
Patient samples
Frozen tumor tissues and adjacent normal tissue from HNSCC patients were obtained from The Ohio State University Medical Center via the Cooperative Human Tissue Network. Surgery was performed on all patients at The Ohio State University Medical Center. All sample collections were done according to the National Institutes of Health guidelines and under a protocol approved by The Ohio State University’s Institutional Review Board. Control samples were collected from morphologically normal tissue located at least 3 cm from the tumor margin. Histopathological evaluation was performed on all samples for verification. For the AP2α RT-PCR expression analysis in HNSCC patient samples, thirteen tumor samples and nine normal tissues were provided from University of Heidelberg in accordance with ethical regulations from the Nationale Centrum fur Tumorerkrankungen, Heidelberg, Germany.
Plasmids and oligonucleotides
The promoter constructs used in the luciferase assay were cloned into the multiple cloning site of pGL3 basic. The promoter sequences spanned from +4 bp (relative to the C/EBPα transcription start site) to −889 bp, −1013 bp, −1256 bp, and −1423 bp. The suppressor constructs contained −1423 bp to −1357 bp (“Sup 1”), −1357 bp to −1258 bp (“Sup 2”), and −1402 bp to −1329 bp (“No Sup”). The E2F3a promoter construct contained 2kb upstream E2F3a promoter sequence removed from pGL2 basic (12) and cloned into the HindIII site within the MCS of pGL3 basic. The full suppressor construct contained −1423 bp to −1258 bp of C/EBPα sequence adjacent to the E2F3a promoter in pGL3. The USF and SP1 mutant promoter constructs were made by site mutagenesis as previously described (13). The control, NFkB responsive promoter, contains three NFkB binding sites in pGL3 basic (14). The dominant negative NFkB, pcDNA IkBα, contains cDNA coding for IkB that is mutant at the phosphorlyation sites necessary for its degradation (i.e. Ser 32 and Ser 36 changed to Ala)(14). The C/EBPα overexpression vector was provided by Dr. Gokhan Hotamisligil (The Harvard School of Public Health). The 30kDa C/EBPα overexpression vector was generated by removing 316 bp of the 5′ cDNA sequence upstream of the 30 kDa translation start site using endogenous SacII restriction sites. The pRS retroviral vector from Origene was used for stable silencing experiments. The AP2α shRNA construct contained an oligo sequence (available upon request) cloned into the BamHI/HindIII sites. The hairpin sequence (available upon request) targeted against C/EBPα (227 bp past the 42kDa translation start site) was cloned into the BamHI and HindIII sites of the plasmid. The C/EBPα double-stranded RNA oligonucleotides (sequence available upon request) (targeted 1,466 bp beyond the 42kDa translation start site) were purchased from Ambion for the transient silencing experiments. The p42 alternate promoter construct contained −1kb to −1423bp of C/EBPα upstream sequence cloned in front of the C/EBPα cDNA in the overexpression vector. The p30 alternate promoter construct contained 0 to −1kb of C/EBPα upstream sequence cloned in front of the C/EBPα cDNA in the overexpression vector.
Transfections
Stable transfections were performed as previously described (15). 120 hrs post-infection, the target cells were collected, counted, and suspended in Laemmeli buffer for western blot confirmation of silencing. For the stable silencing, 30 μg of pRS-C/EBPα along with 60 μl of superfect and 210 μl of DMEM without FBS were incubated together for 10 minutes. This was then suspended in 8 ml of DMEM + 10% FBS and added to a p150 cm plate of Phoenix cells at 50% confluency. After 4 hrs, the media was replaced with 16 ml of DMEM + 10% FBS. After 48 hrs, 8 ml of the Phoenix media was briefly centrifuged with 8 μl of polybrene and added to 1.5 million target cells on a p150 cm plate. 120 hrs post-infection, the target cells were collected, counted and suspended in laemelli buffer for western confirmation of silencing. For the transient silencing experiment, 160,000 C/EBPα-overexpressing SCC22B cells were plated per well in a 6-well plate. The transfections were done in duplicates. The negative control transfection mix contained 100 μl EC-R buffer (Qiagen), 14.4 μl RNAifect (Qiagen), and 2,885.6 μl DMEM + 10% FBS. The 30 nM siRNA transfection mix contained 4.8 μl 20 μM siRNA stock, 95.2 μl EC-R buffer (Qiagen), 14.4 μl RNAifect (Qiagen), and 2,885.6 μl DMEM + 10% FBS. The transfection media was left on the cells for 72 hrs. Then the cells were trypsinized, counted, and suspended in laemelli buffer for western analysis.
Bisulfite Sequencing
−1423bp to −1121 bp upstream of C/EBPα’s transcription start site of bisulfite-treated HNSCC cell line SCC22B (“+” and “−“ AP2α shRNA) was analyzed for methylation as previously described (13).
Growth Curve
10,000 cells were plated in triplicates in a 6-well plate for a 4-day growth curve analysis. For each count, 500 μl of trypsin was added to each well for 3 minutes, after which the cells were suspended in 2 mls of PBS, and 500 μl was counted using a Coulter counter.
Western Blot analysis
The western blots were performed as previously described (13). After AP2α detection, the blot was reprobed with a 1:500 dilution of α-tubulin antibody (1:3,000 secondary mouse antibody) and 1:500 dilution of C/EBPα antibody (Santa Cruz sc-61)(1:1,000 secondary rabbit antibody).
5-aza-2′-deoxycytidine treatment
HNSCC cell lines were incubated for 96 hrs with 3 μM 5-aza-2′-deoxycytidine (5-aza-dC) (Sigma) with medium changed every day. Treated cells were harvested for analysis 1~4 days after the procedure. Cells were trypsinized and suspended in 10 ml DMEM and 270 μl 37% formaldehyde for ChIP analysis.
Chromatin Immunoprecipitation assay (ChIP)
ChIP was performed according to the Upstate Protocol. Cells were grown on p150 mm plates, and then the media was removed and replaced with 10 ml DMEM and 270 μl 37% formaldehyde. Plates were shaken at room temperature for 10 minutes then 1 ml of 1.25 M glycine was added for 5 minutes. Plates were then washed 3 times with cold PBS with 2 μl PIC/ml, centrifuging at 2500 rpm for 5 minutes at 4°C and resuspending the pellet after each wash. PBS was removed and cells were suspended in 7 ml of cell lysis buffer (85 mM KCl, 5 mM PIPES, 2 μl PIC/ml) and dounced on ice. Cells were then centrifuged again and suspended in 500 μl SDS lysis buffer (Upstate) with 1 μl PIC. The cells were then sonicated at setting 7 (Misonix) for 15 pulses of 20 seconds each. Debris was pelleted by brief centrifugation at 4°C, and the supernatant was placed in a new centrifuge tube. 60 μl of agarose beads were added, and the tubes were rotated for 1hr at 4°C. Tubes were then centrifuged, and the supernatant was placed in a clean microcentrifuge tube with 1800 μl ChIP dilution buffer. At this point, 20 μl from each tube was removed as input. Then 5 μg of antibody was added to the tubes and allowed to rotate at 4°C overnight. 60 μl agarose beads were added to the tubes and allowed to rotate 1 hr at 4°C. Tubes were then centrifuged, supernatant removed, and beads were washed with low salt, high salt, LiCl, and TE (2X) rotating for 4 minutes each. DNA was eluted from the beads with 500 μl eluate, and the crosslinks were reversed for 4 hrs at 65°C. The eluate was then treated with RNase, 0.5 M EDTA, 1 M Tris, and PK for 1 hr at 45°C. Samples were then PCR purified with QIAquick PCR purification kit. Quantitative PCR for the ChIP assay was done with 3 μl of ChIP eluate and primers surrounding the promoter binding sites. Promoter enrichment was assessed by normalization of threshold crossing of the samples compared to the threshold crossing of the negative control (no antibody).
RNA isolation and cDNA synthesis
RNA was isolated according to the manufacturer’s protocol. DNase treatment was performed on 1–2 μg of RNA by adding 2U of DNaseI (Invitrogen, Carlsbad, CA), 1 μl DNase buffer, and 0.4 μl RNase Out (Invitrogen, Carlsbad, CA) for 15 min at room temperature. 1 μl ethylenediaminetetraacetic acid (EDTA) was then added to the mix for 10 min at 65°C, followed by an incubation on ice for 5 min. cDNA synthesis was performed by the following reaction: 2 μl random hexamers and 1 μl deoxynucleotide triphosphates (dNTPs) (10 mmol/L) for 5 min at 65°C then 2 min at 4°C; 2 μl of 10X buffer, 4 μl MgCl2, 2 μl dithiothreitol (DTT), and 1 μl RNase Out was added for 2 min at 25°C; 100 U of SuperScript II (Invitrogen) for 50 min at 42°C; 15 min at 70°C, then transferred to 4°C.
Real-time PCR (RT-PCR)
Quantitative mRNA expression was measured using SYBR Green I (BioRad, Hercules, CA) in an I-Cycler (BioRad). Expression of glycosylphosphatidylinositol (GPI) was used as the internal control gene. I-Cycler conditions and RT-PCR primers can be provided upon request. For the RT-PCRs on the HNSCC patient samples, AP2a and three internal controls glycosylphosphatidylinositol (GPI), TATA binding protein (TBP), glyceraldehyde 3-phosphate dehydrogenase (GAPDH) cDNA levels were measured using SYBR Green I (BioRad) in a BioRad I-Cycler. Data were acquired in the format of cycle number crossing the software-generated threshold (Ct). The average of differences in Ct between AP2a and the three internal standards (DCt) from all normal a tissues was defined as normal level (NL). Normalized AP2a cDNA levels in tumors were calculated using the formula 2−(DCtTumor-NL), (2 is the efficiency of the PCRs determined by calibrating experiments). We defined the normal expression range as 1 in normal tissues.
Luciferase Assay
Keratinocytes, SCC11B, or SCC22B cells (20,000) were plated in each well of a 24-well plate. 24 hrs later, the transfection was performed using the Promega manufacturer’s protocol. Briefly, 1 μg of the promoter-pGL3 plasmid DNA was combined with 4 μl superfect, 60 ng renilla TK plasmid, and 60 μl DMEM (without FBS). After complexing for 10 minutes, 200 μl 10% FBS DMEM was added. The transfection media was plated in triplicates, and 48 hrs later the media was removed, cells were lysed in 100 μl of passive lysis buffer for 40 minutes, and 20 μl of lysate was plated in the 96-well opaque plate and analyzed in the luminometer. The resulting luciferase/renilla ratios were normalized to pGL3 basic to obtain the fold increase. For the dominant negative NFkB experiment, the same protocol as mentioned above was used, except the transfection mixture contained also 1 μg of the pcDNAIkBα.
AP2α site mutagenesis
The upstream (−1419bp) and downstream (−311bp) AP2α binding sites in a pGL3 basic construct containing −1618bp to +14bp of upstream C/EBPα sequence were mutagenized using the standard QuickChange®II (Stratagene) protocol with the following oligos: Upstream- AP2SDM.F GCCTTGCCAGAACTAAGGCCACTG and AP2SDM.R CAGTGGCCTTAGTTCTGGCAAGGC, Downstream-AP2SDMctrl.F GATACGGGCAACTAGGGCAGG and AP2SDMctrl.R CCTGCCCTAGTTGCCCGTATCC. The putative AP2α binding sites are underlined and the nucleotide changes are double underlined and in bold. All changes were confirmed by sequencing.
Statistical Analysis
The statistical significance of the results was calculated by unpaired Student’s t test, and P<0.05 was considered to be statistically significant.
Results
C/EBPα promoter activity is greatest −1256 bp upstream of the transcriptional start site
Previous work demonstrated aberrant DNA methylation in the upstream regulatory region (−1399 to −1146 bp) of C/EBPα in HNSCC (7). To investigate whether the upstream methylation seen in HNSCC coincides with gene regulatory regions in HNSCC, we performed luciferase promoter assays using HaCat (normal keratinocytes) and SCC22B cell lines. These assays revealed that the sequence spanning from either −1013 to +4 bp (HaCat) or from −1256 to +4 bp (SCC22B) had the strongest promoter activity. Consistent with previous reports in lung cancer cells (13), the sequence between −1423 and −1256 bp appears to contain a suppressor sequence (Figure 1a). The sequence between −1253 bp and −1146 bp in SCC22B was found to exhibit both high promoter activity and methylation in HNSCC.
Figure 1. C/EBPα methylation and promoter analyses in cell lines.
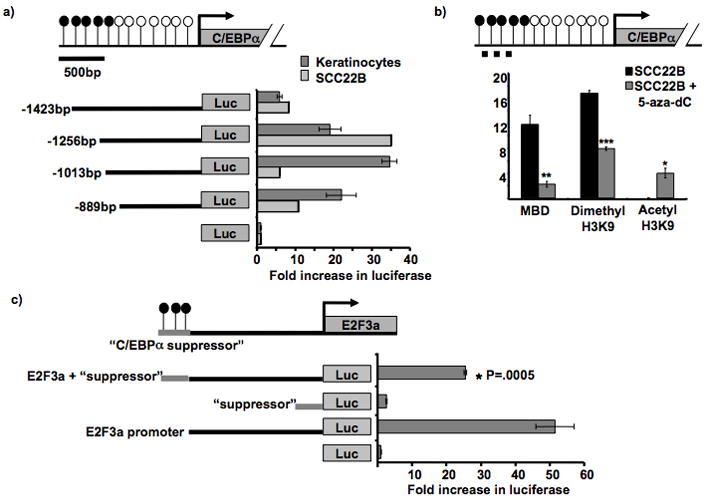
a. C/EBPα promoter assay in HaCat, an immortalized keratinocyte cell line, and SCC22B, a HNSCC cell line. Diagram is drawn to scale and depicts C/EBPα and its upstream sequence. CpG sites’ methylation status is shown by open (unmethylated) and closed (methylated) circles. Constructs contained different lengths of the C/EBPα upstream promoter in front of the luciferase gene in pGL3 basic. Promoter activity (plotted on the x-axis) is measured as the fold increase in the luciferase/renilla ratio in respect to the negative control (pGL3 vector only). The −1423bp construct provided a significant decrease in promoter activity, * P = 0.0028.
b. Quantitative ChIP PCR on HNSCC cells to detect pulldown from MBD and histone dimethylation before and after 5-aza-dC treatment. Diagram depicts C/EBPα and its upstream sequence. CpG sites’ methylation status is shown by open (unmethylated) and closed (methylated) circles. The dashed line represents the region analyzed for promoter pulldown in the ChIP assay. C/EBPα promoter pulldown enrichment (in SCC22B cells before and after 5- aza-2′-deoxycitidine treatment) by MBD2/3, dimethyl H3K9, or acetyl H3K9 antibodies was normalized to the negative (no antibody) control. * P = 0.0003; ** P = 0.0002; *** P = 3 × 10−6.
c. C/EBPα suppressor promoter assay in SCC22B cells. Diagram depicts the “C/EBPα suppressor sequence” (−1423bp to −1256bp) cloned in front of 2kb upstream E2F3a sequence. Promoter activity (plotted on the x-axis) is measured as the fold increase in luciferase/renilla ratio in respect to the negative control (pGL3 basic vector only). The suppressor construct contained −1423bp to −1256bp of upstream C/EBPα sequence cloned into pGL3 basic; the E2F3a/suppressor construct contained −1423bp to −1256bp of upstream C/EBPα sequence cloned in front of a strong E2F3a promoter sequence within pGL3 basic.
Previous studies in lung cancer cell lines indicated SP1 and USF1 binding in this region and activation of C/EBPα (13, 16). We used ChIP analysis to investigate whether or not these proteins play a role in HNSCC cells. A significant increase of USF1 and USF2 bound DNA from the upstream C/EBPα sequence compared to the no-antibody control (P < 0.002)(Supplemental Figure 1a), indicating that SP1 and USF1 sites are required for C/EBPα activation.
The importance of USF and SP1 binding sites was further validated by promoter analysis with C/EBPα promoter constructs containing USF and SP1 binding site mutations. The promoter analysis revealed that mutating the downstream USF1 binding site (−894 bp) did not decrease promoter activity (Supplemental Figure 1b), but mutating the 2 upstream USF1 binding sites (−1201 and −1001 bp) significantly decreased C/EBPα promoter activity (P = 0.004 and 0.010, respectively) compared to the control −1256 bp construct (Supplemental Figure 1b). It is important to note that the most upstream USF binding site (−1201 bp) is within the methylated region in this HNSCC cell line. The construct that contained mutations in the SP1 binding sites also revealed a significant reduction in promoter activity (P = 0.003) (Supplemental Figure 1b). Therefore, it appears that upstream USF1 and SP1 binding is required for enhanced promoter activity.
Computational analysis also predicted NFκB and cRel binding sites in this region, and ChIP analysis revealed they both bind to their predicted binding sites within the upstream C/EBPα sequence (Supplemental Figure 2a). Therefore, we next tested whether cRel and NFκB could affect C/EBPα transcription. We analyzed changes in C/EBPα promoter activity in the HNSCC cell line SCC22B and normal keratinocytes when introducing a construct that overexpresses a dominant negative form to cRel and NFκB (14). An increase in promoter activity would suggest suppressor activity of cRel and NFκB (binding sites shown in Supplemental Figure 2b). This experiment showed that there was a significant increase in promoter activity in normal keratinocytes with the −1423 and −1618 promoter constructs (p= 0.051 and 0.010, respectively), but no increase in activity in the SCC22B cell line (Supplemental Figure 2c). Therefore, NFκB and cRel do not appear to affect C/EBPα promoter activity in HNSCC.
MBD2/3 binding and histone H3Lys9 methylation are relieved by upstream demethylation of C/EBPα, replaced by histone H3 Lys9 acetylation
Methylation is capable of recruiting repressive complexes that can inhibit subsequent binding and activation by transcription factors (17, 18). Therefore, it was investigated whether the upstream methylation seen in HNSCC patient samples and HNSCC cell lines may recruit proteins to C/EBPα’s upstream sequence. Bisulfite sequencing analysis revealed that SCC22B, a HNSCC cell line, is methylated between −1423 bp and −1146 bp (data not shown). The presence of methyl binding domain proteins (MBDs) and dimethyl H3K9 was investigated, because MBDs can be recruited to methylated CpG dinucleotides (17), and the dimethyl H3K9 histone tail modification can coincide with DNA methylation and silenced transcription (19). HNSCC cells were treated with 5-aza-2′-deoxycytidine to investigate whether demethylation was able to relieve MBD binding, decrease histone H3K9 methylation, and increase histone H3K9 acetylation (which correlates with active transcription and the lack of methylation) (19). ChIP analysis was performed using MBD2/3, dimethyl H3K9, and acetyl H3K9 antibodies on SCC22B cells with and without 5-aza-2′deoxycytidine. Quantitative PCR revealed HNSCC cells treated with 5-aza-2′-deoxycytidine showed 5-fold decrease in MBD binding, a 2-fold decrease in dimethyl H3K9, and a >4-fold increase in acetyl H3K9 pulldown of the C/EBPα promoter compared to the untreated SCC22B cells (P = 0.0002, 3 × 10−6, and 0.0003 respectively) (Figure 1b). This emphasizes the importance of DNA methylation in preparing the region for silencing by recruiting MBDs and allowing changes in histone modifications (i.e. replacement of acetyl H3K9 with dimethyl H3K9).
AP2α binds in the upstream promoter and suppresses C/EBPα promoter activity
In order to investigate whether the sequence between −1256 bp and −1423 bp contains true suppressor effects, the 167 bp fragment was cloned in front of a strong promoter, E2F3a, and subjected to a luciferase promoter assay in an HNSCC cell line, SCC22B. This analysis revealed that the “suppressor” fragment decreased E2F3a promoter activity by approximately 50% (Figure 1c).
The suppressor sequence was dissected in order to further define the region providing transcriptional suppression. Two suppressor constructs were created: “Sup1” contained the first portion (−1423bp to −1357bp), and “Sup2” contained the latter portion of the suppressor sequence (−1357bp to −1258bp)(Figure 2a). Promoter analysis of these 2 sequences revealed suppression was maintained with either truncated portion of the suppressor sequence (Figure 2a). The “suppressor sequence” was analyzed for any similarities between the former and the latter halves. This revealed an AP2α site that was contained within both “Sup1” and “Sup2” (Figure 2a). Therefore, an additional construct was formed that contained a large portion of the suppressor sequence but lacked the AP2α sites (“No Sup”). Promoter analysis revealed that the “No Sup” was relieved of its ability to suppress E2F3a promoter activity (Figure 2a). Furthermore, mutagenesis of the upstream AP2α site contained within “Sup1” revealed a significant increase in C/EBPα promoter activity in SCC22B and SCC11B compared to the WT C/EBPα promoter sequence (Figure 3a). However, mutagenesis of a downstream AP2α site (−311bp) that does not suppress C/EBPα promoter activity in HNSCC did not increase C/EBPα promoter activity in SCC22B or SCC11B (Figure 3a).
Figure 2. AP2α suppresses C/EBPα promoter activity.
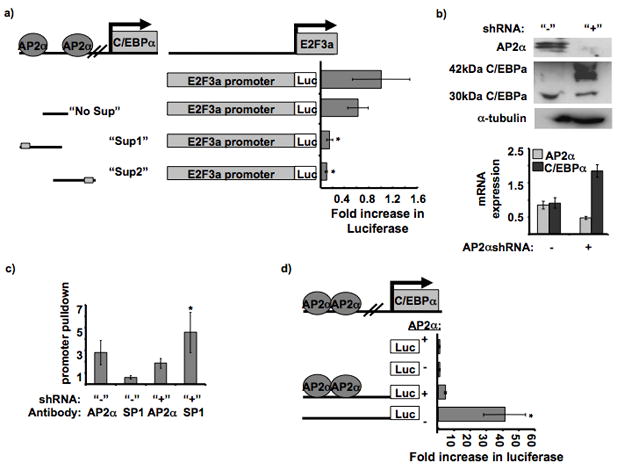
a. Effect of different parts of the C/EBPα suppressor sequence on E2F3a promoter activity. Diagram of the suppressor constructs and location of the AP2α binding sites. The different suppressor parts were cloned in front of the E2F3a promoter sequence in pGL3 basic. The luciferase values were normalized to the E2F3a promoter construct. The “Sup 1” and “Sup 2” constructs provided significantly less promoter activity than the “No Sup” and WT promoter constructs, * P = 0.0043 and 0.0048, respectively.
b. AP2α stable silencing and C/EBPα upregulation in SCC22B cell line. A short hairpin sequence targeting AP2α was cloned into pRS stable silencing vector. The western blot contains 300,000 untransfected SCC22B cells and 300,000 SCC22B cells transfected with the AP2α shRNA. The blot was probed with AP2α antibody,α-tubulin (internal control), and C/EBPα. Also, cDNA was prepared from RNA isolated from the respective cells. Quantitative RT-PCR data is shown for normalized AP2α and C/EBPα expression in SCC22B cells with and without AP2α shRNA. P = 0.0201 and 0.0121, respectively.
c. SP1 binding is inhibited by AP2α binding to the upstream C/EBPα sequence. Quantitative RT-PCR on C/EBPα promoter pulldown (−1423 to −1243 bp) with AP2α and SP1 antibodies in SCC22B cells before and after AP2α silencing. C/EBPαpromoter pulldown enrichment was normalized to the negative (no antibody) control. * P = 0.0451.
d. C/EBPα promoter activity before and after AP2α silencing. The promoter assay was performed in SCC22B cells (“+AP2α”) and SCC22B cells with downregulated AP2α (“−AP2α”). The promoter constructs contained −1423 bp of upstream C/EBPα sequence. Fold increase in luciferase activity was normalized to the negative control in each cell lysate (empty pGL3 basic). Promoter activity was significantly increased in the cell lysates with decreased AP2α expression. * P = 0.0237.
Figure 3. AP2α site mutagenesis or protein downregulation increases C/EBPα promoter activity and decreases upstream methylation, respectively.
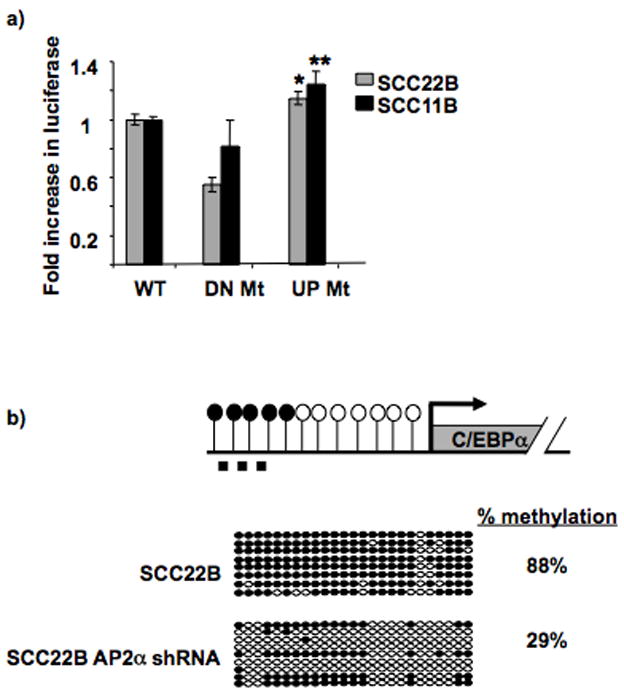
a. AP2α site mutagenesis increases C/EBPα promoter activity. The graph displays the relative fold increase in luciferase activity with the C/EBPα promoter constructs containing the mutation in the upstream AP2α site (−1419 bp; “UP Mt”) compared to the WT promoter (“WT”; which is set as 1) in both SCC11B and 22B. The previously identified downstream AP2α site (−311bp; “DN Mt”) which does not suppress C/EBPα promoter activity in HNSCC was mutated as a control, and it showed no increase in promoter activity compared to “WT”. * P = 0.032; ** P = 0.031. Black bars = SCC11B; Gray bars = SCC22B.
b. AP2α downregulation provides decreased upstream C/EBPα methylation. Diagram depicts C/EBPα and its upstream sequence. CpG sites’ methylation status is shown by open (unmethylated) and closed (methylated) circles. The dashed line represents the region analyzed by bisulfite sequencing (−1423bp to −1121bp). Bisulfite sequencing analysis was performed on SCC22B cells before and after AP2α stable shRNA silencing. The region tested for methylation via bisulfite sequencing spans from −1423 bp to −1121 bp. Bisulfite sequencing was performed by comparing the sequences with 100% and 0% methylated DNA sequence (i.e. CG or TG at CpG sites, respectively). Open circles represent unmethylated CpGs, and closed circles represent methylated CpGs. Each row represents an individual clone. Percentage methylation is shown for the two cell types.
AP2α has previously been shown to suppress C/EBPα promoter activity in adipocytes by binding close to the transcription start site (−311 bp)(16) and blocking SP1 binding. To investigate whether AP2α transcriptional suppression in HNSCC is by inhibiting adjacent SP1 binding to the upstream C/EBPα sequence, AP2α was downregulated in a HNSCC cell line SCC22B (Figure 2b). ChIP analysis was performed using AP2α and SP1 antibodies in these SCC22B cells with and without AP2α shRNA. Quantitative PCR using primers surrounding the AP2α binding sites (−1423 to −1243 bp) revealed a significant ~8 fold increase in SP1 promoter pulldown after AP2α silencing (P = 0.0451) (Figure 2c).
Furthermore, AP2α downregulation resulted in an increase in C/EBPα mRNA and p42C/EBPα protein expression as determined by RT-PCR and western blot analysis, respectively (Figure 2b and Supplemental Figure 3c). Also, the promoter activity of the −1423 bp C/EBPα promoter construct (which contains the 2 upstream AP2α sites) was significantly increased upon AP2α downregulation (Figure 2d). These data further validate that AP2α acts as a suppressor for C/EBPα by inhibiting SP1 binding. Interestingly, AP2α silencing not only increased C/EBPα expression, but also it changed the 42kDa:30kDa C/EBPα isoform ratio (Figure 2b) a result that may point to two different promoters driving the expression of either the p30 or p40 isoform.
AP2α binding to the promoters of retinoicacid responsive genes has previously been shown to initiate transcriptional silencing and recruit HDACs (20). Therefore, AP2α binding to the upstream C/EBPα sequence may initiate its transcriptional silencing and recruit DNA methyltransferase activity. In this manner, eliminating AP2α binding and C/EBPα downregulation via stable shRNA silencing may relieve upstream C/EBPα methylation. Bisulfite sequencing analysis was performed on SCC22B with and without AP2α downregulation via shRNA. This revealed a substantial decrease in methylation upon AP2α downregulation (Figure 3b), which correlates with increased C/EBPα protein expression (Figure 2b). This suggests that AP2α binding to the upstream C/EBPα sequence may preceed and facilitate methylation.
Because AP2α is involved in methylation and suppression of C/EBPα (which is commonly downregulated by methylation in HNSCC), AP2α overexpression in HNSCC was investigated. Quantitative RT-PCR on HNSCC patient tumor and normal samples revealed AP2α mRNA overexpression in ~70% of the samples (Supplemental Figure 3a). Furthermore, western blot analysis of several HNSCC cell lines revealed an inverse correlation between endogenous AP2α expression and full-length C/EBPα expression. Expression of the anti-mitotic p42kDaC/EBPα isoform was only evident in the absence of AP2α expression. In contrast, abundant expression of the dominant negative (p30kDaC/EBPα) correlated with the presence of AP2α expression (Supplemental Figure 3b).
p42C/EBPα silencing in HNSCC restores high proliferative growth rate of the cells
C/EBPα mRNA is translated into two alternative isoforms, p42C/EBPα and p30C/EBPα. This is due to alternative translation initiation at the third in-frame AUG via leaky ribosomal scanning (3). This creates a smaller 30kDa protein that lacks part of the transactivation domain. Therefore, p30C/EBPα has been characterized as a transcriptional activator that lacks antimitotic activity (21). However, the full length 42kDa C/EBPα protein has been shown to exhibit anti-proliferative activity in different cell types such as hepatocytes and adipocytes (22, 23). Therefore, it is not surprising that most of the HNSCC cells (with the exception of SCC8) do not normally express the anti-mitotic 42 kDa isoform (Figure 2b). However, it was interesting that removing the AP2α suppressor in these cells caused an abundant increase in the proportion of the anti-proliferative 42kDa isoform (Figure 2b).
Previously, C/EBPα overexpression in a HNSCC cell line revealed a significant decrease in cell proliferation (7). The growth rate of these overexpressing cells in the presence or absence of stable C/EBPα downregulation was compared to find whether stable silencing would be capable of restoring the normal proliferation rate of the SCC22B cells. Using a retroviral vector containing a hairpin sequence targeting C/EBPα, p42C/EBPα was downregulated by ~75% (according to semi-quantitation by Image Quant) in C/EBPα overexpressing SCC22B cells (Figure 4a). Changes in proliferation were analyzed by growth curve analysis, which revealed that silencing the overexpressed p42C/EBPα restored the proliferation rate back to the normal SCC22B growth rate (Figure 4b).
Figure 4. C/EBPα silencing and p30 C/EBPα-overexpression in SCC22B cells.
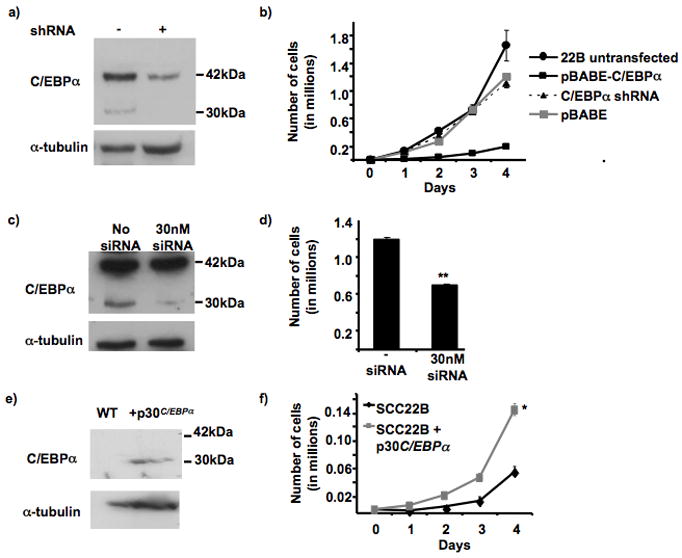
a. Confirmation of C/EBPα silencing in C/EBPα-overexpressing SCC22B cells. Western blot showing C/EBPα-overexpressing SCC22B cells before and after stable C/EBPα silencing. 150,000 cells were loaded in each lane. The blot was first probed with C/EBPα followed by α-tubulin to confirm equal loading. Semi-quantitation was performed using Image Quant computer software, which provided that 75% downregulation of the 42 kDa isoform was attained.
b. Growth curve analysis of C/EBPα-overexpressing SCC22B cells after stable C/EBPα silencing. 10,000 cells (SCC22B; SCC22B + pBABE only; SCC22B C/EBPα-overexpressing cells; and SCC22B C/EBPα-overexpressing + C/EBPα shRNA) were plated in triplicates in a 6 well plate. Cells were permitted to grow three days after plating before taking the first count (Day 1). Growth was assessed by counting the cells every 24 hrs for 4 days. For each count, 500 μl of trypsin was added to each well for 3 min, after which the cells were suspended in 2 mls of PBS, and 500 μl was counted using a Coulter counter. ** P = 0.0002.
c. Confirmation of 30kDa C/EBPα silencing in C/EBPα-overexpressing SCC22B cells. Western blot showing C/EBPα-overexpressing SCC22B cells with and without 30 nM siRNA. 150,000 cells were loaded in each lane. The blot was first probed with C/EBPα followed by α-tubulin to confirm equal loading.
d. Cell count of C/EBPα-overexpressing SCC22B cells after transient 30kDa C/EBPα silencing. Cells were collected and counted 72 hrs after transfection using a Coulter counter. ** p < .0007.
e. Confirmation of p30C/EBPα overexpression in SCC22B cells. Western blot showing SCC22B cells with or without pBABE-p30C/EBPα. 150,000 cells were loaded in each lane. The blot was first probed with C/EBPα followed by α-tubulin to confirm equal loading.
f. Growth curve analysis of p30C/EBPα overexpressing SCC22B cells. 5,000 cells (SCC22B + pBABE and SCC22B + p30C/EBPα) were plated in triplicates in a 6 well plate. Growth was assessed by counting the cells every 24 hrs for 4 days. For each count, 500 μl of trypsin was added to each well for 3 min, after which the cells were suspended in 2 ml of PBS, and 500 μl was counted using a Coulter counter. * p-value < 0.0007.
Silencing p30C/EBPα in HNSCC decreases the proliferation rate of the cells
Next, transient silencing was performed using a double-stranded RNA oligo. In contrast to the stable silencing experiment, now only the p30C/EBPαwas transiently silenced (Figure 4c). When the cells were counted 72 hrs post-transfection, the cells that contained the 30 nM siRNA had a 40% decrease in cell growth compared to the mock-transfected negative control (Figure 4d). Silencing p30C/EBPα in SCC22B apparently relieves the binding competition with p42C/EBPα for C/EBPα binding sites and increases the ability of C/EBPα to provide differentiation and decreased cell proliferation (1). These data suggest that AP2α may be required to foster the tumorigenic potential of HNSCC cells by specifically targeting the anti-mitotic 42kDa isoform.
p30C/EBPα overexpression causes increased cell proliferation in HNSCC
Previously, overexpressed p42C/EBPα in SCC22B revealed decreased cell proliferation and increased differentiation (7). In order to further validate the specificity of p42C/EBPα’s antiproliferative effect, p30C/EBPα was stably overexpressed in SCC22B (Figure 4e). Interestingly, not only did p30C/EBPα overexpression fail to provide the tumor suppressive qualities of p42C/EBPα overexpression, but also it actually provided ~2.5-fold increase in cell proliferation (P = 0.0007) (Figure 4f).
Discussion
C/EBPα is a transcription factor involved in cellular differentiation and cell cycle control, making it a good tumor suppressor candidate (1). In fact, it has already been shown that C/EBPα has tumor suppressor ability in lung cancer and AML (4, 5). Recently, it was shown for the first time that C/EBPα also exhibits tumor suppressor activity in HNSCC. Furthermore, it is epigenetically downregulated by upstream methylation (7). This study suggests that AP2α interferes with SP1 binding, resulting in decreased C/EBPα transcription, and ultimately upstream methylation. The consequent DNA methylation recruits MBD proteins and allows repressive histone modifications. In this way, AP2α initiates C/EBPα downregulation, which is then followed by stable silencing via DNA methylation and chromatin compaction. Furthermore, AP2α silencing effect appears to be specific for the anti-proliferative full length C/EBPα isoform. This may be due to alternate promoter usage for alternative transcription which has yet to be defined.
Overexpression of p30C/EBPα and p42C/EBPα in adipocytes has been shown to provide very different proliferation outcomes (24). Furthermore, mutations in AML have been shown to cause upregulation of p30C/EBPα and revealed its ability to function as a dominant negative (21). Both isoforms can bind to C/CAAT binding sites within promoter sequences, but only p42C/EBPα is capable of providing differentiation and decreased cell proliferation (24). In this manner, p30C/EBPα opposes the p42C/EBPα, because it competes for binding to targets and lacks the ability to function (21). Therefore, it is not surprising that p42C/EBPα and p30C/EBPα also revealed different proliferative properties in HNSCC, similar to adipocytes (24).
C/EBPα is involved in a variety of physiological processes (i.e. cell cycle control, differentiation, metabolism, and inflammation) (1). Alternative translation of C/EBPα mRNA in order to create different isoforms with different domains would accommodate the genetic diversity required for such a multi-functional protein (25). It has been found that the alternative isoforms of both C/EBPα and C/EBPB fluctuate during aging and LPS response (3). For C/EBPα the 42 kDa isoform decreases over time, whereas the amount of 30 kDa isoform remains constant (3). Interestingly, the isoform ratios during LPS response in a young liver mimic the patterns seen in effect to aging (3). Also, previous experiments revealed that p42C/EBPα levels in aged livers were further diminished upon inflammatory insult, suggesting that both age and LPS response can somehow force the scanning ribosome to bypass the first translation start site (3).
p30C/EBPα has been found to be upregulated and act as a dominant negative in AML, relieving the block on cell cycle progression (21). p42C/EBPα has been found to provide tumor suppressor qualities in several cancer types including lung cancer, AML, and more recently HNSCC (13, 26). It is reasonable to propose that carcinogenesis, like aging and inflammation, may also augment the normal C/EBPα isoform ratios and increase translation from the downstream start site. On the other hand, the data could point to the existence of to alternatively transcribed C/EBPα isoforms in HNSCC, under the control of distinct promoters. In this context of alternate promoter usage, AP2α binding and suppression could specifically affect transcription of the 42 kDa isoform. Therefore, further investigation is necessary to decipher how AP2α specifically suppresses the 42 kDa isoform of C/EBPα. Regardless of the mechanism, the resulting increased p30C/EBPα and decreased p42C/EBPα levels provide for increased cell proliferation, which ultimately propagate tumorigenesis. In the case of HNSCC, the suppressor AP2α appears to be involved in this change in C/EBPα isoform ratios.
Supplementary Material
Acknowledgments
The authors would like to thank Dr. Dennis Guttridge (The Ohio State University) for providing the NFκB promoter construct and dominant negative expression plasmid, Dr. Gustavo Leone (The Ohio State University) for the E2F3a promoter construct, Dr. Thomas Carey, University of Michigan, for the HNSCC cell lines used in this study and Plass Lab members for their thoughtful discussions. This work was supported in part by a grant from NIDCR, DE13123 (CP), and an AGGRS scholarship (KB). CE is a recipient of the Doris Duke Distinguished Clinical Scientist Award and is the Sondra J. and Stephen R. Hardis Chair of Cancer Genomic Medicine at the Cleveland Clinic.
References
- 1.Schrem H, Klempnauer J, Borlak J. Liver-enriched transcription factors in liver function and development. Part II: the C/EBPs and D site-binding protein in cell cycle control, carcinogenesis, circadian gene regulation, liver regeneration, apoptosis, and liver-specific gene regulation. Pharmacol Rev. 2004;56:291–330. doi: 10.1124/pr.56.2.5. [DOI] [PubMed] [Google Scholar]
- 2.Johnson PF. Molecular stop signs: regulation of cell-cycle arrest by C/EBP transcription factors. J Cell Sci. 2005;118:2545–55. doi: 10.1242/jcs.02459. [DOI] [PubMed] [Google Scholar]
- 3.Hsieh CC, Xiong W, Xie Q, Rabek JP, Scott SG, An MR, Reisner PD, Kuninger DT, Papaconstantinou J. Effects of age on the posttranscriptional regulation of CCAAT/enhancer binding protein alpha and CCAAT/enhancer binding protein beta isoform synthesis in control and LPS-treated livers. Mol Biol Cell. 1998;9:1479–94. doi: 10.1091/mbc.9.6.1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Porse BT, Bryder D, Theilgaard-Monch K, Hasemann MS, Anderson K, Damgaard I, Jacobsen SE, Nerlov C. Loss of C/EBP alpha cell cycle control increases myeloid progenitor proliferation and transforms the neutrophil granulocyte lineage. J Exp Med. 2005;202:85–96. doi: 10.1084/jem.20050067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Halmos B, Huettner CS, Kocher O, Ferenczi K, Karp DD, Tenen DG. Down-regulation and antiproliferative role of C/EBPalpha in lung cancer. Cancer Res. 2002;62:528–34. [PubMed] [Google Scholar]
- 6.Roepman P, Wessels LF, Kettelarij N, Kemmeren P, Miles AJ, Lijuzaad P, Tilanus MG, Koole R, Hordijk GJ, van der Vliet PC, Reinderg MJ, Slootweg PJ, Holstege FC. An expression profile for diagnosis of lymph node metastases from primary head and neck squamous cell carcinomas. Nat Genet. 2005;37:182–6. doi: 10.1038/ng1502. [DOI] [PubMed] [Google Scholar]
- 7.Bennett KL, Hackanson B, Smith LT, Morrison CD, Lang JC, Schuller DE, Weber F, Eng C, Plass C. Tumor Suppressor Activity of CCAAT/Enhancer Binding Protein {alpha} Is Epigenetically Down-regulated in Head and Neck Squamous Cell Carcinoma. Cancer Res. 2007;67:4657–64. doi: 10.1158/0008-5472.CAN-06-4793. [DOI] [PubMed] [Google Scholar]
- 8.Jiang MS, Lane MD. Sequential repression and activation of the CCAAT enhancer-binding protein-alpha (C/EBPalpha) gene during adipogenesis. Proc Natl Acad Sci U S A. 2000;97:12519–23. doi: 10.1073/pnas.220426097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jiang JG, DeFrances MC, Machen J, Johnson C, Zarnegar R. The repressive function of AP2 transcription factor on the hepatocyte growth factor gene promoter. Biochem Biophys Res Commun. 2000;272:882–6. doi: 10.1006/bbrc.2000.2848. [DOI] [PubMed] [Google Scholar]
- 10.Maytin EV, Lin JC, Krishnamurthy R, Batchvarova N, Ron D, Mitchell PJ, Habener JF. Keratin 10 gene expression during differentiation of mouse epidermis requires transcription factors C/EBP and AP-2. Dev Biol. 1999;216:164–81. doi: 10.1006/dbio.1999.9460. [DOI] [PubMed] [Google Scholar]
- 11.Boukamp P, Petrussevska RT, Breitkreutz D, Hornung J, Markham A, Fusenig NE. Normal Keratinization in a Spontaneously Immortalized Aneuploid Human Keratinocyte Cell Line. J Cell Bio. 1988;106:761–771. doi: 10.1083/jcb.106.3.761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Leone G, Nuckolls F, Ishida S, Adams M, Sears R, Jakoi L, Miron A, Nevins JR. Identification of a novel E2F3 product suggests a mechanism for determining specificity of repression by Rb proteins. Mol Cell Biol. 2000;20:3626–32. doi: 10.1128/mcb.20.10.3626-3632.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tada Y, Brena RM, Hackanson B, Morrison C, Otterson GA, Plass C. Epigenetic modulation of tumor suppressor CCAAT/enhancer binding protein alpha activity in lung cancer. J Natl Cancer Inst. 2006;98:396–406. doi: 10.1093/jnci/djj093. [DOI] [PubMed] [Google Scholar]
- 14.Madrid LV, Wang CY, Guttridge DC, Schottelius AJ, Baldwin AS, Jr, Mayo MW. Akt suppresses apoptosis by stimulating the transactivation potential of the RelA/p65 subunit of NF-kappaB. Mol Cell Biol. 2000;20:1626–38. doi: 10.1128/mcb.20.5.1626-1638.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Smith LT, Lin M, Brena RM, Lang JC, Schuller DE, Otterson GA, Morrison CD, Smiraglia DJ, Plass C. Epigenetic regulation of the tumor suppressor gene TCF21 on 6q23–q24 in lung and head and neck cancer. Proc Natl Acad Sci U S A. 2006;103:982–7. doi: 10.1073/pnas.0510171102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim JW, Monila H, Pandey A, Lane MD. Upstream stimulatory factors regulate the C/EBPalpha gene during differentiation of 3T3-L1 preadipocytes. Biochem Biophys Res Commun. 2007;354:517–21. doi: 10.1016/j.bbrc.2007.01.008. [DOI] [PubMed] [Google Scholar]
- 17.Fatemi M, Wade PA. MBD family proteins: reading the epigenetic code. J Cell Sci. 2006;119:3033–7. doi: 10.1242/jcs.03099. [DOI] [PubMed] [Google Scholar]
- 18.Matarazzo MR, De Bonis ML, Strazzullo M, Cerase A, Ferraro M, Vastarelli P, Ballestar E, Esteller M, Kudo S, D’Esposito M. Multiple binding of methyl-CpG and polycomb proteins in long-term gene silencing events. J Cell Physiol. 2007;210:711–9. doi: 10.1002/jcp.20879. [DOI] [PubMed] [Google Scholar]
- 19.Momparler RL. Cancer epigenetics. Oncogene. 2003;22:6479–83. doi: 10.1038/sj.onc.1206774. [DOI] [PubMed] [Google Scholar]
- 20.Liu H, Tan BC, Tseng KH, Chuang CP, Yeh CW, Chen KD, Lee SC, Yung BY. Nucleophosmin acts as a novel AP2alpha-binding transcriptional corepressor during cell differentiation. EMBO Rep. 2007;8:394–400. doi: 10.1038/sj.embor.7400909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kaeferstein A, Krug U, Tiesmeier J, Aivado M, Faulhaber M, Stadler M, Krauter J, Germing U, Hofmann WK, Koeffler HP, Ganser A, Verbeek W. The emergence of a C/EBPalpha mutation in the clonal evolution of MDS towards secondary AML. Leukemia. 2003;17:343–9. doi: 10.1038/sj.leu.2402805. [DOI] [PubMed] [Google Scholar]
- 22.Cao Z, Umek RM, McKnight SL. Regulated expression of three C/EBP isoforms during adipose conversion of 3T3-L1 cells. Genes Dev. 1991;5:1538–52. doi: 10.1101/gad.5.9.1538. [DOI] [PubMed] [Google Scholar]
- 23.Timchenko NA, Wilde M, Nakanishi M, Smith JR, Darlington GJ. CCAAT/enhancer-binding protein alpha (C/EBP alpha) inhibits cell proliferation through the p21 (WAF-1/CIP-1/SDI-1) protein. Genes Dev. 1996;10:804–15. doi: 10.1101/gad.10.7.804. [DOI] [PubMed] [Google Scholar]
- 24.Lin FT, MacDougald OA, Diehl AM, Lane MD. A 30-kDa alternative translation product of the CCAAT/enhancer binding protein alpha message: transcriptional activator lacking antimitotic activity. Proc Natl Acad Sci U S A. 1993;90:9606–10. doi: 10.1073/pnas.90.20.9606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bismuth K, Maric D, Arnheiter H. MITF and cell proliferation: the role of alternative splice forms. Pigment Cell Res. 2005;18:349–59. doi: 10.1111/j.1600-0749.2005.00249.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Radomska HS, Basseres DS, Zheng R, Zhang P, Dayaram T, Yamamoto T, Sternberg DW, Lokker N, Giese NA, Bohlander SK, Schnittger S, Delmotte MH, Davis RJ, Small D, Hiddemann W, Gilliard DG, Tenen DG. Block of C/EBP alpha function by phosphorylation in acute myeloid leukemia with FLT3 activating mutations. J Exp Med. 2006;203:371–81. doi: 10.1084/jem.20052242. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.