

Abstract
Metabolomics, an omic science in systems biology, is the global quantitative assessment of endogenous metabolites within a biological system. Either individually or grouped as a metabolomic profile, detection of metabolites is carried out in cells, tissues, or biofluids by either nuclear magnetic resonance spectroscopy or mass spectrometry. There is potential for the metabolome to have a multitude of uses in oncology, including the early detection and diagnosis of cancer and as both a predictive and pharmacodynamic marker of drug effect. Despite this, there is lack of knowledge in the oncology community regarding metabolomics and confusion about its methodologic processes, technical challenges, and clinical applications. Metabolomics, when used as a translational research tool, can provide a link between the laboratory and clinic, particularly because metabolic and molecular imaging technologies, such as positron emission tomography and magnetic resonance spectroscopic imaging, enable the discrimination of metabolic markers noninvasively in vivo. Here, we review the current and potential applications of metabolomics, focusing on its use as a biomarker for cancer diagnosis, prognosis, and therapeutic evaluation.
History and Definitions
The omic sciences of systems biology (Fig. 1), including genomics, transcriptomics, proteomics, and metabolomics, have been in existence for decades, whereas much attention has been focused on their development and application in the last several years. Metabolomics is an analytic tool used in conjunction with pattern recognition approaches and bioinformatics to detect metabolites and follow their changes in biofluids or tissue (1-3). Precise numbers of human metabolites is unknown, with estimates ranging from the thousands to tens of thousands. Metabolomics is a term that encompasses several types of analyses, including (a) metabolic fingerprinting, which measures a subset of the whole profile with little differentiation or quantitation of metabolites (4); (b) metabolic profiling, the quantitative study of a group of metabolites, known or unknown, within or associated with a particular metabolic pathway (5, 6); and (c) target isotope-based analysis, which focuses on a particular segment of the metabolome by analyzing only a few selected metabolites that comprise a specific biochemical pathway (7).
Fig. 1.
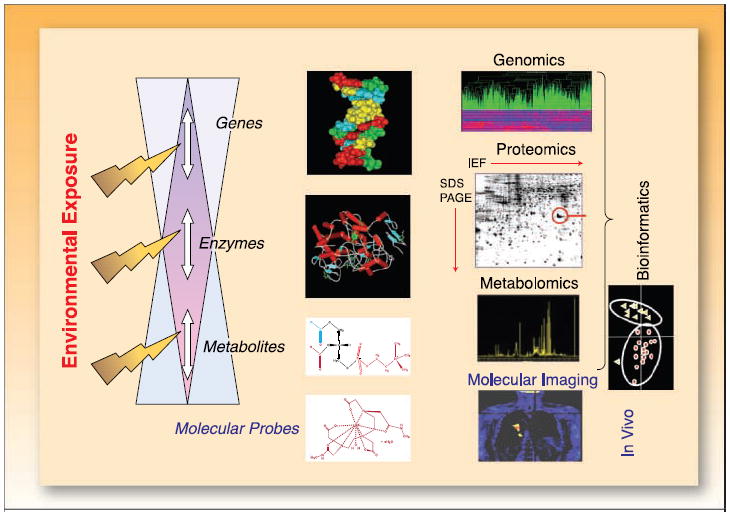
The flow of the “omics”sciences: genomics, proteomics, and metabolomics technologies in individualized medicine for cancer patients. Metabolomics deals with global metabolic profiling and its dynamic changes by monitoring as many as possible endogenous low-weight metabolites in a single analytic assay. Metabolic changes occur through a number of mechanisms, including direct genetic regulation and alterations in enzymatic and metabolic reactions. Techniques applied to metabolic profiling include NMR spectroscopy and MS. Bioinformatics, using techniques developed in the fields of computational science and statistics, remains a key element in data management and analysis of collected data sets. Identified genes, proteins, and metabolites can be assessed by tracer-based molecular imaging using MRI/MRSI and PET.
Metabolomics allows for a global assessment of a cellular state within the context of the immediate environment, taking into account genetic regulation, altered kinetic activity of enzymes, and changes in metabolic reactions (2, 8, 9). Thus, compared with genomics or proteomics, metabolomics reflects changes in phenotype and therefore function. The omic sciences are, however, complementary as “upstream” changes in genes and proteins are measured “downstream” as changes in cellular metabolism (2, 7). The converse, however, is that metabolomics is also a terminal view of the biological system, not allowing for representation of the genes and proteins that are increased or decreased. Other features of metabolomics are similar to those of proteomics and transcriptomics, including the ability to assay biofluids or tumor samples and the relatively inexpensive, rapid, and automated techniques once start-up costs are taken into account.
The origin of metabolomics dates back decades, with initial key applications in the fields of inborn metabolic errors, toxicology, and functional nutrigenomics. Interestingly, modern interest of metabolomics in oncology originally stemmed from the claim in the late 1980s that cancer could be identified by nuclear magnetic resonance (NMR) spectra of blood samples (10). Unfortunately, these data were later found to be falsified and the field of metabolomics was tainted. Despite this, the notion has persisted that correctly applying metabolomics on patient specimens may affect oncologic practice. Recent technological progress in NMR spectroscopy and mass spectrometry (MS), the two most accepted methods used in the measurement of metabolites, has improved the sensitivity and spectral resolution of analytic assays on metabolomic samples in attempts to achieve a comprehensive biochemical assessment. Because cancer cells are known to possess a highly unique metabolic phenotype, development of specific biomarkers in oncology is possible and might be used in identifying fingerprints, profiles, or signatures to detect the presence of cancer, determine prognosis, and/or assess the pharmacodynamic effects of therapy (2, 11-20). In this review, we will present metabolomics methodology and discuss how it is being applied in the field of oncology with particular attention to its application as a biomarker in cancer diagnosis, assessing treatment effects, and in the development of novel therapeutics.
Translational Relevance.
This review article presents metabolomics methodology and focuses on the current and potential applications of metabolomics in oncology with particular attention to its use as a biomarker in cancer diagnosis, prognosis, and therapeutic evaluation. We believe this to be an important and interesting topic that bridges preclinical and clinical oncology. We think this article will appeal to both translational researchers and clinicians as it reviews up-to-date evidence on the utility of metabolomics, an often poorly understood topic. Metabolomics has the potential to influence clinical oncology affecting patient care with benefits already being seen with the use of metabolite imaging in breast and prostate cancer diagnosis and probable future uses as a biomarker for early cancer diagnosis, determination of treatment efficacy, and in developing novel therapeutics.
Metabolomic Methodology
Metabolomic samples
Metabolomic assessment can be pursued both in vitro and in vivo using cells, fluids, or tissues. With regard to acquisition and simplicity of sample preparation, biofluids are the easiest samples to work with and can include serum, plasma, urine, ascitic fluid, saliva, bronchial washes, prostatic secretions, or fecal water. Most experience to date is with serum and urine samples as a surrogate system for tumor biochemistry. Interest is evolving for metabolomic studies directly using tumor tissue; however, such analyses require a more difficult and careful tissue preparation due to tissue heterogeneity. Surrounding stromal and epithelial cells can cause contamination of the resulting metabolic profile, thereby skewing results compared with that obtained from a pure tumor tissue sample. Microdissection techniques could enhance sample purity but also increase the required equipment and expertise.
Information on sample requirements and handling for metabolomics analysis has been published previously (13, 21, 22). Briefly, all biological samples collected for metabolic analysis require careful sample handling, such as special requirements for diet, physical activities, and other patient validation before sample collection. Due to high susceptibility of metabolic pathways to exogenous environment, maintaining low temperature and consistent sample extraction is essential. For biofluids, the standard sample volume is accepted to be in the range of 0.1 to 0.5 mL. For NMR, minimal sample preparation is required for urine and other low-molecular-weight metabolite-containing fluids, whereas blood, plasma, and serum require extraction (using acid, acetonitrile, or two-phase methanol/chloroform protocols) or NMR-weighted techniques to separate polar and lipophilic metabolites (see Table 1; refs. 23, 24). Intact tissue specimens (e.g., biopsies, fine needle aspirates) can be analyzed using high-resolution magic angle spinning (HR-MAS). HR-MAS probes for solid state NMR, as well as cryoprobes and microprobes for liquid NMR, permit quantitative metabolic analysis on samples as small as 3 μL with improved signal-to-noise ratios and solvent suppression (5). MS analysis requires more labor-intensive and destructive tissue preparation than NMR, but has greater sensitivity for metabolite detection (5).
Table 1.
Biofluid and sample preparation requirements
| Biofluid | Required sampling handling |
|---|---|
| Urine | Add deuterated phosphate buffer to 0.2-0.4 mL urine |
| Blood/plasma/serum | For 0.5 mL of heparinized blood product |
| —Add deuterium oxide (to lock) | |
| —Add acetonitrile (for protein precipitation) | |
| —Add methanol/chloroform extraction (for lipid extraction) | |
| CSF | Add deuterium oxide to 0.5 mL of CSF |
| EPS | Add deuterium oxide to 0.03-0.10 mL of EPS |
| Bile | Add deuterium methanol to 0.5 mL of bile |
| BALF | Add deuterium oxide to 0.5 mL of BALF |
| Tissue | —Add 0.01 mL of deuterium oxide to 3-10 g of tissue in MAS rotor |
| —Add perchloric acid extraction on 20-200 g frozen tissue | |
| —Add methanol/chloroform extraction to 20-200 g frozen tissue |
NOTE: Adapted from ref. 13.
Abbreviations: CSF, cerebrospinal fluid; EPS, expressed prostatic secretions; BALF, bronchoalveolar lavage fluid; MAS, magic angle spinning.
Analytic techniques
In general, NMR spectroscopy (mostly 1H-NMR) and MS [particularly liquid chromatography (LC)/MS and Fourier transform ion cyclotron resonance Fourier transform/MS] are the two major spectroscopic techniques used in metabolic analysis. The basic workflow for NMR-based as well as MS-based studies is as follows: quenching/extraction of metabolites → data collection → data processing/analysis (5, 13). NMR exploits the behavior of molecules when placed in a magnetic field, allowing the identification of different nuclei based on their resonant frequency. MS determines the composition of molecules based on the mass-to-charge ratio in charged particles. The resultant metabolite detection and quantification is acquired as a data set called a spectrum. Each technique has distinct advantages and disadvantages (25). For example, LC/MS is highly sensitive, typically at the picogram level, and permits highly specific multiple metabolite identification at low concentrations (21). However, MS sensitivity is dependent on metabolite pK and hydrophobicity (26). Whereas polar molecules may be detected when electrospray ionization is used, nonpolar molecules may require atmospheric pressure chemical ionization. Similarly, the methods of extraction, quenching, and sample storage conditions can affect and potentially modify metabolite structure, thereby confounding already complex data sets and introducing greater sample-to-sample variability. Despite the extensive use of MS to assess small molecules, a widely adopted and validated methodology for sensitive, high-throughput discovery-based LC/MS metabolomics is lacking. Although high-resolution methods exist for gas chromatography (GS)/MS profiling, detectable compounds are limited to those that can be derived, which can be time-consuming, costly, and runs the risk of metabolite loss. Conversely, LC/MS has only recently begun to be applied to metabolic profiling due to advances in chromatography, instrumentation, ionization capabilities, and software. To date, LC/MS–based metabolic profiling experiments have confirmed that metabolic data sets are robust and reproducible and are comparable between different laboratories to reveal pathology-based metabolic differences in human samples (27); these methodologies are currently undergoing validation (28).
Compared with MS, NMR is less sensitive, on the order of 10 μmol/L, and requires more expensive instrumentation. In addition, 1H-NMR spectra are sensitive to pH, ionic content, and temperature, and may require solvent suppression. The major advantages of 1H-NMR include its nonbiased metabolite detection, quantitative nature, and reproducibility. 1H-NMR can also be used for liquid or solid samples, using magic angle spinning (HR-MAS) techniques, with minimal sample preparation (13). 31P-NMR of tissue specimens and cultured cells reflects products of energy or phospholipid metabolism, whereas 13C-NMR measures dynamic carbon fluxes, such as glucose metabolism. 13C-NMR can be performed on tissues and cell extracts following incubation with a 13C-labeled precursor, but is less sensitive than GS/MS–based 13C-assays. Another significant advantage of NMR is that metabolic markers discovered and analyzed in vitro can be measured in vivo, assuming sufficient tissue abundance, using localized magnetic resonance spectroscopy imaging (MRSI). MRSI is an additional technology related to magnetic resonance imaging (MRI) whereby metabolites instead of anatomy are imaged. In essence, MRSI is a composite of traditional NMR spectroscopy and MRI that allows for noninvasive in vivo visualization and determination of spatial distribution of a specific metabolite in patients without exposure to ionizing radiation. Another metabolic imaging alternative is positron emission tomography (PET), which uses radioactively labeled metabolites or their precursors for in vivo imaging. With such tools, preclinical and in vitro assessments can be confirmed in intact living systems.
Data analysis and interpretation
The guiding principle of metabolomics is the global assessment of hundreds of endogenous metabolites in a biological sample simultaneously. Statistical analyses are then applied to provide meaningful information about the metabolic profile of the sample. The three major steps in multivariate metabolomics analyses are depicted in Fig. 2.
Fig. 2.
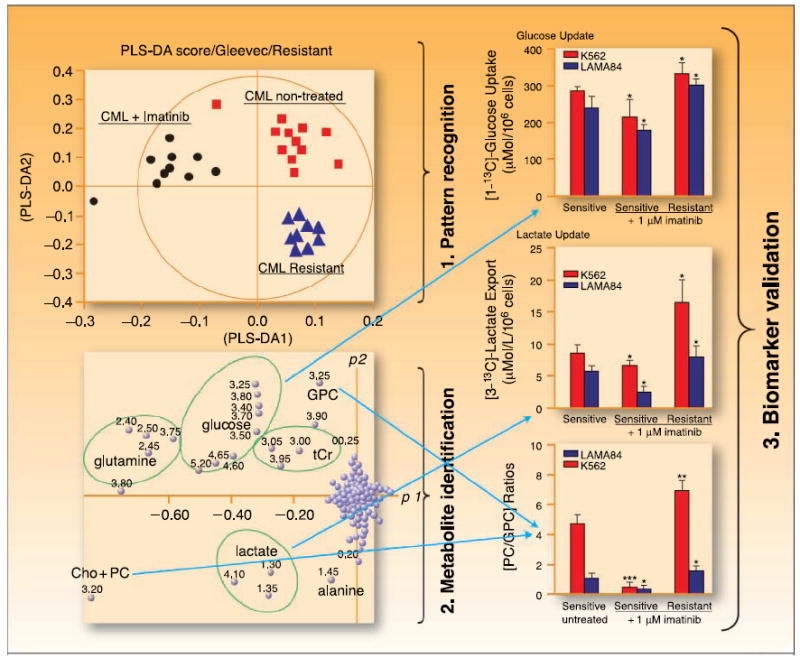
Three major steps of metabolomics analysis. The example is given for imatinib treatment in chronic myeloid leukemia cells using (1) 1H-NMR spectra of cell extracts followed by principal component analysis for pattern recognition → (2) metabolite identification resulting in a biomarker → (3) metabolite quantification and validation. Adapted and reproduced with permission from Thomson Scientific and Serkova NJ, Spratlin JL, Eckhardt SG: NMR-based metabolomics: Translational application and treatment of cancer. Current Opinion in Molecular Therapeutics 2007; 9(6):572–85. Figure 4. ©2007 Thomson Scientific.
Because 1H-NMR or MS spectra from biofluids or tumor tissue contain hundreds of signals from endogenous metabolites and are highly redundant, spectral data sets, reduced to 100 to 500 spectral segments, and their respective signal intensities are directly entered into statistical programs (5, 21, 29). This first step of metabolomics analysis facilitates pattern recognition, or group clustering, such as normal versus cancer or responders versus nonresponders, based on spectral pattern differences. The interpretation of scores reveals information about relationships between samples and illustrates trends, groupings, and/or outliers. In the last 5 years, due to the quantity and complexity of spectroscopic data from NMR and MS studies, the majority of metabolic profiling studies have used computer-aided statistical interpretation of the data. This improves the refining and distilling of complex raw data. Similar to gene array analyses, multivariate statistics have been designed for large data sets, with two major types of pattern recognition processes, unsupervised and supervised. Unsupervised data analysis, such as hierarchical cluster analysis and principal component analysis, measures the innate variation in data sets, whereas the supervised approach, including principal component regression and neural networks, uses prior information to generate the clusters of patterns (30). Although beyond the scope of this review, many other statistical approaches exist, including cluster analysis, linear discriminant analysis, Bayesian spectral decomposition, and several other chemometric methods (31).
In the next step, the specific spectral regions that are responsible for group clustering in step 1 are identified and linked to a specific metabolite based on its NMR chemical shifts. To accomplish this, a database search, for example, at the Human Metabolome Database,3 is often used. The third and final step of analysis includes quantitation and association of putative biomarkers with respect to a particular characteristic or outcome, such as tumor grade or response to therapy. The statistical approach of this step can be represented by a standard Student’s t test or ANOVA, depending on the group number and size.
There has recently been an initiative to form large databases to collect and pool information on the metabolome. A free-access online metabolite database exists that is supported by Genome Alberta and Genome Canada4 (32). This database contains detailed information on nearly 2,500 human metabolites and includes genomic and proteomic links and allows for comparisons of metabolites using search tools for MS and NMR data. Several other databases also exist, or are under construction, including a NIH initiative.5 The goal of these databases is to facilitate links between clinical, chemical, molecular, and biological data that is crucial as metabolomic data accumulates and biomarkers are tested and validated.
Metabolomics as a Biomarker in Oncology
The cancer metabolome
Biomarkers are widely used in clinical medicine for prognostic or predictive purposes. Examples in oncology are the estrogen receptor and Her2/neu status in breast cancer. In general, quantitative metabolomic biomarkers for cancer detection and/or assessment of treatment efficacy are explored preclinically using animal and human cell cultures, followed by validation in biofluid or tumor tissue.
The tumor metabolome is beginning to be characterized. Using standard metabolomic methods, tumors, in general, display elevated phospholipid levels [characterized by an elevation of total choline-containing compounds (tCho) and phosphocholine], increased glycolytic capacity, including increased utilization of glucose carbons to drive synthetic processes, high glutaminolytic function, and overexpression of the glycolytic isoenzyme, pyruvate kinase type M2 (M2-PK; refs. 12, 33, 34). M2-PK may be of particular interest as its inactive dimeric form is dominant in tumors and has been named tumor M2-PK. Interestingly, lipid metabolic profiles have been documented to be 83% accurate at discriminating between cancer patients and controls, using NMR-based metabolomics of blood samples (35). Importantly, in vivo, tCho determination via MRSI has detected breast, prostate, and brain tumors and correlates well with diagnosis via dynamic contrast enhanced-MRI (16, 36-39).
Despite these gains, there are several gaps in knowledge about the tumor metabolome. Among distinct tumor types, profiles vary with respect to many metabolites, including alanine, citrate, glycine, lactate, nucleotides, and lipids, making it difficult to generalize findings across tumor groups (2). There are also technical issues encountered while performing metabolomic analyses that may hinder characterization of a tumor metabolome, including sample-to-sample variation and sensitivity, particularly for extraction-dependent MS-based techniques.
Cancer diagnosis
Pattern recognition technologies in all omics have been used for the diagnosis of several tumor types using a variety of experimental platforms. Perhaps the best application of metabolomics thus far in cancer diagnostics is in breast cancer. Several NMR studies have analyzed breast biopsy samples and have identified over 30 endogenous metabolites in breast tissue. Breast cancers reliably showed elevated tCho levels (resulting from increased phosphocholine), low glycerophosphocholine, and low glucose compared with benign tumors or healthy tissue (17, 40-42). Furthermore, when 91 breast cancers and 48 adjacent normal tissue specimens were examined after surgical resection using HR-MAS 1H-NMR metabolomics, a malignant phenotype could reliably be detected from normal tissue with sensitivity and specificity between 83% and 100% for tumor size, lymph node, and hormonal status, as well as histology (17). In vivo, when MRSI of the breast is performed on patients before biopsy, precise differentiation of cancer and benign tissue is possible based on choline detection, with a sensitivity of 100%. Importantly, a biopsy could have been prevented 68% of the time if only performed on the choline-positive tissue (refs. 36, 43; Fig. 3).
Fig. 3.
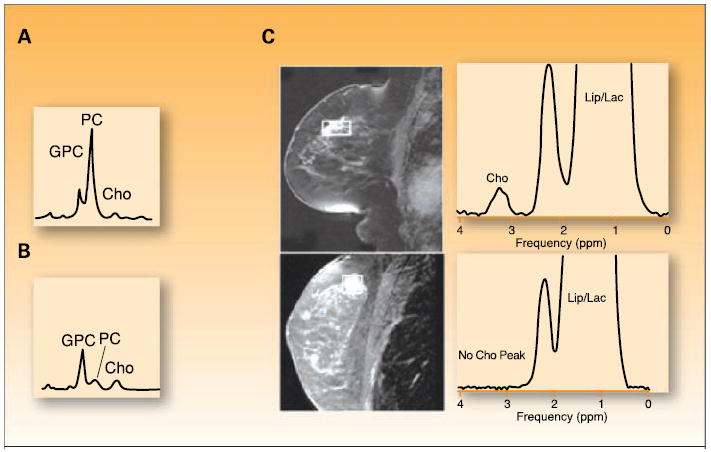
Preclinical to clinical translation of metabolomics discoveries in breast cancer. Proton magnetic resonance spectra distinguishing between (A) orthotopically grown xenograft tumors of malignant human MDA-MB-231 breast cancer and (B) nonmalignant human MCF-12A mammary epithelial cells showing low glycerophosphocholine and high phosphocholine levels in breast cancer compared with high glycerophosphocholine and low phosphocholine in nonmalignant epithelial cells. C, MRSI of a palpable mass in a 56-y-old female, which was a biopsy-proven cancer of the breast with a corresponding Cho peak (top), whereas a suspicious lesion detected at screening MRI in the breast of a 38-y-old female, positive for the BRCA1 gene, shows no spectral Cho peak and was benign at biopsy. GPC, glycerophosphocholine; PC, phosphocholine; tCho, total choline containing metabolites; Cho, choline; Lip, lipid; Lac, lactate. (A and B) Adapted and reproduced with permission from the American Association for Cancer Research (AACR), Inc.: Morvan D, Demidem A. Metabolomics by proton nuclear magnetic resonance spectroscopy of the response to chloroethylnitrosourea reveals drug efficacy and tumor adaptive metabolic pathways. Cancer Research 2007; 67:2150–9. Figure 3. © 2007 AACR, Inc. (C) Reproduced with permission fromThe Radiological Society of North America (RSNA) and Dr. Bartella: Bartella L, Thakur SB, Morris EA, et al. Enhancing nonmass lesions in the breast: evaluation with proton (1H) MR spectroscopy. Radiology 2007; 245:80–7, Figures 3 and 5. © 2007 RSNA.
Similar to breast cancer, prostate cancer exhibits a distinct metabolic profile characterized by high tCho and phosphocholine levels, along with an increase in the glycolytic products lactate and alanine (44, 45). Prostatic fluid from men with prostate cancer exhibit decreased citrate and elevated spermine levels, compared with noncancer patients. Detection of citrate and spermine in prostatic fluid by 1H-NMR correlated well with Gleason score and citrate concentrations in semen or prostatic fluid from 28 cancer patients and 33 normal controls, and outperformed prostate-specific antigen in cancer detection (11, 18). Furthermore, before primary therapy for prostate cancer, MRSI citrate detection increased accuracy and reduced the interobserver variability of determining extracapsular extension, compared with anatomic MRI alone (46, 47).
Data in brain cancers is extensive with defined metabolomic biomarkers established from studies of brain tumor specimens exhibiting discrete 1H-NMR spectra (48, 49). In vitro, cell lines from meningiomas, neuroblastomas, and glioblastomas showed metabolic patterns that reflect differences in alanine, glutamate, creatine, phosphorylcholine, and threonine that are distinct among the histologic subtypes. Clinically, metabolite levels from preoperative MRSI images have been compared with histology results from biopsy specimens in 29 primary glioma patients (50). Interestingly, in this study, the histologic presence of cancer correlated with abnormally elevated tCho and decreased N-acetyl aspartate levels by MRSI. If further validated, MRSI may be able to localize biopsy sites by targeting areas of maximal metabolic abnormality, leading to improved diagnostic accuracy. This approach could result in the ability to “metabolically map” an area for focal ablative procedures.
Metabolomic differences between healthy women and those with epithelial ovarian cancer have been investigated (15). 1H-NMR spectroscopy was done on serum from 38 preoperative epithelial ovarian cancer patients, 12 women with benign ovarian cysts, and 53 samples from healthy women. Serum metabolic profiles correctly separated women with cancer from normal premenopausal women and those with benign ovarian disease in 100% of cases; there was also a 97.4% separation rate for cancer patients versus normal postmenopausal women (Fig. 4). Interestingly, in another study, MS-based metabolic profiling of ovarian tumor tissue showed a statistically significant differentiation between invasive ovarian carcinomas and borderline tumors as reflected by differences in 51 metabolites (P < 0.01; ref. 14). Importantly, the differences noted in these metabolites have previously been linked to prognosis in ovarian cancer and correspond to pathways responsible for regulation of pyrimidine metabolism (51).
Fig. 4.
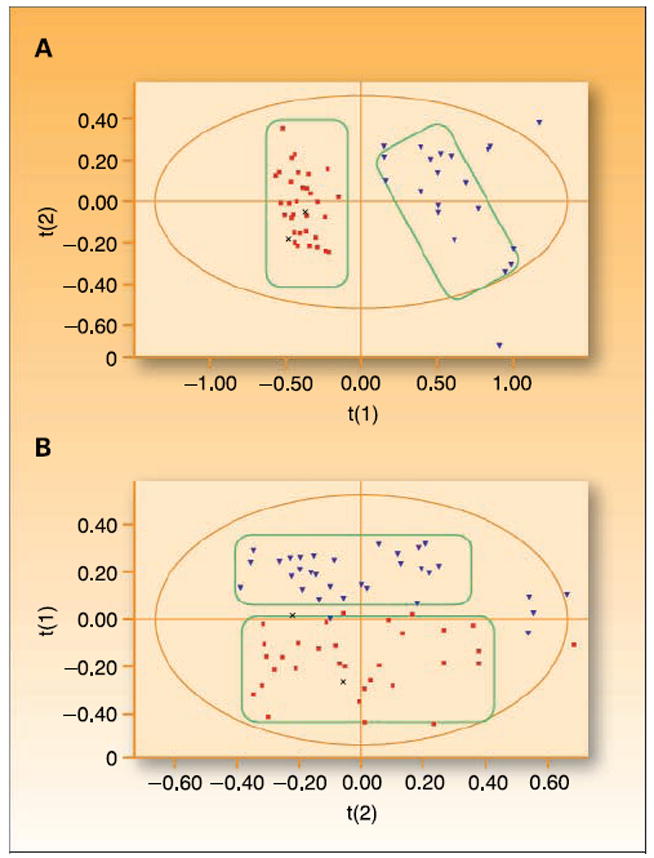
Use of metabolomics for ovarian cancer detection. Use of NMR metabolite detection followed by principal component analysis shows considerable separation between (A) epithelial ovarian cancer (■) and premenopausal women (▼) and (B) epithelial ovarian cancer (■) and postmenopausal women (▼) as depicted by group clustering. Adapted and reproduced with permission of Wiley-Liss, Inc., a subsidiary of JohnWiley & Sons, Inc. Odunsi K, Wollman RM, Ambrosone CB, et al. Detection of epithelial ovarian cancer using 1H-NMR-based metabolomics. Int. J. Cancer; 113:782–88. Figure 3. © 2005 JohnWiley & Sons, Inc.
These results show the potential utility of metabolomics in cancer diagnosis and, in fact, have culminated in the use of MRSI technologies in diagnosing breast and prostate cancer with the cost of these tests being paid for by insurance providers (43, 46).
Assessment of response to traditional therapy
The use of metabolomics for assessment of treatment effect, as both a predictive measure of efficacy and as a pharmacodynamic marker, has been shown in vitro for both traditional chemotherapy and hormonal agents. The use of 1H-NMR on human glioma cell culture successfully predicted separation into drug-resistant and drug-sensitive groups before treatment with 1-(2-chloroethyl)-3-cyclohexyl-1-nitrosourea. Exposure of hormone-responsive Ishikawa human endometrial adenocarcinoma cells to tamoxifen resulted in dose-dependent changes in nucleotides, suggesting that tamoxifen modifies RNA translation (19, 52).
In vivo, 1H-NMR, including HR-MAS, has been used to investigate the metabolic changes associated with nitrosurea treatment of B16 melanoma and 3LL lung carcinoma tumors grown subcutaneously in C57BL6/6J mice (20). During the growth-inhibitory phase, tumor samples showed significant accumulation of glucose, glutamine, aspartate, and serine-derived metabolites, as well as decreased succinate, suggesting the reduction of nucleotide synthesis and induction of DNA repair pathways. Growth recovery reflected metabolic adaptation, including activation of energy production systems and increased nucleotide synthesis (Fig. 5).
Fig. 5.
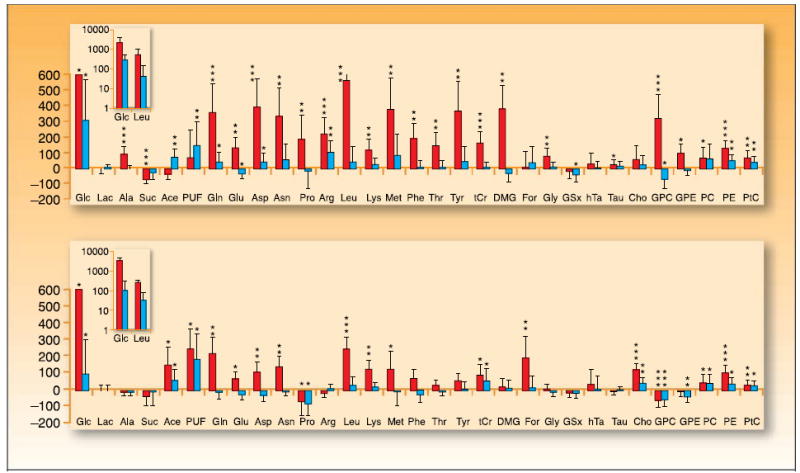
Metabolomic changes as a pharmacodynamic marker of treatment response. Metabolomic profiling of B16 melanoma (top) and 3LL pulmonary carcinoma tumors (bottom) showing variations in multiple metabolites before (black columns) and after (gray columns) chloroethylnitrosurea treatment. Bars, SD. Vertical scale, percentage of change respect to untreated group. Inset, log scale for metabolites with the largest variations. *, P < 0.05; **, P < 0.01; ***, P < 0.001. Glc, glycine; Lac, lactate; Ala, alanine; Suc, succinate; Ace, acetate; PUF, polyunsaturated fatty acid; Gln, glutamine; Glu, glutamate; Asp, aspartate; Pro, proline; Arg, arginine; Leu, leucine; Lys, lysine; Met, methionine;Thr, threonine; Phe, phenylalanine;Tyr, tyronsine; tCr, total creatinine; DMG, dimethylglycine; For, formate; Gly, glycine; hTa, hypotaurine;Tau, taurine; Cho, choline; GPC, glycerophosphocholine; GPE, glycerophosphoethanolamine; PC, phosphocholine; PE, phosphoethanolamine; PtC, phosphotidylcholine. Adapted and reproduced with permission from theAmerican Association for Cancer Research (AACR), Inc.: Glunde K, Jie C, Bhulwalla ZM. Molecular causes of the aberrant choline phospholipid metabolism in breast cancer. Cancer Research 2004; 64:4270–6. Figure 3. © 2007 AACR, Inc.
In prostate cancer, citrate may be a marker of responsiveness to treatment based on a pilot study where 16 high-risk prostate cancer patients were treated with chemotherapy, hormones, radical prostatectomy, or radiotherapy, and subsequently followed with prostate-specific antigen monitoring, MRI, and MRSI (53, 54). A risk score using MRSI was developed based on both tumor volume and metabolic abnormality. The MRSI score and MRI tumor/node stage was then used to determine prostate-specific antigen relapse and was predictive in 15 of 16 cases.
There is an ongoing effort by the National Cancer Institute, researchers, clinicians, and industry to expand the use of metabolomics, with particular attention to MRSI for the assessment of therapeutic response (55). Choline phospholipid metabolism intermediates have been deemed a potential biomarker for monitoring treatment efficacy in a variety of human cancers (reviewed in ref. 12). In general, a decrease in the tCho signal on 1H-NMR equates to a response to chemotherapy or radiation and may be an early marker of effect as it can be detected before changes on conventional imaging in breast and prostate cancer, brain tumors, and non–Hodgkin’s lymphoma.
Novel therapeutics
Therapeutics in oncology is moving toward the use of drugs that specifically target aberrant pathways involved in growth, proliferation, and metastases. Biomarkers are being increasingly used in the early clinical development of such agents to identify, validate, and optimize therapeutic targets and agents; determine and confirm mechanism of drug action, as a pharmacodynamic end point; and in predicting or monitoring responsiveness to treatment, toxicity, and resistance (56). Current examples of using metabolomics in developmental therapeutics are with tyrosine kinase inhibitors, proapoptotic agents, and heat shock protein inhibitors (57-63).
One hypothesis explored was that treatment with targeted therapies, such as signal transduction inhibitors, would result in a distinct metabolic profile between sensitive and resistant cells. Imatinib, a tyrosine kinase inhibitor of the BCR-ABL oncogene, decreases cell proliferation and induces apoptosis in human chronic myeloid leukemia (64-66). Metabolically, imatinib interrupts the synthesis of macromolecules required for cell survival by deprivation of key substrates (58). Investigating glucose metabolism changes in imatinib-treated human leukemia BCR-ABL – positive cell lines with NMR showed decreased glucose uptake by inhibition of glycolysis, but unlike classic therapeutics, stimulated mitochondrial metabolism leading to cell differentiation (57). Imatinib also led to a significant decrease in phosphocholine in imatinib-sensitive cells that correlated with a decrease in cell proliferation rate (57). Metabolomic detection of imatinib resistance has also been reported; a decrease in mitochondrial glucose oxidation and a nonoxidative ribose synthesis from glucose, as well as highly elevated phosphocholine levels, was indicative of drug resistance and disease progression (58). These data indicate that NMR metabolomics may provide a method for monitoring changes in cellular metabolism that reflect early resistance to novel targeted agents. This could be particularly useful in hematologic malignancies where frequent tissue sampling is feasible and early metabolomic markers of resistance may dictate therapy adjustments that prevent overt phenotypic progression.
Apoptosis has an established role in chemotherapy and radiation-induced cell death and its absence correlates with treatment resistance and induction of prosurvival pathways (67, 68). Multiple new agents targeting apoptosis are currently in early clinical development, including tumor necrosis factor–related apoptosis-inducing ligand, agonist death receptor antibodies, and inhibitors of the antiapoptotic proteins. FK866 is a novel agent that purportedly induces apoptosis independent of anti-DNA effects by decreasing NAD+ levels (59). The metabolic effects of FK866 on mouse mammary carcinoma cells using 1H-NMR showed a significant increase in fructose-biphosphate with a subsequent decrease in pH and NAD+ resulting from an incomplete glycolytic cycle (60). Other alterations observed included changes in guanylate synthesis, pyridine nucleotide levels, and phospholipid metabolism, indicating there are multiple aberrant cellular pathways resulting in apoptosis (60). High-resolution MAS-NMR has showed that apoptotic activity can be characterized in cervical carcinoma biopsies before and during treatment with external beam radiation, brachytherapy, and weekly chemotherapy (61). In this study, 44 cervical cancer biopsies indicated that lipid metabolism differed both in tumor cell fraction (percentage of tumor cells per tissue biopsy) and tumor cell density (number of carcinoma cell nuclei per mm2 of tumor tissue). Ratios of fatty acid -CH2 to -CH3, specifically, a lengthening of the fatty acid -CH2 chain, were associated with apoptosis, a finding substantiated in another study of acute lymphoblastic leukemia cell cultures treated with doxorubicin (62). These early studies suggest that metabolomics may be a useful biomarker in the development and validation of proapoptotic agents.
Another interesting application of metabolomics is in the area of heat shock protein 90 (Hsp90) inhibitors. Although their mechanism of action is not fully elucidated, current data suggest that this family of agents increase the cellular destruction of client oncogenic proteins (69). In one study, colon cancer xenografts were treated with an Hsp90 inhibitor and extracts of these tumors were analyzed by 31P-NMR, reflecting a significant increase in phosphocholine, phosphomonoester/phosphodiester ratio, valine, and phosphoethanolamine levels, indicating altered phospholipid metabolism (63). These results, although preliminary, address that metabolic changes could be used as pharmacodynamic biomarkers of Hsp90 inhibitors, a class of agents that do not seem to result in classic antitumor effects.
The Future of Metabolomics in Oncology
Metabolomics is a novel discipline encompassing comprehensive metabolite evaluation, pattern recognition, and statistical analyses. Biomarkers are widely used in clinical medicine for prognostic or predictive interpretation of disease status. Metabolomics should be used for identifying multivariate biomarkers, including fingerprints, profiles, or signatures, the patterns of which characterize a state of cancer. By using this technology, we might eventually be able to diagnose cancer earlier when it is still amenable to cure, determine aggressiveness of cancer to help direct prognosis and therapy, and predict drug efficacy. These signatures can be practical and accurate although they also require sophisticated analytic techniques (70, 71).
The use of metabolomics as a diagnostic tool has been validated using citrate and choline in prostate and breast cancer, respectively, both of which are now covered by health insurance providers (11, 43, 46). The precedent for this type of omics technology includes colon cancer diagnosis and prognosis by gene microarrays and ovarian cancer diagnosed with serum protein profiles (72, 73). Conceivably, metabolomics may play a role in tumors posing a diagnostic challenge. Evidence suggests that metabolomic profiles are already used in diagnosing ovarian cancer by analyzing either serum or tumor tissue (14, 15). Detection of pancreatic cancer has also been possible in vivo by analyzing cellular glucose use via GS/MS (7). Future studies should evaluate the use of bodily fluids, for example ascitic fluid in ovarian cancer, pancreatic secretions in pancreatic cancer, and/or bronchoalveolar or pleural fluid in lung cancer. If pathognomonic profiles can be identified and validated in these fluids, metabolomics may save time, cost, and effort in obtaining a definitive diagnosis in situations where no other test can provide answers. Additionally, there could be a future role for metabolomics as a screening tool, particularly in those tumors that readily produce or secrete easily accessible fluid.
Similar to other strategies currently being investigated to individualize therapy, such as the assessment of mutations or amplification of receptor tyrosine kinase genes in GIST, metabolomic studies should be integrated into preclinical and clinical research and assessed for predictive value (74-76). A form of in vivo metabolomics, PET imaging, with the use of radioactive glucose, choline, or thymidine as metabolic end points, has already been evaluated as a predictor of drug efficacy in some tumor types. In recurrent GIST, compared with standard computed tomography scanning, [18F]fluor-deoxyglucose (FDG) PET was superior in predicting early response to imatinib therapy when evaluated in 56 patients before and after initiating imatinib therapy (77, 78). Furthermore, changes on [18F]FDG PET have been predictive of response to standard cytotoxic treatments in patients with breast cancer, locally advanced or metastatic non–small cell lung cancer, ovarian cancer, after high-dose salvage chemotherapy in relapsed germ-cell cancer, and in treatment-naïve patients with cervical cancer (79-83). In hematologic malignancies not amenable to [18F]FDG PET imaging, metabolomic analysis on circulating tumor cells after [13C]glucose administration could be used in assessing treatment effects, thereby providing biological response information noninvasively. This could also be applied to circulating tumor cells from solid tumors.
The principal objectives of early clinical trials are to determine the maximum tolerated dose of new drugs or drug combinations while also collecting information on drug tolerability, pharmacokinetics, and pharmacodynamics. Increasingly, biomarkers are being used preclinically and in early clinical development to identify, validate, and optimize therapeutic targets, to confirm mechanism of drug action, and as pharmacodynamic end points (56). As discussed earlier, metabolomics is already being assessed as a pharmacodynamic marker of novel agents, whereas another application is in the characterization of toxic effects. For example, metabolomics can be used as a biomarker of hepatic, renal, and lung toxicity with various metabolites, including glucose, lactate, lipoproteins, and amino acids, increasing or decreasing providing a recognizable pattern associated with organ dysfunction (84-91). Much of these data have not been validated and there is some overlap between various toxins but the pattern, temporal rate of change, and extent of change in metabolites can still provide toxicity assessments (13). Such patterns may be used for preclinical drug screening or as a means of following a patient clinically to monitor target organ effects.
Although metabolomic technology has improved and evidence is accumulating to support its use in clinical decision making, the discipline is still in its infancy and metabolomics has somewhat lagged behind other omic sciences due to technical limitations, database challenges, and costs. Future development and application will be dependent on several factors, such as the establishment of spectral databases of metabolites and associated biochemical identities, as well as cross-validation of NMR- or MS-obtained metabolites and correlation with other quantitative assays. Lastly, it will be important to integrate the results of metabolomic assessments with other omics technology so that the entire spectrum of the malignant phenotype can be characterized.
Acknowledgments
We thank Shana Spears for her help with figure editing.
Grant support: J. Spratlin is a Developmental Therapeutics/GI Oncology Senior Fellow at the University of Colorado at Denver being mentored by S. Gail Eckhardt. Her fellowship is funded by the National Cancer Institute of Canada, through a research grant supported by The Terry Fox Foundation, and by the Alberta Heritage Foundation for Medical Research.
Footnotes
Disclosure of Potential Conflicts of Interest No potential conflicts of interest were disclosed.
References
- 1.Fiehn O. Metabolomics—the link between genotypes and phenotypes. Plant Mol Biol. 2002;48:155–71. [PubMed] [Google Scholar]
- 2.Griffin JL, Shockcor JP. Metabolic profiles of cancer cells. Nat Rev Cancer. 2004;4:551–61. doi: 10.1038/nrc1390. [DOI] [PubMed] [Google Scholar]
- 3.Kell DB, Mendes P. Snapshots of systems: metabolic control analysis and biotechnology in the post-genomic era. In: Cornish-Bowden A, Cardenas ML, editors. Technological and medical implications of metabolic control analysis. Dordrecht (The Netherlands): Kluwer Academic Publishers; 2000. pp. 3–25. [Google Scholar]
- 4.Ryan D, Robards K. Metabolomics: The greatest omics of them all? Anal Chem. 2006;78:7954–8. doi: 10.1021/ac0614341. [DOI] [PubMed] [Google Scholar]
- 5.Dunn WB, Bailey NJ, Johnson HE. Measuring the metabolome: current analytical technologies. Analyst. 2005;130:606–25. doi: 10.1039/b418288j. [DOI] [PubMed] [Google Scholar]
- 6.Roessner U, Luedemann A, Brust D, et al. Metabolic profiling allows comprehensive phenotyping of genetically or environmentally modified plant systems. Plant Cell. 2001;13:11–29. doi: 10.1105/tpc.13.1.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boros LG, Lerner MR, Morgan DL, et al. [1,2-13C2]-d-glucose profiles of the serum, liver, pancreas, and DMBA-induced pancreatic tumors of rats. Pancreas. 2005;31:337–43. doi: 10.1097/01.mpa.0000186524.53253.fb. [DOI] [PubMed] [Google Scholar]
- 8.Mendes P, Kell DB, Westerhoff HV. Why and when channeling can decrease pool size at constant net flux in a simple dynamic channel. Biochim Biophys Acta. 1996;1289:175–86. doi: 10.1016/0304-4165(95)00152-2. [DOI] [PubMed] [Google Scholar]
- 9.Mendes P, Kell DB, Westerhoff HV. Channelling can decrease pool size. Eur J Biochem. 1992;204:257–66. doi: 10.1111/j.1432-1033.1992.tb16632.x. [DOI] [PubMed] [Google Scholar]
- 10.Fossel ET, Carr JM, McDonagh J. Detection of malignant tumors. Water-suppressed proton nuclear magnetic resonance spectroscopy of plasma. N Engl J Med. 1986;315:1369–76. doi: 10.1056/NEJM198611273152201. [DOI] [PubMed] [Google Scholar]
- 11.Serkova NJ, Spratlin JL, Eckhardt SG. NMR-based metabolomics: translational application and treatment of cancer. Curr Opin Mol Ther. 2007;9:572–85. [PubMed] [Google Scholar]
- 12.Glunde K, Serkova NJ. Therapeutic targets and biomarkers identified in cancer choline phospholipid metabolism. Pharmacogenomics. 2006;7:1109–23. doi: 10.2217/14622416.7.7.1109. [DOI] [PubMed] [Google Scholar]
- 13.Serkova NJ, Niemann CU. Pattern recognition and biomarker validation using quantitative 1H-NMR-based metabolomics. Expert Rev Mol Diagn. 2006;6:717–31. doi: 10.1586/14737159.6.5.717. [DOI] [PubMed] [Google Scholar]
- 14.Denkert C, Budczies J, Kind T, et al. Mass spectrometry-based metabolic profiling reveals different metabolite patterns in invasive ovarian carcinomas and ovarian borderline tumors. Cancer Res. 2006;66:10795–804. doi: 10.1158/0008-5472.CAN-06-0755. [DOI] [PubMed] [Google Scholar]
- 15.Odunsi K, Wollman RM, Ambrosone CB, et al. Detection of epithelial ovarian cancer using 1H-NMR-based metabonomics. Int J Cancer. 2005;113:782–8. doi: 10.1002/ijc.20651. [DOI] [PubMed] [Google Scholar]
- 16.Howe FA, Barton SJ, Cudlip SA, et al. Metabolic profiles of human brain tumors using quantitative in vivo1H magnetic resonance spectroscopy. Magn Reson Med. 2003;49:223–32. doi: 10.1002/mrm.10367. [DOI] [PubMed] [Google Scholar]
- 17.Bathen TF, Jensen LR, Sitter B, et al. MR-determined metabolic phenotype of breast cancer in prediction of lymphatic spread, grade, and hormone status. Breast Cancer Res Treat. 2007;104:181–9. doi: 10.1007/s10549-006-9400-z. [DOI] [PubMed] [Google Scholar]
- 18.Kline EE, Treat EG, Averna TA, et al. Citrate concentrations in human seminal fluid and expressed prostatic fluid determined via 1H nuclear magnetic resonance spectroscopy outperform prostate specific antigen in prostate cancer detection. J Urol. 2006;176:2274–9. doi: 10.1016/j.juro.2006.07.054. [DOI] [PubMed] [Google Scholar]
- 19.El-Deredy W, Ashmore SM, Branston NM, et al. Pretreatment prediction of the chemotherapeutic response of human glioma cell cultures using nuclear magnetic resonance spectroscopy and artificial neural networks. Cancer Res. 1997;57:4196–9. [PubMed] [Google Scholar]
- 20.Morvan D, Demidem A. Metabolomics by proton nuclear magnetic resonance spectroscopy of the response to chloroethylnitrosourea reveals drug efficacy and tumor adaptive metabolic pathways. Cancer Res. 2007;67:2150–9. doi: 10.1158/0008-5472.CAN-06-2346. [DOI] [PubMed] [Google Scholar]
- 21.Dettmer K, Aronov PA, Hammock BD. Mass spectrometry-based metabolomics. Mass Spectrom Rev. 2007;26:51–78. doi: 10.1002/mas.20108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shulaev V. Metabolomics technology and bioinformatics. Brief Bioinform. 2006;7:128–39. doi: 10.1093/bib/bbl012. [DOI] [PubMed] [Google Scholar]
- 23.Reo NV. NMR-based metabolomics. Drug Chem Toxicol. 2002;25:375–82. doi: 10.1081/dct-120014789. [DOI] [PubMed] [Google Scholar]
- 24.Aranibar N, Ott KH, Roongta V, et al. Metabolomic analysis using optimized NMR and statistical methods. Anal Biochem. 2006;355:62–70. doi: 10.1016/j.ab.2006.04.014. [DOI] [PubMed] [Google Scholar]
- 25.Pan Z, Raftery D. Comparing and combining NMR spectroscopy and mass spectrometry in metabolomics. Anal Bioanal Chem. 2007;387:525–7. doi: 10.1007/s00216-006-0687-8. [DOI] [PubMed] [Google Scholar]
- 26.Want EJ, Nordstrom A, Morita H, et al. From exogenous to endogenous: the inevitable imprint of mass spectrometry in metabolomics. J Proteome Res. 2007;6:459–68. doi: 10.1021/pr060505+. [DOI] [PubMed] [Google Scholar]
- 27.Want EJ, O’Maille G, Smith CA, et al. Solvent-dependent metabolite distribution, clustering, and protein extraction for serum profiling with mass spectrometry. Anal Chem. 2006;78:743–52. doi: 10.1021/ac051312t. [DOI] [PubMed] [Google Scholar]
- 28.Gika HG, Theodoridis GA, Wingate JE, et al. Within-day reproducibility of an HPLC-MS-based method for metabonomic analysis: application to human urine. J Proteome Res. 2007;6:3291–303. doi: 10.1021/pr070183p. [DOI] [PubMed] [Google Scholar]
- 29.Buchholz A, Hurlebaus J, Wandrey C, et al. Metabolomics: quantification of intracellular metabolite dynamics. Biomol Eng. 2002;19:5–15. doi: 10.1016/s1389-0344(02)00003-5. [DOI] [PubMed] [Google Scholar]
- 30.Lee JK, Williams PD, Cheon S. Data mining in genomics. Clin Lab Med. 2008;28:145–66. doi: 10.1016/j.cll.2007.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Holmes E, Antti H. Chemometric contributions to the evolution of metabonomics: mathematical solutions to characterising and interpreting complex biological NMR spectra. Analyst. 2002;127:1549–57. doi: 10.1039/b208254n. [DOI] [PubMed] [Google Scholar]
- 32.Wishart D. HMDB: human metabolome database. Nucleic Acids Research. 2007;35:D521–6. doi: 10.1093/nar/gkl923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mazurek S, Eigenbrodt E. The tumor metabolome. Anticancer Res. 2003;23:1149–54. [PubMed] [Google Scholar]
- 34.Ackerstaff E, Glunde K, Bhujwalla ZM. Choline phospholipid metabolism: a target in cancer cells? J Cell Biochem. 2003;90:525–33. doi: 10.1002/jcb.10659. [DOI] [PubMed] [Google Scholar]
- 35.Bathen TF, Engan T, Krane J, et al. Analysis and classification of proton NMR spectra of lipoprotein fractions from healthy volunteers and patients with cancer or CHD. Anticancer Res. 2000;20:2393–408. [PubMed] [Google Scholar]
- 36.Jacobs MA, Barker PB, Bottomley PA, et al. Proton magnetic resonance spectroscopic imaging of human breast cancer: a preliminary study. J Magn Reson Imaging. 2004;19:68–75. doi: 10.1002/jmri.10427. [DOI] [PubMed] [Google Scholar]
- 37.Yeung DK, Cheung HS, Tse GM. Human breast lesions: characterization with contrast-enhanced in vivo proton MR spectroscopy—initial results. Radiology. 2001;220:40–6. doi: 10.1148/radiology.220.1.r01jl0240. [DOI] [PubMed] [Google Scholar]
- 38.Bolan PJ, Meisamy S, Baker EH, et al. In vivo quantification of choline compounds in the breast with 1H MR spectroscopy. Magn Reson Med. 2003;50:1134–43. doi: 10.1002/mrm.10654. [DOI] [PubMed] [Google Scholar]
- 39.Stanwell P, Gluch L, Clark D, et al. Specificity of choline metabolites for in vivo diagnosis of breast cancer using 1H MRS at 1.5 T. Eur Radiol. 2005;15:1037–43. doi: 10.1007/s00330-004-2475-1. [DOI] [PubMed] [Google Scholar]
- 40.Gribbestad IS, Sitter B, Lundgren S, et al. Metabolite composition in breast tumors examined by proton nuclear magnetic resonance spectroscopy. Anticancer Res. 1999;19:1737–46. [PubMed] [Google Scholar]
- 41.Sitter B, Lundgren S, Bathen TF, et al. Comparison of HR MAS MR spectroscopic profiles of breast cancer tissue with clinical parameters. NMR Biomed. 2006;19:30–40. doi: 10.1002/nbm.992. [DOI] [PubMed] [Google Scholar]
- 42.Glunde K, Jie C, Bhujwalla ZM. Molecular causes of the aberrant choline phospholipid metabolism in breast cancer. Cancer Res. 2004;64:4270–6. doi: 10.1158/0008-5472.CAN-03-3829. [DOI] [PubMed] [Google Scholar]
- 43.Bartella L, Thakur SB, Morris EA, et al. Enhancing nonmass lesions in the breast: evaluation with proton (1H) MR spectroscopy. Radiology. 2007;245:80–7. doi: 10.1148/radiol.2451061639. [DOI] [PubMed] [Google Scholar]
- 44.Cheng LL, Wu C, Smith MR, et al. Non-destructive quantitation of spermine in human prostate tissue samples using HRMAS 1H NMR spectroscopy at 9.4 T. FEBS Lett. 2001;494:112–6. doi: 10.1016/s0014-5793(01)02329-8. [DOI] [PubMed] [Google Scholar]
- 45.Swanson MG, Zektzer AS, Tabatabai ZL, et al. Quantitative analysis of prostate metabolites using 1H HR-MAS spectroscopy. Magn Reson Med. 2006;55:1257–64. doi: 10.1002/mrm.20909. [DOI] [PubMed] [Google Scholar]
- 46.Scheidler J, Hricak H, Vigneron DB, et al. Prostate cancer: localization with three-dimensional proton MR spectroscopic imaging – clinicopathologic study. Radiology. 1999;213:473–80. doi: 10.1148/radiology.213.2.r99nv23473. [DOI] [PubMed] [Google Scholar]
- 47.Yu KK, Scheidler J, Hricak H, et al. Prostate cancer: prediction of extracapsular extension with endorectal MR imaging and three-dimensional proton MR spectroscopic imaging. Radiology. 1999;213:481–8. doi: 10.1148/radiology.213.2.r99nv26481. [DOI] [PubMed] [Google Scholar]
- 48.Maxwell RJ, Martinez-Perez I, Cerdan S, et al. Pattern recognition analysis of 1H NMR spectra from perchloric acid extracts of human brain tumor biopsies. Magn Reson Med. 1998;39:869–77. doi: 10.1002/mrm.1910390604. [DOI] [PubMed] [Google Scholar]
- 49.Florian CL, Preece NE, Bhakoo KK, et al. Cell type-specific fingerprinting of meningioma and meningeal cells by proton nuclear magnetic resonance spectroscopy. Cancer Res. 1995;55:420–7. [PubMed] [Google Scholar]
- 50.Dowling C, Bollen AW, Noworolski SM, et al. Pre-operative proton MR spectroscopic imaging of brain tumors: correlation with histopathologic analysis of resection specimens. AJNR Am J Neuroradiol. 2001;22:604–12. [PMC free article] [PubMed] [Google Scholar]
- 51.Fujiwaki R, Hata K, Nakayama K, et al. Gene expression for dihydropyrimidine dehydrogenase and thymidine phosphorylase influences outcome in epithelial ovarian cancer. J Clin Oncol. 2000;18:3946–51. doi: 10.1200/JCO.2000.18.23.3946. [DOI] [PubMed] [Google Scholar]
- 52.Griffin JL, Pole JC, Nicholson JK, et al. Cellular environment of metabolites and a metabonomic study of tamoxifen in endometrial cells using gradient high resolution magic angle spinning 1H NMR spectroscopy. Biochim Biophys Acta. 2003;1619:151–8. doi: 10.1016/s0304-4165(02)00475-0. [DOI] [PubMed] [Google Scholar]
- 53.Mueller-Lisse UG, Swanson MG, Vigneron DB, et al. Time-dependent effects of hormone-deprivation therapy on prostate metabolism as detected by combined magnetic resonance imaging and 3D magnetic resonance spectroscopic imaging. Magn Reson Med. 2001;46:49–57. doi: 10.1002/mrm.1159. [DOI] [PubMed] [Google Scholar]
- 54.Pucar D, Koutcher JA, Shah A, et al. Preliminary assessment of magnetic resonance spectroscopic imaging in predicting treatment outcome in patients with prostate cancer at high risk for relapse. Clin Prostate Cancer. 2004;3:174–81. doi: 10.3816/cgc.2004.n.028. [DOI] [PubMed] [Google Scholar]
- 55.Evelhoch J, Garwood M, Vigneron D, et al. Expanding the use of magnetic resonance in the assessment of tumor response to therapy: workshop report. Cancer Res. 2005;65:7041–4. doi: 10.1158/0008-5472.CAN-05-0674. [DOI] [PubMed] [Google Scholar]
- 56.Park JW, Kerbel RS, Kelloff GJ, et al. Rationale for biomarkers and surrogate end points in mechanism-driven oncology drug development. Clin Cancer Res. 2004;10:3885–96. doi: 10.1158/1078-0432.CCR-03-0785. [DOI] [PubMed] [Google Scholar]
- 57.Gottschalk S, Anderson N, Hainz C, et al. Imatinib (STI571)-mediated changes in glucose metabolism in human leukemia BCR-ABL-positive cells. Clin Cancer Res. 2004;10:6661–8. doi: 10.1158/1078-0432.CCR-04-0039. [DOI] [PubMed] [Google Scholar]
- 58.Serkova N, Boros LG. Detection of resistance to imatinib by metabolic profiling: clinical and drug development implications. Am J Pharmacogenomics. 2005;5:293–302. doi: 10.2165/00129785-200505050-00002. [DOI] [PubMed] [Google Scholar]
- 59.Hasmann M, Schemainda I. FK866, a highly specific noncompetitive inhibitor of nicotinamide phosphori-bosyltransferase, represents a novel mechanism for induction of tumor cell apoptosis. Cancer Res. 2003;63:7436–42. [PubMed] [Google Scholar]
- 60.Muruganandham M, Alfieri AA, Matei C, et al. Metabolic signatures associated with a NAD synthesis inhibitor-induced tumor apoptosis identified by 1H-decoupled-31P magnetic resonance spectroscopy. Clin Cancer Res. 2005;11:3503–13. doi: 10.1158/1078-0432.CCR-04-1399. [DOI] [PubMed] [Google Scholar]
- 61.Lyng H, Sitter B, Bathen TF, et al. Metabolic mapping by use of high-resolution magic angle spinning 1H MR spectroscopy for assessment of apoptosis in cervical carcinomas. BMC Cancer. 2007;7:11. doi: 10.1186/1471-2407-7-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Blankenberg FG, Katsikis PD, Storrs RW, et al. Quantitative analysis of apoptotic cell death using proton nuclear magnetic resonance spectroscopy. Blood. 1997;89:3778–86. [PubMed] [Google Scholar]
- 63.Chung YL, Troy H, Banerji U, et al. Magnetic resonance spectroscopic pharmacodynamic markers of the heat shock protein 90 inhibitor 17-allylamino,17-demethoxygeldanamycin (17AAG) in human colon cancer models. J Natl Cancer Inst. 2003;95:1624–33. doi: 10.1093/jnci/djg084. [DOI] [PubMed] [Google Scholar]
- 64.Fang G, Kim CN, Perkins CL, et al. CGP57148B (STI-571) induces differentiation and apoptosis and sensitizes Bcr-Abl-positive human leukemia cells to apoptosis due to antileukemic drugs. Blood. 2000;96:2246–53. [PubMed] [Google Scholar]
- 65.Deininger MW, Goldman JM, Lydon N, et al. The tyrosine kinase inhibitor CGP57148B selectively inhibits the growth of BCR-ABL-positive cells. Blood. 1997;90:3691–8. [PubMed] [Google Scholar]
- 66.Vigneri P, Wang JY. Induction of apoptosis in chronic myelogenous leukemia cells through nuclear entrapment of BCR-ABL tyrosine kinase. Nat Med. 2001;7:228–34. doi: 10.1038/84683. [DOI] [PubMed] [Google Scholar]
- 67.Schmitt CA, Lowe SW. Apoptosis and therapy. J Pathol. 1999;187:127–37. doi: 10.1002/(SICI)1096-9896(199901)187:1<127::AID-PATH251>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 68.Hockel M, Schlenger K, Hockel S, et al. Hypoxic cervical cancers with low apoptotic index are highly aggressive. Cancer Res. 1999;59:4525–8. [PubMed] [Google Scholar]
- 69.Neckers L. Heat shock protein 90: the cancer chaperone. J Biosci. 2007;32:517–30. doi: 10.1007/s12038-007-0051-y. [DOI] [PubMed] [Google Scholar]
- 70.van der Greef J, Stroobant P, van der Heijden R. The role of analytical sciences in medical systems biology. Curr Opin Chem Biol. 2004;8:559–65. doi: 10.1016/j.cbpa.2004.08.013. [DOI] [PubMed] [Google Scholar]
- 71.Ardekani AM, Liotta LA, Petricoin EF. Clinical potential of proteomics in the diagnosis of ovarian cancer. Expert Rev Mol Diagn. 2002;2:312–20. doi: 10.1586/14737159.2.4.312. [DOI] [PubMed] [Google Scholar]
- 72.Buckhaults P, Rago C, St Croix B, et al. Secreted and cell surface genes expressed in benign and malignant colorectal tumors. Cancer Res. 2001;61:6996–7001. [PubMed] [Google Scholar]
- 73.Petricoin EF, Ardekani AM, Hitt BA, et al. Use of proteomic patterns in serum to identify ovarian cancer. Lancet. 2002;359:572–7. doi: 10.1016/S0140-6736(02)07746-2. [DOI] [PubMed] [Google Scholar]
- 74.Prenen H, Cools J, Mentens N, et al. Efficacy of the kinase inhibitor SU11248 against gastrointestinal stromal tumor mutants refractory to imatinib mesylate. Clin Cancer Res. 2006;12:2622–7. doi: 10.1158/1078-0432.CCR-05-2275. [DOI] [PubMed] [Google Scholar]
- 75.Weisberg E, Wright RD, Jiang J, et al. Effects of PKC412, nilotinib, and imatinib against GIST-associated PDGFRA mutants with differential imatinib sensitivity. Gastroenterology. 2006;131:1734–42. doi: 10.1053/j.gastro.2006.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Heinrich MC, Maki RJ, Corless CL, et al. Sunitinib (SU) response in imatinib-resistant (IM-R) GIST correlates with KIT and PDGFRA mutation status. J Clin Oncol; 2006 ASCO Annual Meeting Proceedings Part I; 2006. p. 9502. [Google Scholar]
- 77.Gayed I, Vu T, Iyer R, et al. The role of 18F-FDG PET in staging and early prediction of response to therapy of recurrent gastrointestinal stromal tumors. J Nucl Med. 2004;45:17–21. [PubMed] [Google Scholar]
- 78.Holdsworth CH, Badawi RD, Manola JB, et al. CT and PET: early prognostic indicators of response to imatinib mesylate in patients with gastrointestinal stromal tumor. AJR Am J Roentgenol. 2007;189:W324–30. doi: 10.2214/AJR.07.2496. [DOI] [PubMed] [Google Scholar]
- 79.Pio BS, Park CK, Pietras R, et al. Usefulness of 3′-[F-18]fluoro-3′-deoxythymidine with positron emission tomography in predicting breast cancer response to therapy. Mol Imaging Biol. 2006;8:36–42. doi: 10.1007/s11307-005-0029-9. [DOI] [PubMed] [Google Scholar]
- 80.de Geus-Oei LF, van der Heijden HF, Corstens FH, et al. Predictive and prognostic value of FDG-PET in nonsmall-cell lung cancer: a systematic review. Cancer. 2007;110:1654–64. doi: 10.1002/cncr.22979. [DOI] [PubMed] [Google Scholar]
- 81.Avril N, Sassen S, Schmalfeldt B, et al. Prediction of response to neoadjuvant chemotherapy by sequential F-18-fluorodeoxyglucose positron emission tomography in patients with advanced-stage ovarian cancer. J Clin Oncol. 2005;23:7445–53. doi: 10.1200/JCO.2005.06.965. [DOI] [PubMed] [Google Scholar]
- 82.Bokemeyer C, Kollmannsberger C, Oechsle K, et al. Early prediction of treatment response to high-dose salvage chemotherapy in patients with relapsed germ cell cancer using [(18)F]FDG PET. Br J Cancer. 2002;86:506–11. doi: 10.1038/sj.bjc.6600122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kidd EA, Siegel BA, Dehdashti F, et al. The standardized uptake value for F-18 fluorodeoxyglucose is a sensitive predictive biomarker for cervical cancer treatment response and survival. Cancer. 2007;110:1738–44. doi: 10.1002/cncr.22974. [DOI] [PubMed] [Google Scholar]
- 84.Robertson DG, Reily MD, Sigler RE, et al. Metabonomics: evaluation of nuclear magnetic resonance (NMR) and pattern recognition technology for rapid in vivo screening of liver and kidney toxicants. Toxicol Sci. 2000;57:326–37. doi: 10.1093/toxsci/57.2.326. [DOI] [PubMed] [Google Scholar]
- 85.Nicholls AW, Holmes E, Lindon JC, et al. Metabonomic investigations into hydrazine toxicity in the rat. Chem Res Toxicol. 2001;14:975–87. doi: 10.1021/tx000231j. [DOI] [PubMed] [Google Scholar]
- 86.Holmes E, Bonner FW, Nicholson JK. Comparative studies on the nephrotoxicity of 2-bromoethanamine hydrobromide in the Fischer 344 rat and the multimammate desert mouse (Mastomys natalensis) Arch Toxicol. 1995;70:89–95. doi: 10.1007/BF02733668. [DOI] [PubMed] [Google Scholar]
- 87.Holmes E, Nicholson JK, Tranter G. Metabonomic characterization of genetic variations in toxicological and metabolic responses using probabilistic neural networks. Chem Res Toxicol. 2001;14:182–91. doi: 10.1021/tx000158x. [DOI] [PubMed] [Google Scholar]
- 88.Lenz EM, Bright J, Knight R, et al. Cyclosporin A-induced changes in endogenous metabolites in rat urine: a metabonomic investigation using high field 1H NMR spectroscopy, HPLC-TOF/MS and chemometrics. J Pharm Biomed Anal. 2004;35:599–608. doi: 10.1016/j.jpba.2004.02.013. [DOI] [PubMed] [Google Scholar]
- 89.Azmi J, Connelly J, Holmes E, et al. Characterization of the biochemical effects of 1-nitronaphthalene in rats using global metabolic profiling by NMR spectroscopy and pattern recognition. Biomarkers. 2005;10:401–16. doi: 10.1080/13547500500309259. [DOI] [PubMed] [Google Scholar]
- 90.Coen M, Lenz EM, Nicholson JK, et al. An integrated metabonomic investigation of acetaminophen toxicity in the mouse using NMR spectroscopy. Chem Res Toxicol. 2003;16:295–303. doi: 10.1021/tx0256127. [DOI] [PubMed] [Google Scholar]
- 91.Beckwith-Hall BM, Nicholson JK, Nicholls AW, et al. Nuclear magnetic resonance spectroscopic and principal components analysis investigations into biochemical effects of three model hepatotoxins. Chem Res Toxicol. 1998;11:260–72. doi: 10.1021/tx9700679. [DOI] [PubMed] [Google Scholar]