

Abstract
Mitochondrial reactive oxygen species (ROS) production is recognized as a major pathogenic event in a number of human diseases, and mitochondrial scavenging of ROS appears a promising therapeutic approach. Recently, two mitochondrial antioxidants have been developed; conjugating α-tocopherol and the ubiquinol moiety of coenzyme Q to the lipophilic triphenylphosphonium cation (TPP+), denominated MitoE2 and MitoQ10, respectively. We have investigated the effect of these compounds on mitochondrial Ca2+ homeostasis, which controls processes as diverse as activation of mitochondrial dehydrogenases and pro-apoptotic morphological changes of the organelle. We demonstrate that treatment of HeLa cells with both MitoE2 and MitoQ10 induces (albeit with different efficacy) a major enhancement of the increase in matrix Ca2+ concentration triggered by cell stimulation with the inositol 1,4,5-trisphosphate-generating agonist histamine. The effect is a result of the inhibition of Ca2+ efflux from the organelle and depends on the TPP+ moiety of these compounds. Overall, the data identify an effect independent of their antioxidant activity, that on the one hand may be useful in addressing disorders in which mitochondrial Ca2+ handling is impaired (e.g., mitochondrial diseases) and on the other may favor mitochondrial Ca2+ overload and thus increase cell sensitivity to apoptosis (thus possibly counteracting the benefits of the antioxidant activity).
Keywords: ROS, calcium, mitochondria
Introduction
Mitochondria contribute to cellular energy balance, cell death, and cell survival pathways in different ways.1-4 First, the most established function of the organelle is to synthesize ATP through oxidative phosphorylation, which is associated with reactive oxygen species (ROS) production, including primarily superoxide (O2·-) formation by the respiratory chain complexes I and III;5,6 O2·- is converted to other potent oxidants, such as peroxynitrite, hydrogen peroxide (H2O2), and the most reactive hydroxyl radical (OH·-). While various antioxidant mechanisms usually inactivate ROS, their relative inefficiency at high levels of ROS production trigger both a nonspecific increase of mitochondrial membrane permeability and the activation of mitochondrial apoptotic pathways through the release of pro-apoptotic factors from the mitochondrial intermembrane space.4
Increasing evidence suggests that mitochondrial dysfunction by oxidative damage plays a crucial role in major pathologies, such as neurodegenerative diseases (Parkinson’s disease, Huntington’s disease, Friedrich’s ataxia, and Alzheimer’s disease)7-11 and diabetes.12,13 Innovative therapeutic molecules are mitochondrial antioxidants generated by conjugating α-tocopherol (vitamin E) or the ubiquinol moiety of coenzyme Q to the lipophilic triphenylphosphonium cation (TPP+) in novel compounds named MitoE2 and MitoQ10, respectively.2,14-17 These compounds are able to localize almost exclusively to mitochondria, to permeate through the plasma membrane bilayer, and then to accumulate into the negatively charged mitochondrial matrix.16,18-23
Moreover, these molecules have been shown to reduce oxidized lipids in the inner mitochondrial membrane (IMM) in numerous cellular models, providing protection from lipid peroxidation and nonspecific membrane damage.24-29 These observations led to the introduction of these antioxidants in Phase I and II clinical trials in neurodegenerative diseases.
However, the extensive accumulation of lipophilic cations within isolated mitochondria at concentrations approaching millimolar levels can disrupt membrane integrity and impair respiration and ATP synthesis.17,30-32 Indeed, MitoQ10 starts to increase the respiration of isolated mitochondria at low micromolar concentrations. These effects may represent an unspecific effect on membrane permeability but may also be the consequence of the interaction of the antioxidant or TPP+ moieties with specific IMM enzymes and transporters, as was previously reported for TPP+.33 Understanding these interactions is important for the evaluation of therapeutic strategies using MitoE2 and MitoQ10 as well as for the development of newer generations of compounds based on the mitochondrial targeting approach.
The electrical gradient (Δψm) across the IMM provides a huge driving force for accumulation of inorganic cations into the mitochondrial matrix. Indeed, direct measurement of Ca2+ concentration in the matrix ([Ca2+]m) with targeted recombinant probes demonstrated rapid fluctuations of [Ca2+]m upon cell stimulation, strongly amplifying the parallel cytosolic Ca2+ ([Ca2+]c) changes. This leads to an extremely efficient increase of [Ca2+]m (100-500 μmol/L), as measured by recombinant low-affinity Ca2+ sensors and determined by three principal processes: Ca2+ uptake through the mitochondrial Ca2+ uniporter (MCU), driven by Δψm; (ii) Ca2+ efflux through the Na+/Ca2+ (mNCX) and H+/Ca2+ (mHCX) exchangers; (iii) Ca2+-buffering activity in the mitochondrial matrix through the formation of insoluble xCa2+-xPO4x--xOH- complexes. The exact role of such a large elevation of [Ca2+]m is not yet clear, but recent data indicate that it might be related to the regulation of mitochondrial dynamics and sensibility to apoptotic challenges.34
Given the correlation between mitochondrial Ca2+ transport and respiratory chain activity and the role of mitochondrial Ca2+ accumulation and ROS production in cell fate regulation, we investigated the effects of TPP+-based antioxidants on mitochondrial Ca2+ homeostasis, using targeted recombinant aequorin (AEQ) probes. We observed that TPP+, as well as MitoE2 and MitoQ10 although with different efficiency, specifically interact with the Na+/Ca2+ or H+/Ca2+Ca2+-exchangers (mCX), leading to profound alteration of mitochondrial Ca2+ homeostasis.
Results
The Mitochondrially Targeted Antioxidants MitoQ10 and MitoE2 Modify the Kinetics of Agonist-induced Ca2+ Uptake into the Organelle
To determine the effect of MitoQ10 and MitoE2 on mitochondrial Ca2+ handling, we measured histamine-evoked mitochondrial Ca2+ transients in HeLa cells. Cells were transfected with a mitochondrially targeted low-affinity aequorin probe (mtAEQmut).35 After reconstitution of the probe with coelenterazine, the transfected cells were pretreated for 10 min with 10 μmol/L MitoQ10 and 5 μmol/L MitoE2. Cells were then stimulated with 10 μmol/L histamine, an agonist acting on G-protein-coupled receptors and leading to the production of inositol 1,4,5 trisphosphate (IP3), in the continuous presence of the antioxidant. The consequent Ca2+ release from the intracellular stores induced a mitochondrial Ca2+ transient, which was recorded.
As shown in Figure 1A, MitoE2 caused a profound change in the mitochondrial Ca2+ transient. In MitoE2 pretreated cells, [Ca2+]m reached a much higher peak than control cells (peak Ca2+ response was 36.8 ± 5.9 μmol/L for MitoE2, n = 13 versus 20.1 ± 1.6 μmol/L for controls, n = 11, p = 0.005) and [Ca2+]m returned to basal level at a much slower rate, suggesting an effect on Ca2+ extrusion from mitochondria. On the other hand, as shown in Figure 1B, while the effect of MitoQ10 was similar to that of MitoE2, it was not so pronounced, suggesting that mitochondrial Ca2+ extrusion was more modestly inhibited (22.1 ± 5.2 μmol/L for MitoQ10, n = 18 versus 21.0 ± 1.6 μmol/L for controls, n = 19, p = 0.7).
Figure 1.

Effect of two different mitchondrial targeted antioxidants, mitochondrial vitamin E (mtVitE) and MitoQ, on mitochondrial Ca2+ signaling in HeLa cells. HeLa cells were transfected with a mitochondrially targeted low-affinity aequorin (mtAeqmut) probe, treated with (A) 5 μmol/L MitoE2, (B) 10 μmol/L MitoQ10, or (C) 20 μmol/L CGP37157 for 10 min, and then stimulated with histamine 10 μmol/L. Agonist stimulation induced a rapid Ca2+ uptake into mitochondria and a consequent release, which is strongly inhibited in MitoE2- and CGP37157-pretreated cells. MitoQ10 has a milder effect. These and the following traces are representative of more than five independent experiments that gave similar results.
Since the above changes suggested the inhibition of the Na+/Ca2+ exchanger, we compared the effect of the mitochondrial antioxidant MitoE2 to that of CGP37157, a specific inhibitor of mCXs.36,37 Figure 1C shows that the effect of 20 μmol/L CGP37157 on [Ca2+]m kinetics was almost identical to those observed in cells treated with MitoE2 (peak [Ca2+]m response was 40.3 ± 2.4 μmol/L for 20 μmol/L CGP37157, n = 17 versus 27.5 ± 2.3 μmol/L for controls, n = 11, p = 0.002). Importantly, the effects of CGP37157 and MitoE2 were not additive (data not shown).
MitoQ10 and MitoE2 Specifically Inhibit the Mitochondrial Na+/Ca2+ Exchanger
In the next series of experiments we verified whether the effects of MitoQ10 and MitoE2 are specific to mitochondria. We first analyzed for comparison the effect on mitochondrial Ca2+ homeostasis of untargeted antioxidants. For this purpose, we pretreated HeLa cells with vitamin E (α-tocopherol) at different concentrations (5 and 20 μmol/L) that were shown to exert a potent antioxidant effect in whole-cell systems.38-40 The application of the experimental protocol of Figure 1 showed that there were no alterations in the peak of mitochondrial Ca2+ uptake following histamine stimulation (at 5 μmol/L vitamin E, the peak rise was 17.3 ± 1.4 μmol/L, n = 15 versus 21.5 ± 1.5 μmol/L in controls, n = 16, p = 0.043; and at 20 μmol/L vitamin E, the peak was 17.1 ± 1.4 μmol/L, n = 19 vs. 19.8 ± 1.6 μmol/L in controls, p = 0.39 Fig. 2). The kinetics of the decay phase was identical in vitamin E-treated and control cells. Similarly, the use of other nontargeted antioxidants was without effect on mitochondrial Ca2+ homeostasis (ascorbic acid 1 mmol/L, 20 min and trolox 750 μmol/L, 20 min; data not shown).
Figure 2.
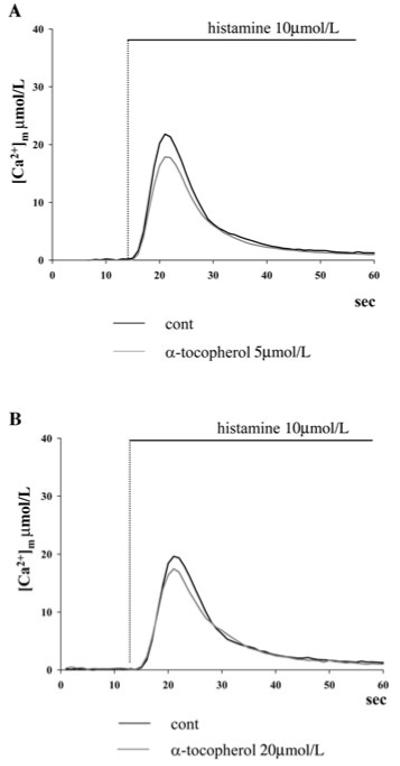
Effect of different doses of cytosolic vitamine E on mitochondrial Ca2+ homeostasis. HeLa cells were transfected with the mtAeqmut probe, treated with (A) 5 μmol/L or (B) 20 μmol/L unmodified vitamine E (α-tocopherol), and then stimulated with 10 μmol/L histamine. No major difference on the amplitude and kinetics of the [Ca2+]m rise is observed compared to untreated cells.
We then investigated the effect of MitoE2 (which had the stronger effect on mitochondrial Ca2+ signals) on the cytosolic Ca2+ signal. For this purpose, we transfected HeLa cells with the cytosolic form of the aequorin probe35 and again we analyzed the [Ca2+]c rise triggered by histamine stimulation. As shown in Figure 3, the [Ca2+]c rise following the application of 10 μmol/L histamine was not significantly altered in cells treated with MitoE2 (2.00 ± 0.09 μmol/L for 5 μmol/L MitoE2, n = 16 versus 1.79 ± 0.07 μmol/L for controls, n = 17, p = 0.08), in agreement with previous observations using the mCX inhibitor CGP37157.41
Figure 3.

Effect of MitoE2 on cytosolic Ca2+ homeostasis. HeLa cells were transfected with cytosolic Aeq probe, treated with 5 μmol/L MitoE2, and than stimulated with 10 μmol/L histamine. No major effect on the amplitude and kinetics of the [Ca2+]c rise is observed.
Lastly, we performed experiments in cells in which the plasma membrane was permeabilized in order to apply a defined extramitochondrial buffer. For this purpose HeLa cells, expressing the mtAeqmut probe, were incubated for 1 min with 25 μmol/L digitonin and then perfused with a buffer mimicking the ionic composition of the intracellular milieu [(intracellular buffer ([IB]): 130 mmol/L KCl, 10 mmol/L NaCl, 1 mmol/L MgSO4, 0.5 mmol/L K2HPO4, 5 mmol/L succinic acid, 1 mmol/L pyruvic acid, 3 mmol/L MgCl2, glucose 5.5 mmol/L, and 20 mmol/L HEPES, pH = 7.4) supplemented with 2 mmol/L ethylene glycol tetraacetic acid (IB/EGTA). Mitochondrial Ca2+ uptake was initiated by replacing IB/EGTA with IB containing a buffered Ca2+ concentration of 1 μmol/L (IB/Ca2+). When [Ca2+]m reached a steady state level, the kinetics of Ca2+ extrusion were analyzed by blocking Ca2+ influx through the application of 10 μmol/L ruthenium red (RR), an inhibitor of the MCU. As shown in Figure 4A, the addition of RR causes a rapid return of mitochondrial [Ca2+] to basal levels because of the activity of the mCX. With this procedure we compared the effect on the mCX of the mitochondrially targeted antioxidant with that of CGP37157. The rate of Ca2+ calcium extrusion was measured as a function of [Ca2+]m. As shown in the histograms of Figure 4B and C, MitoE2 and CGP37157 induced a comparable inhibition of Ca2+ extrusion (0.050 0.004 μmol/L/s for 5 μmol/L MitoE2, n ± 15 vs. 0.100 ± 0.004 μmol/L/s for controls, n = 25, p = 7.8e-6; 0.06 ± 0.01 μmol/L/s for 20 μmol/L CGP37157, n = 17, p=1.1e-6). To rule out a possible effect of MitoE2 on a MCU-independent uptake pathway, RR-insensitive Ca2+ uptake has been directly measured in permeabilized cells. For this purpose cells were perfused with 10 μmol/L RR from the very beginning of the experiments. As shown in Figure 4D under those conditions mitochondrial Ca2+ uptake is strongly reduced (less than 1% of control cells, plateau [Ca2+]μmol/L 144.9 ± 6.3 cont vs. 0.88 ± 0.05 RR). Moreover the initial rate of Ca4 uptake showed no differences in cells perfused with RR ** μmol/L and MitoE2 5 μmol/L compared to cells perfused only with RR 109 μmol/L (0.020 + 0.04 [μmol/L])/s for controls, 0.0205 + 0.04 [μmol/L]/s for 5 μmol/L Mito E2, p > 0.05). Thus, summarizing the last three experimental approaches, we concluded that MitoQ10 and MitoE2 exert a specific inhibitory effect on the mitochondrial Na+/Ca2+ exchanger independently of changes in global cellular Ca2+ homeostasis, and that for this effect, their mitochondrial targeting is essential.
Figure 4.
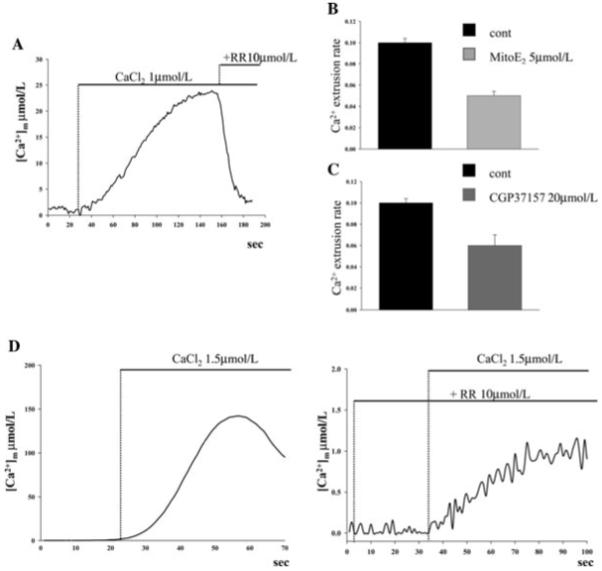
Effect of ruthenium red on mitochondrial Ca2+ homeostasis in permeabilized HeLa cells. HeLa cells were transfected with the mtAeqmut probe, permeabilized by a 1-min treatment with 25 μmol/L digitonin, and then perfused with intracellular buffer (IB)/ethylene glycol tetraacetic acid (EGTA) buffer (composition in the text). When indicated the medium was switched to IB/Ca2+. When a plateau [Ca2+]m level was reached, the perfusion medium was supplemented with (A) 10μmol/L ruthenium red (RR). The rate of Ca2+ extrusion in control cells versus (B) MitoE2- and (C) CGP37157-treated cells was then calculated. (D) When 10 μmol/L RR was added (right panel) both to IB/EGTA and to IB/Ca2+, mitochondrial Ca2+ uptake was strongly reduced as compared to nontreated cells (left panel). Note the different scales on the two panels. Traces are representative of > nine measurements.
The Effect of MitoQ10 and MitoE2 Relies on the Presence of Their Triphenylphosphonium Moiety
In the next set of experiments we investigated the possible mechanism of mCX inhibition by MitoQ10 and MitoE2. These molecules are composed of the lipophilic cation tetraphenilphosphonium bromide (TPP+) and ubiquinol (MitoQ10) or vitamin E (MitoE2), and the subscript in the compound refers to the number of carbons in the alkyl chain connecting the TPP and antioxidant moieties. Transport studies have shown that these complex molecules are well absorbed by the IMM, having their mostly lipophilic part embedded in the membrane42 and TPP+ interacting with the hydrophilic lipid surface. Thus, we hypothesized two scenarios. In the first, the antioxidant activity of MitoQ10 and MitoE2, by changing the lipid oxidation state in the membrane, might change the microenvironment of the mCX and thus its activity. Alternatively, the membrane surface-associated TPP+ moiety may interact directly with the protein responsible for the Na+/Ca2+ (or H+/Ca2+)-exchange activity, leading to its inhibition.
To distinguish between these possibilities, we performed two set of experiments. First, we assessed the effect of TPP+ on agonist-induced mitochondrial Ca2+ signals in mtAEQmut-transfected HeLa cells. Cells were treated with 5 μmol/L TPP+ and then stimulated with 10 μmol/L histamine. Strikingly, the effects on [Ca2+]m were the same as of those of MitoE2, that is, TPP+ induced an increase of the [Ca2+]m peak and reduced the rate of Ca2+ extrusion ([Ca2+]m peak TPP+ = 38.9 ± 3.0 μmol/L, n = 11 vs. controls = 19.4 ± 1.5 μmol/L, n = 11, p=8.1e-6) (Fig. 5). These results demonstrated that TPP+ in itself can inhibit the mitochondrial Ca2+ extrusion machinery. However, it did not exclude that the antioxidant moiety of MitoQ10 and MitoE2 could induce a similar effect. To examine this possibility, we assumed that for the effect of antioxidants on the oxidative state of membrane lipid, a relatively long-term accumulation and reaction time is necessary,16,17 thus the effect on Ca2+ kinetics should not be immediate. Therefore, using the above described permeabilized cell model, we applied MitoE2 directly in the perfusion buffer and carried out a longer measurement of [Ca2+]m. Under those conditions, the [Ca2+]plateauwas followed by a slow [Ca2+]m decline, dependent on Ca2+ extrusion and possibly buffering. Application of both MitoE2 and TPP+ during this decay phase caused a rapid transient inversion of the [Ca2+]m decline (Δ[Ca2+]m = 8.04 ± 1.24 μmol/L and 6.77 ± 1.06 μmol/L, respectively p = 0.46), indicative of inhibition of Ca2+ extrusion (Fig. 6). Altogether, our results strongly suggest that the MitoQ10 and MitoE2 mitochondrially targeted antioxidants inhibit the Na+/Ca2+ (or H+/Ca2+) exchange mechanism through the TPP+ moiety present in both molecules.
Figure 5.

Effect of triphenylphosphonium cation (TPP+) on mitochondrial Ca2+ homeostasis. HeLa cells were transfected with mtAeqmut probe, treated with TPP+ 5 μmol/L, and then stimulated with 10 μmol/L histamine. The amplitude of the [Ca2+]m rise is increased in TPP+ treated cells and the return to basal levels significantly delayed.
Figure 6.
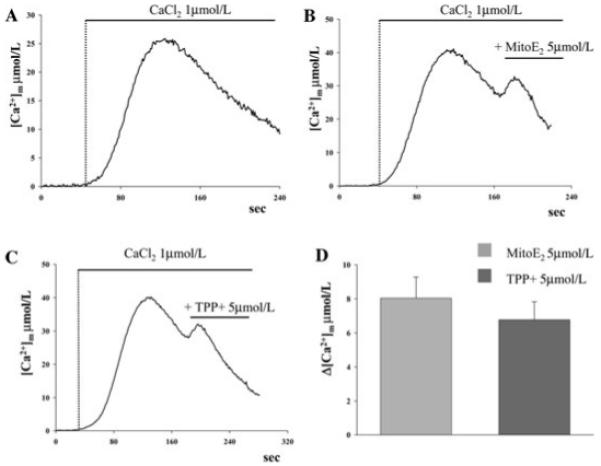
Effect on [Ca2+]m of perfused MitoE2- and TPP+-permeabilized cells. Permeabilization of the plasma membrane with digitonin and initiation of mitochondrial Ca2+ uptake through perfusion of 1 μmol/L Ca2+ were carried out as in Figure 4A. Panel (A) shows the slow decay of the [Ca2+]m plateau. During the decay phase the perfusion medium was supplemented with (B) 5 μmol/L MitoE2 or (C) 5 μmol/L TPP+. In both cases, a rapid inversion of the decay phase is observed. Statistical analysis of Δ[Ca2+]m (μmol/L, measured from the application of the drugs until the signal initiated decay again) is shown in panel D.
Discussion and Perspectives
Our results disclosed a specific interaction of the mitochondrially targeted antioxidants MitoE2 and MitoQ10 with the Na+ (H+)/Ca2+ exchanger. Moreover, they provide evidence that this interaction is mediated by the TPP+ moiety of the compounds. Even if analyses of steady-state tetramethyl rhodamine methyl ester (TMRM) accumulation showed a slight reduction of mitochondrial membrane potential (about 10%) induced by MitoE2 and TPP+, the interaction resulted in increased Ca2+ accumulation in the mitochondria following both IP3-mediated Ca2+ release from the ER or incubation of permeabilized cells with Ca2+. Interestingly, the effect of MitoE2 by far exceeded that of MitoQ10 and was comparable to the effect of TPP+ alone. These differences in the efficiency of the three molecules may provide some clues for identifying the site of TPP+ interaction with the mCX. Indeed, MitoQ10 has a long alkyl chain, which was shown to increase the tendency of the lipophilic cations to absorb to the inner surface of the IMM. In addition, a longer alkyl chain increases the extent to which the attached molecule (TPP+ in this case) penetrates into the membrane. Thus, we can speculate that loose attachment of TPP+ to the inner surface of the IMM promotes its interaction with the mCX while its penetration into the membrane counteracts it and diminishes the inhibitory effect. A further hypothesis arises by taking into consideration that TPP+ is a cation, making it possible that it directly interacts with the site of Ca2+ or Na+ binding on the transporter.
To be pharmaceutically manageable an antioxidant should be a small molecule, should have a high rate of mitochondrial localization in order to selectively protect mitochondria from oxidative stress, should be recycled back to its active form after its antioxidant action, and last, but not least, should be orally bioavailable. MitoE2, the triphenylphosphonium-conjugated form of α-tocopherol, was shown to be taken up rapidly by isolated mitochondria because of the TPP+ ability to pass through phospholipid bilayers, and it has been demonstrated that in mice after intravenous injection it can diffuse from the bloodstream to most tissues. If we add its capacity to be cleared from all organs approximately in 1.5 days, oral administration of the mitochondrial form of vitamin E appears a reasonable prospective.
However, the side effects shown by our study raise a series of important questions. How does the alteration of mitochondrial calcium signaling modify cell metabolism? Will this effect influence (or even reverse) the anti-apoptotic effect of the antioxidants? Mitochondrial Ca2+ accumulation is determined by three principal processes: (i) Ca2+ uptake through the MCU, driven by Δψm and activated in a cooperative manner by external [Ca2+] ([Ca2+]e); (ii) Ca2+ efflux through the mNCX and mHCX exchangers, saturated at ≈ 1 μmol/L [Ca2+]m; (iii) Ca2+-buffering activity in the mitochondrial matrix through the formation of insoluble xCa2+-xPO4x--xOH- complexes, driven by HxPO4x- uptake accompanying Ca2+ accumulation and the alkaline pH of the mitochondrial matrix. As a result of these processes, in isolated mitochondria two patterns of [Ca2+]m changes were observed following elevation of [Ca2+]e. At submicromolar [Ca2+]e, [Ca2]m increases in a range (0.2-3 μmol/L), which allows the parallel activation of Ca2+-dependent enzymes of the Krebs cycle, leading to increased supply of reducing equivalents (NADH+/NADPH+).43-45 This [Ca2+]m increase activates mitochondrial metabolism, i.e., the supply of ATP under aerobic conditions.34,46 At [Ca2+]e above the μmol/L level, the mitochondrial efflux mechanisms, assisted by the matrix Ca2+-buffering activity, keep [Ca2+]m relatively stable, allowing mitochondria to accumulate as much as 700-1000 nmol Ca2+/mg mitochondrial protein. On the other hand, mitochondria positioned at the cytoplasmic face of the ER Ca2+ release channels (inositol 1,4,5-trisphosphate receptors and ryanodine receptors) or close to plasma membrane (PM) Ca2+ influx channels (e.g., capacitative Ca2+ entry or ionotropic glutamate receptors) are exposed to Ca2+ concentrations well above those measured in the bulk cytosol. Such a large elevation of [Ca2+]m might be related to the regulation of mitochondrial dynamics and sensibility to apoptotic challenges.34
As detailed in the introduction, both mitochondrial ATP production and Ca2+-mediated cell death can be positively modified by the increased mitochondrial Ca2+ uptake induced by TPP+ and its derivatives. The former effect, that is, increased mitochondrial ATP production, might enhance the protective effect of the antioxidant moiety in cell types with high ATP demand, such as the cardiomyocytes. On the other hand, increased cell death by mitochondrial Ca2+ overload might cancel the benefits from the antioxidant effect. Further work in cell types of pathophysiological interest and using experimental approaches for investigating cell metabolism and apoptotic death (ATP measurements with targeted luciferases, enzymatic assays of caspase activation, monitoring of mitochondrial structure with targeted green fluorescent proteins (GFPs) will be needed to clarify this issue.
Matherial and Methods
Cell Culture and Transfection
HeLa cells were grown in Dulbecco’s modified Eagle’s medium (DMEM), supplemented with 10% fetal calf serum (FCS), in 75 cm2 Falcon flasks. For aequorin measurements, the cells were seeded before transfection onto 13-mm glass coverslips and allowed to grow to 50% confluence. At this stage, transfection with 4 μg of plasmid DNA (3 μg mt-GFP + 1 μg mtAEQ or cytAEQ) was carried out as previously described47 and aequorin measurements were performed 36 h after transfection.
Aequorin Measurements
For cytosolic aequorin (cytAEQ) and mtAEQ measurements, the coverslip with the cells was incubated with 5 μmol/L coelenterazine for 1-2 h in DMEM, supplemented with 1% FCS, and then transferred to the perfusion chamber. All aequorin measurements were carried out in Krebs-Ringer modified buffer (KRB): 125 mmol/L NaCl, 5 mmol/L KCl, 1 mmol/L Na3PO4, 1 mmol/L MgSO4, 5.5 mmol/L glucose, 20 mmol/L HEPES, pH 7.4, 37 °C) supplemented with 1 mmol/L CaCl2 (KRB/Ca2+). Agonists and other drugs were added to the same medium, as specified in the figure legends. The experiments were terminated by lysing the cells with 100 μmol/L digitonin in a hypotonic Ca2+-rich solution (10 mmol/L CaCl2 in H2O), thus discharging the remaining aequorin pool. The light signal was collected and calibrated into [Ca2+] values as previously described.48,49 All the results are expressed as means ± standard error.
Experiments in permeabilized HeLa cells were performed as previously described,50 except that 25 μmol/L digitonin was used in order to preserve mitochondrial integrity.
Acknowledgments
We thank Antipodean for kindly supplying MitoE2 and MitoQ10 as well as Professors Michael R. Duchen and Michael P. Murphy for valuable advice. This work was supported by Telethon grant GGP05284, the Italian Association for Cancer Research, local funds from the University of Ferrara, the Italian University Ministry, the European Union (fondi strutturali Obiettivo 2), the PRRIITT program of the Emilia Romagna Region, the Italian Space Agency, and National Institute of Health (grant: A mitochondrial longevity pathway: p66shc mechanisms).
Footnotes
Conflicts of Interest
The authors declare no conflicts of interest.
References
- 1.Scheffler IE. Mitochondria. Wiley-Liss; New York: 1999. [Google Scholar]
- 2.Murphy MP, Smith RAJ. Drug delivery to mitochondria: the key to mitochondrial medicine. Adv. Drug Deliv. Rev. 2000;41:235–2350. doi: 10.1016/s0169-409x(99)00069-1. [DOI] [PubMed] [Google Scholar]
- 3.Nicholls DG, Ferguson SJ. Bioenergetics 3. Academic; London: 2002. [Google Scholar]
- 4.Green DR, Kroemer G. The pathophysiology of mitochondrial cell death. Science. 2004;305:626–629. doi: 10.1126/science.1099320. [DOI] [PubMed] [Google Scholar]
- 5.Raha S, Robinson BH. Mitochondria, oxygen free radicals, disease and ageing. Trends Biochem. Sci. 2000;25:502–508. doi: 10.1016/s0968-0004(00)01674-1. [DOI] [PubMed] [Google Scholar]
- 6.Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. 2005;120:483–495. doi: 10.1016/j.cell.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 7.Finkel T. Radical medicine: treating ageing to cure disease. Nat. Rev. Mol.Cell Biol. 2005;6:971–976. doi: 10.1038/nrm1763. [DOI] [PubMed] [Google Scholar]
- 8.Mattson MP, Magnus T. Ageing and neuronal vulnerability. Nat. Rev.Neurosci. 2006;7:278–294. doi: 10.1038/nrn1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Abou-Sleiman PM, Muqit MMK, Wood NW. Expanding insights of mitochondrial dysfunction in Parkinson’s disease. Nat. Rev. Neurosci. 2006;7:207–219. doi: 10.1038/nrn1868. [DOI] [PubMed] [Google Scholar]
- 10.Squitieri F, et al. Severe ultrastructural mitochondrial changes in lymphoblasts homozygous for Huntington disease mutation. Mech. Ageing Dev. 2006;127:217–220. doi: 10.1016/j.mad.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 11.Beal MF. Mitochondria take center stage in aging and neurodegeneration. Ann. Neurol. 2005;58:495–505. doi: 10.1002/ana.20624. [DOI] [PubMed] [Google Scholar]
- 12.Green K, Brand MD, Murphy MP. Prevention of mitochondrial oxidative damage as a therapeutic strategy in diabetes. Diabetes. 2004;53:S110–S118. doi: 10.2337/diabetes.53.2007.s110. [DOI] [PubMed] [Google Scholar]
- 13.Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414:813–820. doi: 10.1038/414813a. [DOI] [PubMed] [Google Scholar]
- 14.Murphy MP. Development of lipophilic cations as therapies for disorders due to mitochondrial dysfunction. Exp. Opin. Biol. Therapy. 2001;1:753–764. doi: 10.1517/14712598.1.5.753. [DOI] [PubMed] [Google Scholar]
- 15.Smith RAJ, et al. Using mitochondria-targeted molecules to study mitochondrial radical production and its consequences. Biochem. Soc. Trans. 2003;31:1295–1299. doi: 10.1042/bst0311295. [DOI] [PubMed] [Google Scholar]
- 16.Smith RAJ, et al. Targeting coenzyme Q derivatives to mitochondria. Meth. Enzymol. 2004;382:45–67. doi: 10.1016/S0076-6879(04)82003-2. [DOI] [PubMed] [Google Scholar]
- 17.Murphy MP. Targeting bioactive compounds to mitochondria. Trends Biotechnol. 1997;15:326–330. doi: 10.1016/S0167-7799(97)01068-8. [DOI] [PubMed] [Google Scholar]
- 18.Ketterer B, Neumcke B, Laeuger P. Transport mechanism of hydrophobic ions across through lipid bilayers. J. Membr. Biol. 1971;5:225–245. doi: 10.1007/BF01870551. [DOI] [PubMed] [Google Scholar]
- 19.Flewelling RF, Hubbell WL. Hydrophobic ion interactions with membranes. Biophys. J. 1986;49:531–540. doi: 10.1016/S0006-3495(86)83663-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Honig BH, Hubbell WL, Flewelling RF. Electrostatic interactions in membranes and proteins. Annu. Rev. Biophys. Biophys. Chem. 1986;15:163–193. doi: 10.1146/annurev.bb.15.060186.001115. [DOI] [PubMed] [Google Scholar]
- 21.Cafiso DS, Hubbell WL. EPR determination of membrane potentials. Annu. Rev. Biophys. Bioeng. 1981;10:217–244. doi: 10.1146/annurev.bb.10.060181.001245. [DOI] [PubMed] [Google Scholar]
- 22.Ono A, et al. Activation energy for permeation of phosphonium cations through phospholipid bilayer membrane. Biochemistry. 1994;33:4312–4318. doi: 10.1021/bi00180a027. [DOI] [PubMed] [Google Scholar]
- 23.Demura M, Kamo N, Kobatake Y. Determination of membrane potential with lipophilic cations: correction of probe binding. Biochim. Biophys. Acta. 1985;820:207–215. [Google Scholar]
- 24.Smith RAJ, et al. Targeting an antioxidant to mitochondria. Eur. J. Biochem. 1999;263:709–716. doi: 10.1046/j.1432-1327.1999.00543.x. [DOI] [PubMed] [Google Scholar]
- 25.Kelso GF, et al. Selective targeting of a redoxactive ubiquinone to mitochondria within cells. J. Biol. Chem. 2001;276:4588–4596. doi: 10.1074/jbc.M009093200. [DOI] [PubMed] [Google Scholar]
- 26.James AM, et al. Interactions of mitochondria-targeted and untargeted ubiquinones with the mitochondrial respiratory chain and reactive oxygen species. Implications for the use of exogenous ubiquinones as therapies and experimental tools. J. Biol.Chem. 2005;280:21295–21312. doi: 10.1074/jbc.M501527200. [DOI] [PubMed] [Google Scholar]
- 27.Maguire JJ, Wilson DS, Packer L. Mitochondrial electron transportlinked tocoperoxyl radical reduction. J. Biol. Chem. 1989;264:21462–21465. [PubMed] [Google Scholar]
- 28.James AM, Smith RAJ, Murphy MP. Antioxidant and prooxidant properties of mitochondrial coenzyme Q. Arch. Biochem. Biophys. 2004;423:47–56. doi: 10.1016/j.abb.2003.12.025. [DOI] [PubMed] [Google Scholar]
- 29.Asin-Cayuela J, et al. Finetuning the hydrophobicity of a mitochondria-targeted antioxidant. FEBS Lett. 2004;571:9–16. doi: 10.1016/j.febslet.2004.06.045. [DOI] [PubMed] [Google Scholar]
- 30.Azzone GF, Pietrobon D, Zoratti M. Determination of the proton electrochemical gradient across biological membranes. Curr.Topics Bioenerg. 1984;13:1–77. [Google Scholar]
- 31.Brand MD. Measurement of mitochondrial protonmotive force. In: Brown GC, Cooper CE, editors. Bioenergetics—A Practical Approach. IRL; Oxford: 1995. pp. 39–62. [Google Scholar]
- 32.Bakeeva LE, et al. Conversion of biomembrane-produced energy into electric form. II. Intact mitochondria. Biochim. Biophys. Acta. 1970;216:13–21. doi: 10.1016/0005-2728(70)90154-4. [DOI] [PubMed] [Google Scholar]
- 33.Wingrove DE, Gunter TE. Kinetics of mitochondrial calcium transport II. A kinetic description of the sodium-dependent calcium efflux mechanism of liver mitochondria and inhibition by ruthenium red and by tetraphenylphosphonium. J. Biol. Chem. 1986;261:15166–15171. [PubMed] [Google Scholar]
- 34.Jouaville LS, et al. Regulation of mitochondrial ATP synthesis by calcium: evidence for a long-term metabolic priming. Proc. Natl. Acad. Sci. USA. 1999;96:13807–13812. doi: 10.1073/pnas.96.24.13807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chiesa A, et al. Recombinant aequorin and green fluorescent protein as valuable tools in the study of cell signalling. Biochem. J. 2001;355:1–12. doi: 10.1042/0264-6021:3550001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cox DA, Matlib MA. A role for the mitochondrial Na+/Ca2+ exchanger in the regulation of oxidative phosphorylation in isolated heart mitochondria. J. Biol. Chem. 1993;268:938–947. [PubMed] [Google Scholar]
- 37.Cox DA, et al. Selectivity of inhibition of Na+/Ca2+ exchange of hearth mitochondria by benzothiazepine CGP-37157. J. Cardiovasc. Pharmacol. 1993;21:595–599. doi: 10.1097/00005344-199304000-00013. [DOI] [PubMed] [Google Scholar]
- 38.Gulec M, Gurel A, Armutcu F. Vitamin E protect against oxidative damage caused by formaldehyde in the liver and plasma of rats. Mol. Cell. Biochem. 2006;290:61–67. doi: 10.1007/s11010-006-9165-z. [DOI] [PubMed] [Google Scholar]
- 39.Thews O, et al. Possible protective effects of alpha-tocopherol on enhanced induction of reactive oxygen species by 2-methoxyestradiol in tumors. Adv. Exp. Med. Biol. 2005;566:349–355. doi: 10.1007/0-387-26206-7_46. [DOI] [PubMed] [Google Scholar]
- 40.Pathania V, et al. Vitamin E suppresses the induction of reactive oxygen species release by lipopolysaccharide, interleukin-1beta and tumor necrosis factor-alpha in rat alveolar macophages. J. Nutr. Sci. Vitaminol. (Tokyo) 1999;46:675–686. doi: 10.3177/jnsv.45.675. [DOI] [PubMed] [Google Scholar]
- 41.Brini M, et al. A calcium signaling defect in the pathogenesis of a mitochondrial DNA inherited oxidative phosphorylation deficiency. Nat. Med. 1999;5:951–954. doi: 10.1038/11396. [DOI] [PubMed] [Google Scholar]
- 42.Murphy MP, Smith RA. Targetin antioxidants to mitochondria by conjugation to lipophilic cations. Annu. Rev. Pharmcol. Toxicol. 2007;47:629–656. doi: 10.1146/annurev.pharmtox.47.120505.105110. [DOI] [PubMed] [Google Scholar]
- 43.Pitter JG, et al. Mitochondria respond to Ca2+ already in the submicromolar range: correlation with redox state. Cell Calcium. 2002;31:97–104. doi: 10.1054/ceca.2001.0264. [DOI] [PubMed] [Google Scholar]
- 44.McCormack JG, Halestrap AP, Denton RM. Role of calcium ions in regulation of mammalian intramitochondrial metabolism. Physiol. Rev. 1990;70:391–425. doi: 10.1152/physrev.1990.70.2.391. [DOI] [PubMed] [Google Scholar]
- 45.Pralong WF, et al. Pyridine nucleotide redox state parallels production of aldosterone in potassium-stimulated adrenal glomerulosa cells. Proc. Natl. Acad. Sci. USA. 1992;89:132–136. doi: 10.1073/pnas.89.1.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rutter GA, Rizzuto R. Regulation of mitochondrial metabolism by ER Ca2+ release: an intimate connection. Trends Biochem. Sci. 2000;25:215–221. doi: 10.1016/s0968-0004(00)01585-1. [DOI] [PubMed] [Google Scholar]
- 47.Rittuto R, et al. Photoprotein-mediated measurement of calcium ion concentration in mitrochondria of living cells. Methods Enzymol. 1995;260:417–428. doi: 10.1016/0076-6879(95)60155-4. [DOI] [PubMed] [Google Scholar]
- 48.Brini M, et al. Transfected Aequorin in the measurement of cytosolic Ca2 concentration ([Ca2]c). a critical evaluation. J. Biol. Chem. 1995;270:9896–9903. doi: 10.1074/jbc.270.17.9896. [DOI] [PubMed] [Google Scholar]
- 49.Barrero MJ, Montero M, Alvarez J. Dynamics of [Ca2] in the endoplasmic reticulum and cytoplasm of intact Hela cells: a comparative study. J. Biol. Chem. 1997;272:27694–27699. doi: 10.1074/jbc.272.44.27694. [DOI] [PubMed] [Google Scholar]
- 50.Rapizzi E, et al. Recombinant expression of the voltage-dependent anion channel enhances the transfer of Ca2+ microdomains to mitochondria. J. Cell. Biol. 2002;159:613–624. doi: 10.1083/jcb.200205091. [DOI] [PMC free article] [PubMed] [Google Scholar]