

Abstract
Analogs of the TRα-specific thyromimetic CO23 were synthesized and analyzed in vitro using competitive binding and transactivation assays. Like CO23, all analogs bind to both thyroid hormone receptor subtypes with about the same affinity; however, modification of CO23 by derivatization of the 3’ position of the outer-ring or replacement of the inner-ring iodides with bromides attenuates binding. Despite lacking a preference in binding to TRα, all analogs display TRα-specificity in transactivation assays using U2OS and HeLa cells. At best, several agonists exhibit an approximately 6−12 fold preference in transactivation when tested with TRα in HeLa cells. One analog, CO24, showed in vivo TRα-specific action in a tadpole metamorphosis assay.
Introduction
Thyroid hormone is a classic endocrine signaling hormone that mediates a wide variety of regulatory events affecting growth, development, and metabolism1-3. In circulation, thyroid hormone exists as a pro-hormone, 3,5,3’,5’-tetraiodo-L-thyronine (T4, Fig. 1), but is converted to its principal active form, 3,5,3’-triiodo-L-thyronine (T3, Fig. 1), by deiodination of one outer-ring position by deiodinases4-5. T3 exerts its actions by translocating into the nucleus of target cells and binding to the ligand binding domain (LBD) of thyroid hormone receptors (TRs) which are members of the nuclear receptor superfamily of ligand responsive transcriptional regulators4. There are two genes for TR, TRα and TRβ, that give rise to an ensemble of four different isoforms by means of alternative splicing or differential promoter usage: TRα1, TRα2, TRβ1, and TRβ24,6. Ligand binding to TR induces a conformational change in the LBD allowing it to induce or repress gene expression by recruitment of coactivator or corepressor proteins4. Selective thyromimetics are T3 analogs that unlike T3, have tissue selective actions1,2. A current guiding hypothesis is that TR subtype selectivity may correlate with tissue selective actions and TRβ-selective compounds such as GC-1 (Fig. 1) are being developed as potential therapeutic agents for hyperlipidemia and obesity. Until recently, little success had beeen reported on the development of TRα-selective thyromimetics.
Figure 1.

Structures of T3, T4, CO23, and GC-1.
We recently reported on the synthesis and characterization of CO23, the first potent thyromimetic with TRα-specific effects in vitro and in vivo. This compound demonstrated 3 to 5-fold TRα-specificity in transactivation assays using U2OS and HeLa cells respectively7. Despite not having an overwhelming preference for TRα activation, CO23 has profound effects on precocious X. laevis tadpole metamorphosis that correlates with the selective activation of TRα. In this study, we have prepared a focused panel of CO23 analogs and evaluated them for TRα selectivity in vitro and in vivo.
Chemistry
The hydantoin moiety attached to position one of the inner-ring by a methylene linker was deemed necessary for conferring TRα-specificity7, and hence to improve TRα-specificity, additional modifications to inner- and outer-ring substituents were examined. Substitution of the outer-ring was achieved by preparation or purchase of para-brominated phenols with varying groups in the ortho-position followed by TIPS protection (Scheme 1). One of the rare para-brominated phenols was generated by protection of 2-bromophenol (1) using allyl bromide which gives rise to allyl 2-bromophenyl ether (2). Lithiation of 2 causes it to undergo an intramolecular carbolithiation/1,3-elimination reaction that gives rise to 2-cyclopropylphenol (3a)8. After generation of TIPS protected bromophenols (4a-4f), they were all converted to boronic acids (5a-5f) by treatment with n-butyllithium followed by addition of triisopropylborate and 3N hydrochloric acid and used at a later stage for the generation of biaryl ethers (Scheme 2)7.
Scheme 1.

Synthetic route used for the synthesis of TIPS-protected, 4-hydroxyphenyl boronic acid.
Scheme 2.

Synthesis of CO24 and CO26-CO32
At this stage, another level of thyromimetic diversity is achieved by starting with either diiodo-L-tyrosine or dibromo-L-tyrosine (6a-6b) and converting them to N-Boc-3,5-dihalo-L-tyrosine methyl esters (7a-7b)7. 7a-7b and boronic acids 5a-5f were coupled under Evan's conditions using cupric acetate as a catalyst leading to biaryl ethers (8a-8f)7. Amidation of 8a-8f in methanol saturated with ammonia gas and Boc-deprotection yields biaryl ethers with amino acid amide side chains (9a-9f)7. This side chain undergoes cyclization to form the imidazolidinedione after treatment with para-nitrophenylchloroformate, sodium bicarbonate, and water7. Deprotection of the TIPS groups with tetrabutylammonium fluoride leads to several CO23 analogs (CO24 and CO26-CO30). The iodination9 or bromination7 of the 3’ position of CO30 leads to two further analogs, CO31 and CO32.
Biological Evaluation
The biological activity of the aforementioned CO23 analogs was measured in vitro using 125I-T3 competitive binding and transactivation assays. Replacement of inner-ring iodides with bromides causes a ∼10-fold decrease in binding (CO24 vs. CO23). TR ligand activation in U2OS cells (Table 1) showed that CO24 was not TRα-specific compared to the T3 control. In U2OS cells it is important to compare the potencies of test ligands to that of T3 as thyroid hormone shows a difference in activation of TRα1 and TRβ1 using a synthetic thyroid hormone response element driven luciferase reporter construct. However, in HeLa cells, a cell line where thyroid hormone consistently shows equal activation of both TR subtypes, not only does CO24 show four-fold TRα-selectivity in potency, it is TRα-selective in terms of efficacy in that it causes transcriptional activity to plateau at a level that is twice as high as T3 (Table 1 and Fig. 2a). In vivo, X. laevis tadpoles precociously induced to undergo metamorphosis revealed gross morphological changes compared to untreated tadpoles, some of which are consistent with enhanced TRα activity. Tadpoles treated with 30 nM T3 and 300 nM CO24 both experienced resorption of tissue in the head and tail, experienced an overall decrease in size, and developed Meckel's cartilage (lower jaw); however, CO24 treated tadpoles exhibited massive hind leg and fore leg development, a noticeably larger body size, and less resorption of tissue in the head compared to T3 treated tadpoles (Fig. 3). Compared to CO23 induced metamorphosis, CO24 had similar affects on tadpole metamorphosis with some exceptions. For example, both CO24 and CO23 treated tadpoles developed more massive fore and hind limbs than T3 treated tadpoles, but tadpoles treated with CO24 developed the most massive limbs overall. Furthermore, CO24 treated tadpoles experienced slightly greater resorption of larval tissue compared to CO23 treated tadpoles, particularly in the head and tail, but this may be due to a slight decrease in TRα-specificity compared to CO237.
Table 1.
Binding affinity and potency of CO23 analogues
|
Kd and EC50 values (nM) |
||||||
|---|---|---|---|---|---|---|
|
Binding affinity (Kd)a |
Transactivation in U2OS cells (EC50)b |
Transactivation in HeLa cells (EC50)b |
||||
| Compound | TRα | TRβ | TRα | TRβ | TRα | TRβ |
| T3c | 0.058 | 0.081 | 2.4 ± 0.4 | 11 ± 2 | 2.4 ± 0.5 | 2.4 ± 0.5 |
| CO23c | 1.2 ± 0.2 | 1.7± 0.3 | 34 ± 4 | 390 ± 3 | 11 ± 1 | 58 ± 1 |
| CO24 | 17 ± 1 | 18.4 ± 1 | 128 ± 5 | 421 ± 3 | 8.4 ± 3 | 32 ± 3 |
| CO26 | 82 ± 21 | 119 ± 30 | 870 ± 160 | 8000 ± 1800 | 42 ± 6 | 265 ± 10 |
| CO27 | 42 ± 13 | 53 ± 17 | 146 ± 28 | 4000 ± 1000 | 27 ± 6 | 180 ± 30 |
| CO28 | 15 ± 6 | 18 ± 2 | 87 ± 14 | 1400 ± 170 | 10 ± 1 | 27 ± 6 |
| CO29 | 25 ± 7 | 49 ± 5 | 145 ± 10 | 2000 ± 290 | 39 ± 4 | 100 ± 14 |
| CO30 | 1700 ± 10 | 2000 ± 360 | >20000d | >20000d | >10000d | >10000d |
| CO31 | 24 ± 3 | 36 ± 1 | 105 ± 8 | 2000 ± 170 | 21 ± 3 | 216 ± 80 |
| CO32 | 43 ± 6 | 68 ± 10 | 206 ± 41 | 5200 ± 670 | 44 ± 1 | 530 ± 170 |
Determined by means of an 125I-T3 competitive binding assay and data is reported as the mean Kd ± standard error of the mean, n=3.
Determined through use of a TRE-driven dual-luciferase reporter assay in U2OS or HeLa cells and the data is reported as the mean EC50 value ± standard error of the mean, n=3.
See reference 36.
Dose-response curves generated using CO30 in transactivation assays did not plateau, and hence the EC50 value is an approximated value based on extrapolation.
Figure 2.

TRE-driven dual-luciferase reporter assays showing transactivation curves for T3, (a) CO24, (b) CO26, and (c) CO32 against hTRα1 and hTRβ1 in HeLa cells. Plots show mean of triplicates with s.d.
Figure 3.
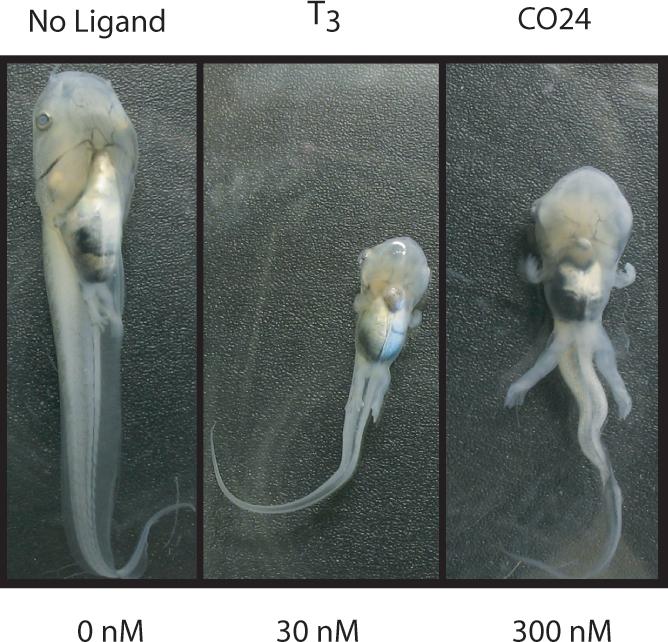
In vivo analysis of CO24. Induced metamorphosis of stage-53/54 tadpoles, n=3, treated for 4-days with DMSO, 30 nM T3, and 300 nM CO24.
CO23 analogs with outer-ring substitutions all displayed a decrease in binding affinity and potency when tested in U2OS cells compared to CO23 (Table 1). In U2OS cells, the potency of transactivation of the following substituents against TRα decreases from left to right: Ethyl > iodo > cyclopropyl > methyl > bromo > methoxy > hydrogen (Table 1). Unlike CO23, displaying only modest transcriptional activity in U2OS cells, they activate in the presence of TRβ very poorly. The best outer-ring analogue had an EC50 value of 1.4 μM compared to 390 nM for CO23 when tested in U2OS cells in the presence of TRβ (Table 1).
Like CO24, in order to get a more direct measure of TRα-specificity, all analogs were assayed in HeLa cells. With the exception of CO30-only because it's lack of activity in both U2OS and HeLa cells made it impossible to calculate and compare EC50 values from their dose-response curves-all analogs demonstrated TRα-specificity with a few proving superior to CO23. CO26, CO27, CO31, and CO32 demonstrated 6-, 6-, 10- and 12-fold TRα-specificity respectively, all of which are an improvement over CO23's 5 fold TRα-specificity. Dose-response curves for CO26 and CO32 showing transactivation in the presence of TRα and TRβ clearly demonstrate TRα-specificity despite the compounds' inferior potencies compared to CO23 (Figs. 2b and 2c). The rank order potency from left to right in HeLa cells is also impressive as some compounds are about equipotent to CO23-mediated TRα transactivation: Ethyl > iodo > methyl > cyclopropyl > methoxy > bromo > hydrogen.
Discussion
CO23, the first potent thyromimetic to demonstrate TRα-specificity in vitro and in vivo7, was derivatized at the 3, 5, and 3’ positions in order to generate thyromimetics with enhanced TRα-specificity. Small substituents with varying electronic properties were selected to be placed on the 3’ position as the structure activity data correlate with a decrease in potency with substituents larger than an isopropyl group5. In terms of the 3 and 5 positions, bromides were selected as previous studies show a dramatic decrease in potency with methyl groups on the inner-ring7. In HeLa cells, four of eight ligands showed greater TRα-specificity with CO32 achieving greater than a twofold gain in specificity. Although all analogs bound poorly to both TRα1 and TRβ1 and with about equal binding affinity to both receptor subtypes, analogs CO24 and CO28 were about equipotent to CO23 in their ability to cause TRα-mediated transcription. This phenomenon brings attention to a mode of subtype specificity which has gone unnoticed for some time. Analogs reported herein are not the first to demonstrate functional specificity. Considering that there is only one amino acid side chain difference in the TR LBD, Ser277 (TRα) to Asn331 (TRβ), the similarity in affinity is not surprising10. However, previous studies of related estrogen receptors demonstrate that subtle differences in the induced conformations of amino acid side chains may result in this selectivity11. In this case, the same ligand may cause minor differences in one receptor LBD that causes amino acid side chains to perturb the conformationally mobile helix-12, an important mediator of coactivator recruitment, gene regulation, and potentiation of interactions with the transcriptional machinery10.
In vivo, CO24 led to changes that are consistent with enhanced TRα-activity compared to T3 treated controls, particularly as demonstrated by the massive hind and fore leg emergence in precociously induced X. laevis tadpoles. The inner-ring bromides probably confer resistance to dehalogenation by deiodinases, and therefore provide one explanation for CO24's potency and efficacy in vivo and in vitro. Although it is a leap to suggest from studies on amphibian metamorphosis that these analogs would have beneficial effects in mammals and possibly humans, other studies with TRα and cardiac-selective ligands suggest that TRα-specific thyromimetics may have therapeutic utility in the area of heart disease. Finally, TRα-specific thyromimetics, like their predecessor TRβ-selective counterparts, may be useful probes of TR biology.
Experimental
General
All chemicals used for organic synthesis were purchased from Aldrich, Sigma-Aldrich, Fluka, or Acros and were used without further purification. Anhydrous conditions were maintained under argon using standard schlenk line techniques and oven-dried glassware. Anhydrous THF, DCM, pyridine, and diisopropyl ethylamine were available in house and dispensable from a solvent purification system. Compounds were purified by either flash chromatography using silica gel (VWR Scientific) or through preparatory thin layer chromatography (prep TLC) using Analtech prep-TLC plates (20 × 20 cm, 1000 μm). 1H NMR spectra were taken on the Varian Utility 400 MHz spectrometer in CDCl3 or DMSO-d6 solvents and chemical shifts were reported as δ (part per million) downfield of the internal control trimethylsilane (TMS) for all solvents. High resolution mass spectrometry (HRMS) using electrospray ionization was performed by the National Bio-Organic, Biomedical Mass Spectrometry Resource at UCSF. All compounds were at least 95% pure as determined by HPLC analysis using a Waters 2695 instrument and an Xterra 3.5 μM reverse-phase C18 2.1 × 50-mm column. HPLC grade Acetonitrile and H2O were purchased from Fisher.
125I-T3 Competitive Binding Assay
Full-length hTRα1 and hTRβ1 were expressed using a TNT T7 quick-coupled transcription translation system (Promega). Competition assays for binding of unlabeled T3 and CO23 analogs were performed using 1 nM 125I-T3 in a gel filtration binding assay as described12.
Transactivation Assay
Human bone osteosarcoma epithelial (U2OS) cells or human uterine cervix cancer (HeLa) cells (Cell Culture Facility, UCSF) were grown to ∼80% confluency in Dulbecco's modified Eagles (DME) /H-21, 4.5 g/liter glucose medium containing 10% newborn calf serum (NCS) or fetal bovine serum (FBS) respectively (both heat-inactivated), 2 mM glutamine, 50 units ml−1 penicillin, and 50 μg ml−1 streptomycin. Cells (∼1.5−2 × 106) were collected and resuspended in 0.5 ml of electroporation buffer (Dulbecco's phosphate-buffered saline containing 0.1% glucose and 10 mg/ml bioprene) with 1.5 μg of a TR expression vector full-length hTRα1-CMV or hTRβ1-CMV), 0.5 μg of pRL-TK constitutive Renilla luciferase reporter plasmid (Promega), 5 μg of a reporter plasmid containing a synthetic TR response element (DR-4) containing two copies of a direct repeat spaced by four nucleotides (AGGTCAcaggAGGTCA) cloned immediately upstream of a minimal thymidine kinase promoter linked to a luciferase coding sequence13. Cells were electroporated using a Bio-Rad gene pulser at 350 V and 960 microfarads in 0.4-cm cuvettes, pooled in DME/F-12 Ham's 1:1 without phenol red (U2OS) or DME/H-21 (HeLa) supplemented as above except that NCS and FBS were hormone stripped using dextrose-coated charcoal, and plated in 96-well (U2OS) or 12-well (HeLa) plates to a final density of 20,000 cells/well and 100,000 cells per well respectively. After a 2-h incubation period, compounds in 1% dimethyl sulfoxide (DMSO) were added to the cell culture medium in triplicate. After an additional 16-h incubation period, cells were harvested and assayed for luciferase activity using the Promega dual-luciferase kit (Promega) and an Analyst AD (Molecular Devices). Data normalized to the Renilla internal control were analyzed with GraphPad Prism, v4, using the sigmoid-dose response model to generate EC50 values; EC50 values were obtained by fitting data to the following equation: Y=Bottom + (Top-Bottom)/(1+10^((LogEC50-X)*HillSlope)).
Preparation of Chemicals
Stocks of T3 and CO23 analogs were prepared with DMSO at a concentration of 10 mM and stored at −20° C until use. All other chemicals were purchased from Sigma unless otherwise indicated. 0.1% aminobenzoic acid ethyl ester (Tricaine or MS222) was made fresh in sterile ddH2O and kept at 4° C for no longer than 1 week.
General Xenopus Laevis Tadpole Procedures
Xenopus laevis stage-53/54 tadpoles were purchased from NASCO, Inc. and staged according to Nieuwkoop and Faber14. Upon receipt, tadpoles were allowed to set over night at room temperature (18−25° C) in order to recover from shipping shock, after which half of the initial rearing water was replaced with 0.1× Marc's Modified Ringer's (MMR) buffer (10× solution consists of 100 mM NaCl (Fisher), 2 mM KCl (Fisher), 1 mM MgCl2, 2 mM CaCl2, 0.1 mM EDTA, and 5 mM Hepes, pH 7.8). Tadpoles were ultimately maintained in fresh 0.1× MMR buffer, changed every 2-days. After completion of experiments, live tadpoles were euthanized by treatment with 0.01% Tricaine, exposure to an ice-bath, and either fixed in phosphate-buffered saline containing 3.5% formalin or decapitated in order to ensure death. Animals were photographed with a Canon PowerShot A510 and images were processed with Adobe Photoshop CS, v8, and Adobe Illustrator CS, v11. All tadpole experiments were conducted in accordance with Institutional Animal Care and Use Committee approval (animal protocol #: A7228−23070−01).
Induced metamorphosis experiments
Stage-53/54 tadpoles were added to Extra-Deep petri dishes (Fisher) in triplicate containing 50 mL of 0.1× MMR buffer and vehicle or the appropriate concentration of ligand (T3 or CO23 analogue). The final DMSO concentration was 0.1%. Induced metamorphosis experiments were repeated at least threefold.
Chemistry
2-Cyclopropylphenol (3a)
To 2-bromophenol 1 (8 g, 46.2 mmol) in 100 ml of dimethylformamide at 0° C was added NaH (2.6 g of a 60% suspension, 64.7 mmol). The reaction mixture was stirred for about 10 min. after which allyl bromide was added dropwise. After 30 min., the reaction mixture was treated with water and extracted with diethyl ether. The organic portion was dried over MgSO4, filtered, and concentrated in vacuo to give the crude product, which was purified by flash chromatography (silica gel, hexane:ethyl acetate, 5:95) to give allyl 2-bromophenyl ether 2 (9.7 g, 45.5 mmol, 99%) as a white solid. This material was carried on to the next reaction to make 3a. A dry round-bottom flask was charged with 2 (11 g, 51.6 mmol) and 260 ml of anhydrous diethyl ether and stirred at −78° C. To this solution was added drop-wise 1.7 M tert-BuLi in hexanes (60.7 ml, 103.3 mmol) after which stirring commenced for 30 min. To this solution was added N,N,N’,N’-tetramethylethylenediamine (17 ml, 113.5 mmol) and stirring commenced for 45 min. before warming to room temperature. The reaction was allowed to stir overnight before addition of water. The aqueous phase was extracted with EtOAc. The combined organic layers were washed with water, brine, and 3 N HCl, dried over MgSO4, concentrated in vacuo, an purified by flash chromatography (silica gel, hexane:ethyl acetate, 20:80 ) to give 3a (5.1 g, 38.0 mmol, 74%) as a white solid. 1H NMR (CDCl3) δ 7.36 (d, J = 8 Hz, 1H), 7.06 (m, 2H), 6.85 (d, J = 8 Hz), 5.45 (s, 1H), 1.81 (m, 1H), 0.95 (m, 2H), 0.64 (m, 2H).
General procedure for preparation of TIPS-protected, 4-hydroxyphenyl boronic acids substituted at the 3-position (5a-5f)
2-Isopropylphenol 4 (10.0 g, 73.4 mmol) was added to a dry three-neck round-bottom flask fitted with an addition funnel and an exhaust line that runs into a base trap (6 M KOH). This solution was allowed to stir at 0° C after which Br2 (4.5 ml, 88.1 mmol) was added drop-wise over a period of 15 min. Stirring commenced for an additional 3-h before addition of sat. NaHCO3, water, and EtOAc. The aqueous phase was extracted with EtOAC and the combined organic layers were washed with brine and then dried over MgSO4. After concentration of the organic phase in vacuo, the crude product was purified by flash chromatography (silica gel, hexane:ethyl acetate, 10:90) to give a slightly yellow clear oil (11.8 g, 55.1 mmol, 75%). This material (5 g, 23.3 mmol) was combined with TIPS-Cl (5.9 ml, 27.8 mmol) in a dry round-bottom flask containing 50 ml of anhydrous DCM and stirred at 0° C. To this solution was added imidazole (3.9 g, 58 mmol) and stirring commenced for an additional 18-h. The next day, the reaction was quenched with water and the aqueous phase extracted with EtOAc. The combined organic layers were washed with brine, dried over MgSO4, concentrated in vacuo, and then purified with a short-path distillation column under high vacuum (0.5 mtorr). Pure fractions were collected at 130° C to give 4b as a clear white solid (6.9 g, 18.6 mmol, 80%). This material was carried on to make 5b. A dry round-bottom flask was charged with 4b (6.9 g, 18.6 mmol) and 100 ml of anhydrous THF and stirred at −78° C. To this solution was added drop-wise 2.5 M n-BuLi in hexanes (9.7 ml, 24.2 mmol) after which stirring commenced for 30 min. To this solution was added triisopropyl borate (8.7 ml, 37.2 mmol) and stirring commenced for 45 min. before warming to room temperature. After 1-h, the reaction was quenched with 3 N HCl and the aqueous phase extracted with EtOAc. The combined organic layers were washed with water and brine, dried over MgSO4, concentrated in vacuo, and purified by flash chromatography (silica gel, hexane:ethylacetate, 10−40%) to give 5b (5.5 g, 16.4 mmol, 88%) as a white solid.
General procedure for preparation of N-Boc-3,5-dihalo-L-tyrosine methyl esters (7a-7b)
L-diiodotyrosine (6a) (5 g, 11.5 mmol) was added to a round-bottom flask and dissolved in MeOH/H2O (2:1). To this mixture was added NaHCO3 (2.9 g, 34.5 mmol) followed by Boc2O (3.97 ml, 17.3 mmol). The reaction mixture was allowed to stir until completion as determined by TLC analysis (product should turn blue when tested with p-anisaldehyde). Upon completion, the mixture was acidified to pH 4.5 and extracted with EtOAc. The combined organic layers were washed with water and brine and then dried over MgSO4. The crude material (5.9 g, 11.1 mmol, 96%) was then utilized in the next reaction. To this material (5.9 g, 11.1 mmol) in toluene/MeOH (9:1) was added TMSCHN2 (0.5 M, 22 ml, 11.6 mmol) drop-wise over 30 min. at room temperature using a syringe pump. The reaction was allowed to stir until completion as determined by TLC analysis then washed with 0.5 M HCl and water. The aqueous phase was extracted with EtOAc and the organic layers washed with brine, dried over MgSO4, and concentrated in vacuo to give N-Boc-3,5-diiodo-L-tyrosine methyl ester (7a) (5.4 g, 9.9 mmol, 89%).
N-Boc-3,5-diiodo-L-tyrosine methyl ester
The preparation of 7a was effected using the general procedure for the preparation of Boc-protected dihalo-L-tyrosine methyl esters to give 5.4 g (89%) of the titled compound as a white solid. 1H NMR (CDCl3) δ 7.44 (s, 1H), 5.01 (s, 1H), 4.49 (dd, J = 4.0 Hz, J = 8.0 Hz, 1H), 3.74 (s, 3H), 3.00 (dd, J = 4.0 Hz, J = 14.0 Hz, 1H), 2.91 (dd, J = 8.0 Hz, J = 14.0 Hz, 1H), 1.45 (s, 9H).
N-Boc-3,5-dibromo-L-tyrosine methyl ester
The preparation of 7b was effected using the general procedure for the preparation of Boc-protected dihalo-L-tyrosine methyl esters to give 1.1 g (87%) of the titled compound as a white solid. 1H NMR (CDCl3) δ 7.22 (s, 1H), 5.02 (s, 1H), 4.50 (dd, J = 4.0 Hz, J = 8.0 Hz, 1H), 3.74 (s, 3H), 3.05 (dd, J = 4.0 Hz, J = 14.0 Hz, 1H), 2.93 (dd, J = 8.0 Hz, J = 14.0 Hz, 1H), 1.44 (s, 9H).
General procedure for the preparation of biary ethers (8a-8f)
4 Ǻ molecular sieves were flame dried under high vacuum in a dry round-bottom flask. To this flask was added boronic acid (3 mmol) and copper acetate (dried to a verdigris color). These components were dissolved in 10 ml anhydrous DCM after which anhydrous pyridine (5 mmol) and diisopropyl ethylamine (5 mmol) were added. This mixture was then allowed to stir at room temperature for 5 min. before addition of phenol (1 mmol) in three portions separated by 5 min. each. At this point, the flask was fitted with a drying tube containing drierite and allowed to stir under ambient air over night, ∼16−24-h. After this time, the reaction mixture was concentrated in vacuo and purified by flash chromatography to give products 8a-8f (yield generally from 55−83%).
General procedure for the preparation of 5-(4-(4-hydroxyphenoxy)-3,5-dihalobenzyl)imidazolidine-2,4-diones (CO24 and CO26-CO30)
Biaryl ether 8b (510 mg, 0.69 mmol) was dissolved in 20 ml MeOH and saturated with ammonia gas. After 16−18-h, the mixture was purged with argon, concentrated in vacuo, and dissolved in anhydrous 3 N HCl in EtOAc/Ether. After 3-h, the mixture was quenched with water, the pH was adjusted to 4.5, and the aqueous phase extracted with EtOAc. The organic layers were washed with water and brine, dried over MgSO4, and the crude material 9b (410 mg, 0.67 mmol, 99%) was carried on to make CO24. 9b (410 mg, 0.67 mmol), 4-nitrophenyl chloroformate (160 mg, 0.78 mmol), and NaHCO3 (218 mg, 2.6 mmol) were added to a dry round-bottom flask containing 10 ml anhydrous MeCN. The reaction was allowed to stir overnight followed by addition of 6.5 ml of H2O. The solution should quickly turn yellow due to generation of nitrophenol. After 6-h, the reaction mixture was roto-vapped in order to remove MeCN, acidified to pH 5, and then extracted with EtOAc. The combined organic layers were washed with brine, dried over MgSO4, and reconstituted in 10 ml THF. Deprotection of the TIPS group afforded CO24 (204 mg, 0.41 mmol, 63%, 2-steps from 9b) after purification by prep TLC (silica gel, hexane:ethyl acetate, 40:60).
Preparation of CO24
The preparation of CO24 was effected using the general procedure for the preparation of 5-(4-(4-hydroxyphenoxy)-3,5-dihalobenzyl)imidazolidine-2,4-diones to give 204 mg (63%, 2-steps from 9b) of the titled compound as a white solid. 1H NMR (DMSO-d6) δ 10.65 (s, 1H), 9.06 (s, 1H), 7.99 (s, 1H), 7.58 (s, 2H), 6.64 (d, J = 8.0 Hz, 1H), 6.63 (d, J = 4.0 Hz, 1H), 6.20 (dd, J = 4.0 Hz, J = 8.0 Hz, 1H), 4.39 (dd, J = 4.0 Hz, J = 8.0 Hz, 1H), 3.15 (heptet, J = 8.0 Hz, 1H), 2.99 (dd, J = 4.0Hz, J = 14.0Hz, 1H), 2.87 (dd, J = 8.0Hz, J = 14.0Hz, 1H), 1.10 (d, J = 8.0 Hz, 6H). HPLC (MeCN/water, 50−100%, 15 min.): retention time 3.1 min. HR-MS calcd for C19H18Br2N2O4: 497.9613. Found: 497.9607.
Preparation of CO26
The preparation of CO26 was effected using the general procedure for the preparation of 5-(4-(4-hydroxyphenoxy)-3,5-dihalobenzyl)imidazolidine-2,4-diones to give 80 mg (41%, 2-steps from 9e) of the titled compound as a white solid. 1H NMR (DMSO-d6) δ 10.61 (s, 1H), 8.65 (s, 1H), 7.99 (s, 1H), 7.75 (s, 2H), 6.64 (d, J = 8.0 Hz, 1H), 6.50 (d, J = 4.0 Hz, 1H), 5.90 (dd, J = 4.0 Hz, J = 8.0 Hz, 1H), 4.37 (dd, J = 4.0 Hz, J = 8.0 Hz, 1H), 3.72 (s, 3H), 2.94 (dd, J = 4.0Hz, J = 14.0Hz, 1H), 2.81 (dd, J = 8.0Hz, J = 14.0Hz, 1H). HPLC (MeCN/water, 50−100%, 15 min.): retention time 1.8 min. HR-MS calcd for C17H14I2N2O5: 579.8992. Found: 579.9004.
Preparation of CO27
The preparation of CO27 was effected using the general procedure for the preparation of 5-(4-(4-hydroxyphenoxy)-3,5-dihalobenzyl)imidazolidine-2,4-diones to give 50 mg (25%, 2-steps from 9c) of the titled compound as a white solid. 1H NMR (DMSO-d6) δ 10.65 (s, 1H), 9.09 (s, 1H), 7.97 (s, 1H), 7.77 (s, 2H), 6.66 (d, J = 8.0 Hz, 1H), 6.48 (d, J = 4.0 Hz, 1H), 6.26 (dd, J = 4.0 Hz, J = 8.0 Hz, 1H), 4.40 (dd, J = 4.0 Hz, J = 8.0 Hz, 1H), 2.94 (dd, J = 4.0Hz, J = 14.0Hz, 1H), 2.81 (dd, J = 8.0Hz, J = 14.0Hz, 1H), 2.07 (s, 3H). HPLC (MeCN/water, 50−100%, 15 min.): retention time 1.9 min. HR-MS calcd for C17H14I2N2O4: 563.9043. Found: 563.9045.
Preparation of CO28
The preparation of CO28 was effected using the general procedure for the preparation of 5-(4-(4-hydroxyphenoxy)-3,5-dihalobenzyl)imidazolidine-2,4-diones to give 91 mg (35%, 2-steps from 9d) of the titled compound as a white solid. 1H NMR (DMSO-d6) δ 10.60 (s, 1H), 8.94 (s, 1H), 7.98 (s, 1H), 7.75 (s, 2H), 6.64 (d, J = 8.0 Hz, 1H), 6.53 (d, J = 4.0 Hz, 1H), 6.24 (dd, J = 4.0 Hz, J = 8.0 Hz, 1H), 4.36 (dd, J = 4.0 Hz, J = 8.0 Hz, 1H), 3.16 (q, 2H), 2.95 (dd, J = 4.0Hz, J = 14.0Hz, 1H), 2.85 (dd, J = 8.0Hz, J = 14.0Hz, 1H), 1.11 (t, 3H). HPLC (MeCN/water, 50−100%, 15 min.): retention time 2.1 min. HR-MS calcd for C18H16I2N2O4: 577.9199. Found: 577.9198.
Preparation of CO29
The preparation of CO29 was effected using the general procedure for the preparation of 5-(4-(4-hydroxyphenoxy)-3,5-dihalobenzyl)imidazolidine-2,4-diones to give 1.0 g (67%, 2-steps from 9a) of the titled compound as a white solid. 1H NMR (DMSO-d6) δ 10.60 (s, 1H), 9.00 (s, 1H), 7.98 (s, 1H), 7.74 (s, 2H), 6.62 (d, J = 8.0 Hz, 1H), 6.24 (d, J = 4.0 Hz, 1H), 6.13 (dd, J = 4.0 Hz, J = 8.0 Hz, 1H), 4.37 (dd, J = 4.0 Hz, J = 8.0 Hz, 1H), 3.17 (m, 1H), 2.94 (dd, J = 4.0Hz, J = 14.0Hz, 1H), 2.85 (dd, J = 8.0Hz, J = 14.0Hz, 1H), 0.84 (m, 2H), 0.53 (m, 2H). HPLC (MeCN/water, 50−100%, 15 min.): retention time 2.0 min. HR-MS calcd for C19H16I2N2O4: 589.9199. Found: 589.9209.
Preparation of CO30
The preparation of CO30 was effected using the general procedure for the preparation of 5-(4-(4-hydroxyphenoxy)-3,5-dihalobenzyl)imidazolidine-2,4-diones to give 1.3 g (65%, 2-steps from 9f) of the titled compound as a white solid. 1H NMR (DMSO-d6) δ 10.61 (s, 1H), 9.09 (s, 1H), 7.98 (s, 1H), 7.76 (s, 2H), 6.67 (d, J = 8.0 Hz, 2H), 6.51 (d, J = 8.0 Hz, 2H), 4.36 (dd, J = 4.0 Hz, J = 8.0 Hz, 1H), 2.95 (dd, J = 4.0Hz, J = 14.0Hz, 1H), 2.84 (dd, J = 8.0Hz, J = 14.0Hz, 1H). HPLC (MeCN/water, 50−100%, 15 min.): retention time 1.7 min. HR-MS calcd for C16H12I2N2O4: 549.8886. Found: 549.8900.
Preparation of 5-(4-(4-hydroxy-3-iodophenoxy)-3,5-diiodobenzyl)imidazolidine-2,4-dione (CO31)
5-(4-(4-hydroxyphenoxy)-3,5-diiodobenzyl)imidazolidine-2,4-dione (CO30, 100 mg, 0.2 mmol) in 0.2 ml of MeOH was added to a round-bottom flask at −5° C and dissolved in 5 ml of a 70% solution of aqueous ethylamine. To this mixture was added drop-wise Iodine (I2) as a 1N aqueous solution saturated with KI (0.24 ml, 0.24 mmol). After 6-h, the reaction was acidified to pH 4.5, extracted with EtOAc, concentrated in vacuo, and purified by flash chromatography (silica gel, hexane:ethyl acetate, 40:60) to give CO31 (85 mg, 0.13 mmol, 62%). 1H NMR (DMSO-d6) δ 10.62 (s, 1H), 9.97 (s, 1H), 8.00 (s, 1H), 7.77 (s, 2H), 7.02 (d, J = 4.0 Hz, 1H), 6.81 (d, J = 8.0 Hz, 1H), 6.58 (dd, J = 4.0 Hz, J = 8.0 Hz, 1H), 4.37 (dd, J = 4.0 Hz, J = 8.0 Hz, 1H), 2.95 (dd, J = 4.0Hz, J = 14.0Hz, 1H), 2.84 (dd, J = 8.0Hz, J = 14.0Hz, 1H). HPLC (MeCN/water, 50−100%, 15 min.): retention time 2.1 min. HR-MS calcd for C16H11I3N2O4: 675.7853. Found: 675.7877.
Preparation of 5-(4-(4-hydroxy-3-iodophenoxy)-3,5-diiodobenzyl)imidazolidine-2,4-dione (CO32)
5-(4-(4-hydroxyphenoxy)-3,5-diiodobenzyl)imidazolidine-2,4-dione (CO30, 100 mg, 0.2 mmol) was added to a round-bottom flask and dissolved in 2 ml of DCM and 0.25 ml of glacial acetic acid at 0° C. To this mixture was added drop-wise bromine (12.3 μl, 0.24 mmol) in 1 ml of DCM. After 1-h, the reaction was extracted with EtOAc, concentrated in vacuo, and purified by flash chromatography (silica gel, hexane:ethyl acetate, 40:60) to give CO32 (109 mg, 0.18 mmol, 88%). 1H NMR (DMSO-d6) δ 10.61 (s, 1H), 9.90 (s, 1H), 7.98 (s, 1H), 7.76 (s, 2H), 6.87 (d, J = 8.0 Hz, 1H), 6.82 (d, J = 4.0 Hz, 1H), 6.55 (dd, J = 4.0 Hz, J = 8.0 Hz, 1H), 4.36 (dd, J = 4.0 Hz, J = 8.0 Hz, 1H), 2.96 (dd, J = 4.0Hz, J = 14.0Hz, 1H), 2.79 (dd, J = 8.0Hz, J = 14.0Hz, 1H). HPLC (MeCN/water, 50−100%, 15 min.): retention time 2.0 min. HR-MS calcd for C16H11BrI2N2O4: 627.7992. Found: 627.7981.
Acknowledgements
We would like to thank Professor J. David Furlow, Eric Neff, and Cindy Chen for their advice, guidance, and taking time to critically analyze the Xenopus laevis induced metamorphosis experiments. We are also grateful to Suzana T. Cunha Lima, Ph.D., for her technical expertise with the 125I-T3 competitive binding assay. Finally, we are grateful to the NIH (DK-52798, T.S.S.) and the Ford Foundation for financial support.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Legends Graphical Table of Contents. CO23 displays TRα-specificity in vitro and in vivo. CO23 analogs with greater specificity may prove superior to CO23 in treating heart disease or as a pharmacological probe of TR biology.
References
- 1.Scanlan TS, Yoshihara H, Nguyen N-H, Chiellini G. Curr. Opin. Drug Discov. Devel. 2001;4:614. [PubMed] [Google Scholar]
- 2.Ocasio CA, Scanlan TS. Curr. Opin. Endocrinol. Diabetes. 2005;12:363. [Google Scholar]
- 3.Morkin E, Ladenson P, Goldman S, Adamson CJ. Mol. Cell. Cardiol. 2004;37:1137. doi: 10.1016/j.yjmcc.2004.09.013. [DOI] [PubMed] [Google Scholar]
- 4.Greenspan FS, Gardner DG. In: In Basic and Clinical Endocrinology. 6th ed. Greenspan FS, editor. Lange Medical Books/McGraw-Hill; New York: 2001. [Google Scholar]
- 5.Jorgensen E. In: In Hormonal Proteins and Peptides. Li CH, editor. Academic Press; New York: 1978. [Google Scholar]
- 6.Adamson C, Maitra N, Bahl J, Greer K, Klewer S, Hoying J, Morkin JJ. Pharmacol. Exp. Ther. 2004;311:164. doi: 10.1124/jpet.104.069153. [DOI] [PubMed] [Google Scholar]
- 7.Ocasio CA, Scanlan TS. ACS Chem. Biol. 2006;1:585. doi: 10.1021/cb600311v. [DOI] [PubMed] [Google Scholar]
- 8.Barluenga J, Fañanás FJ, Sanz R, Marcos C. Chemistry - A European Journal. 2005;11:5397. doi: 10.1002/chem.200500377. [DOI] [PubMed] [Google Scholar]
- 9.Hart M, Suchland K, Miyakawa M, Bunzow J, Grandy D, Scanlan TS. J. Med. Chem. 2006;49:1101. doi: 10.1021/jm0505718. [DOI] [PubMed] [Google Scholar]
- 10.Peterson BR. ACS Chem. Biol. 2006;1:559. doi: 10.1021/cb600398a. [DOI] [PubMed] [Google Scholar]
- 11.Shiau AK, Barstad D, Radek JT, Meyers MJ, Nettles KW, Katzenellenbogen BS, Katzenellenbogen JA, Agard DA, Greene GL. Nat. Struct. Biol. 2002;9:359. doi: 10.1038/nsb787. [DOI] [PubMed] [Google Scholar]
- 12.Apriletti J, Baxter J, Lau K, West B. Protein Express. Purif. 1995;6:363. doi: 10.1006/prep.1995.1048. [DOI] [PubMed] [Google Scholar]
- 13.Chiellini G, Apriletti J, Yoshihara H, Baxter J, Ribeiro R, Scanlan TS. Chem. Biol. 1998;5:299. doi: 10.1016/s1074-5521(98)90168-5. [DOI] [PubMed] [Google Scholar]
- 14.Nieuwkoop PD, Faber J. In Normal Table of Xenopus Laevis (Daudin): A Systematical and Chronological Survey of the Development From the Fertilized Egg Till the End of Metamorphosis. 2nd ed. Garland Publishing; New York and London: 1994. [Google Scholar]